
Beyond Low Prevalence: Exploring Antibiotic Resistance and Virulence Profiles in Sri Lankan Helicobacter pylori with Comparative Genomics

 , ,
, ,  ,
,  , , , ,
, , , ,  , , and
, , and 
Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Collection and Bacterial Strains
2.2. Next-Generation Sequencing and H. pylori Virulence Factors
2.3. Comparative Genomic Analysis
2.4. Adherence Test and Hummingbird Phenotypes
2.5. Antibiotic Susceptibility Test
3. Results
3.1. H. pylori Infection and Gastric Inflammation Severity
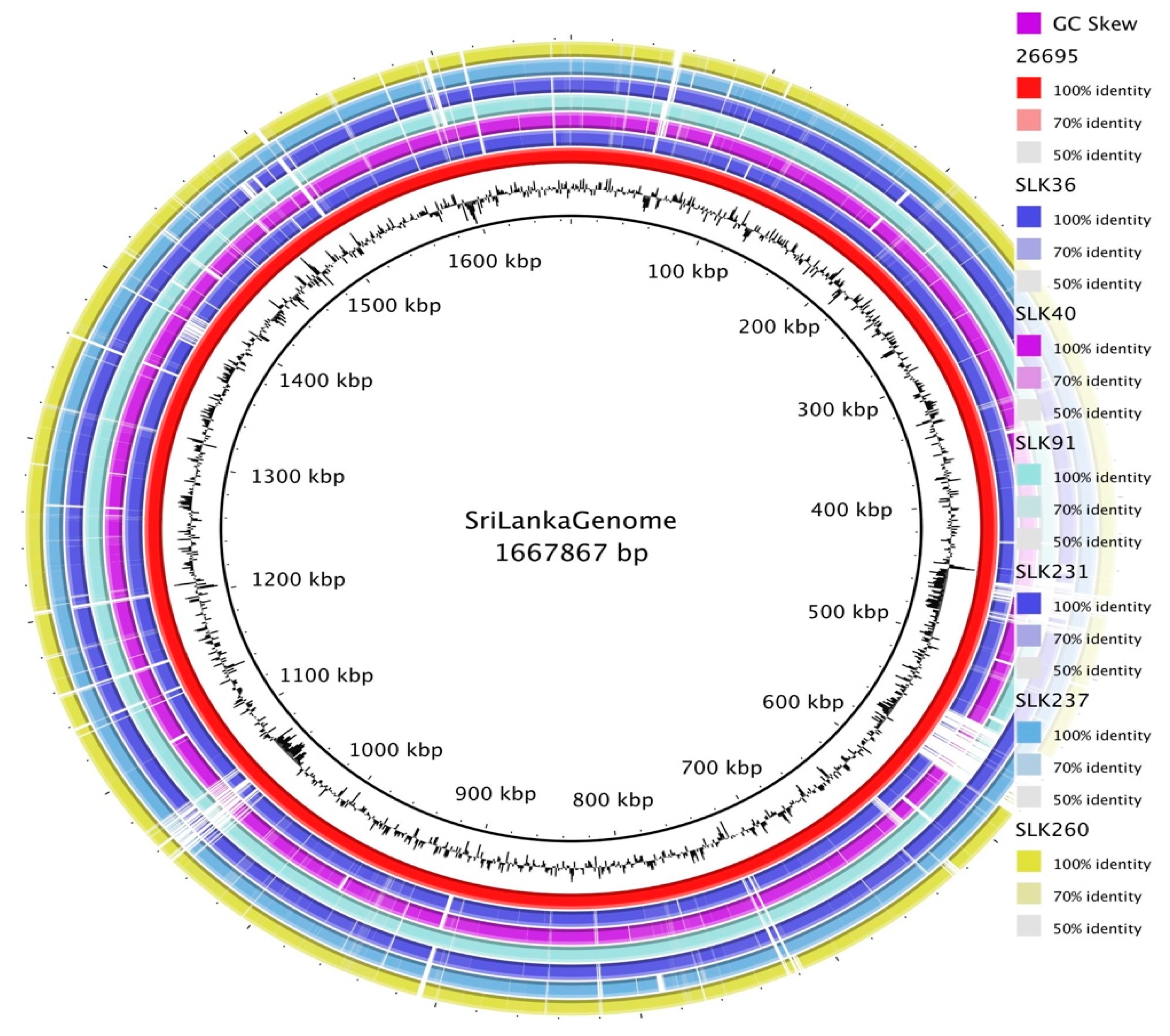
3.2. General Genomic Features of Sri Lankan Strains
3.3. Pan-Genome Analysis of Sri Lankan Strains
3.4. Presence of H. pylori Virulence Factors and Genotypes
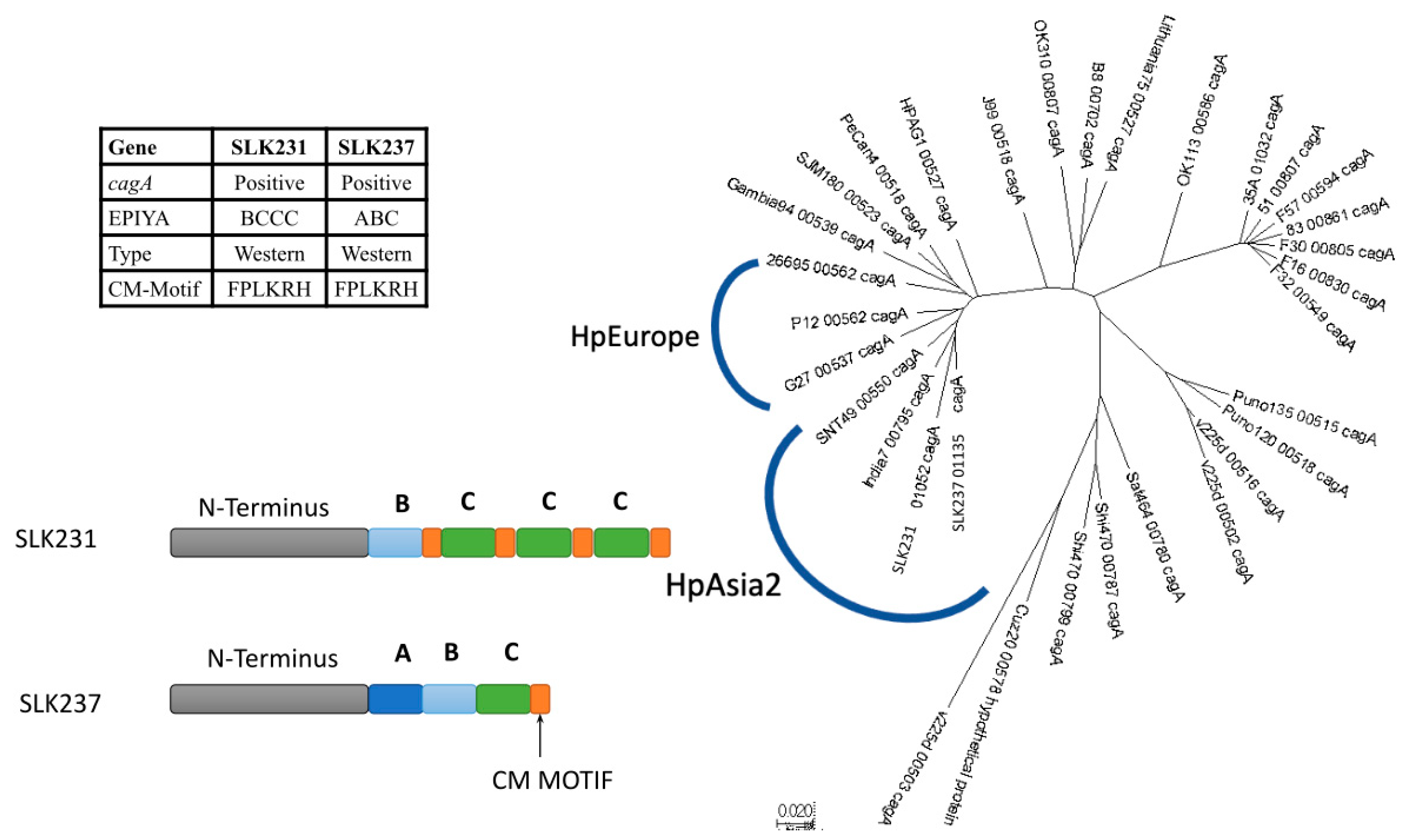
3.5. CagA Genotype Specific to Strains from South Asia
3.6. Other Virulence Determinants: Screening for Plasmids, Phages, and Secretion Systems
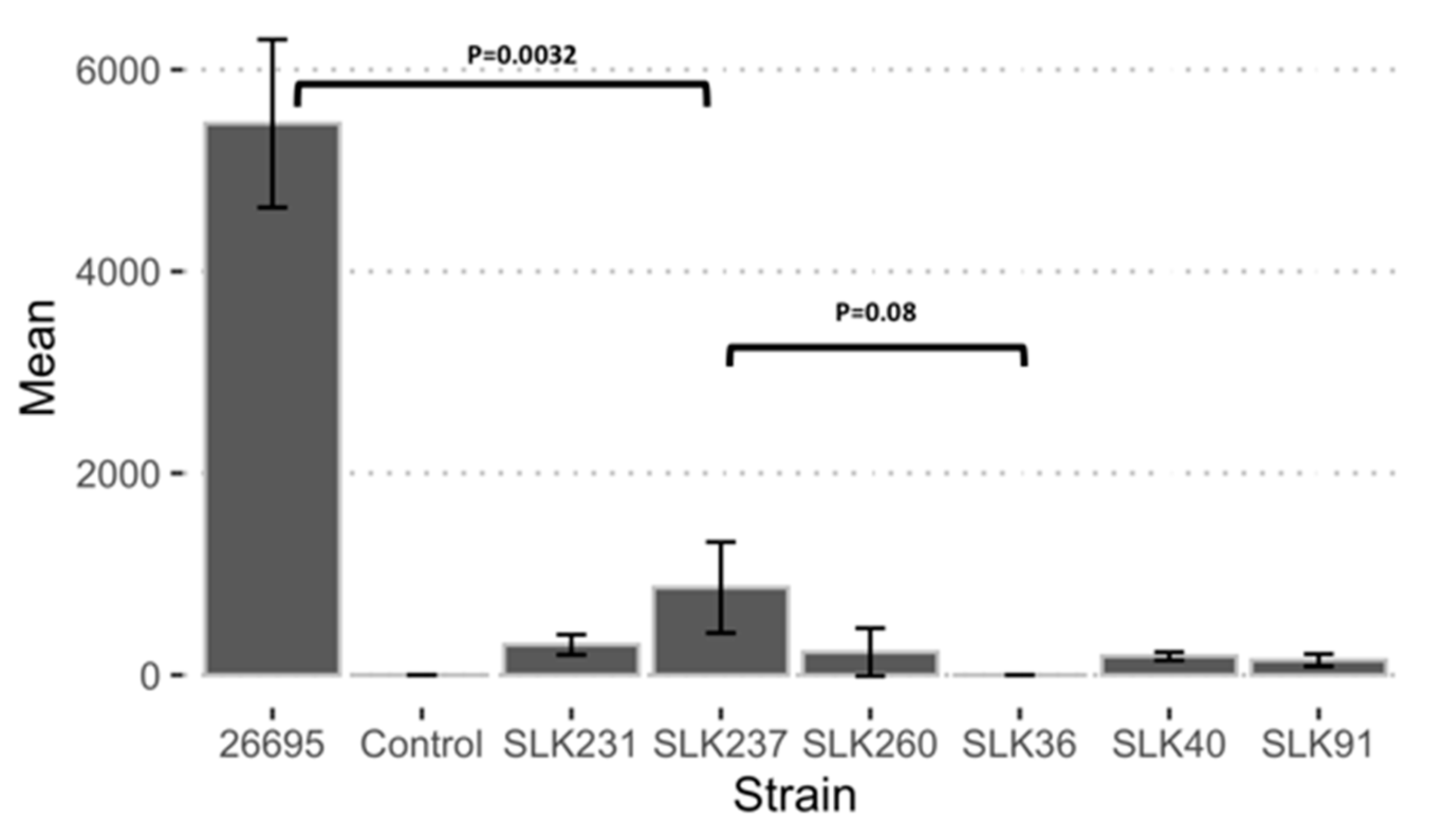
3.7. Adherence Ability of Sri Lankan Strains
3.8. Antibiotic Susceptibility Test Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Mera, R.M.; Bravo, L.E.; Camargo, M.C.; Bravo, J.C.; Delgado, A.G.; Romero-Gallo, J.; Yepez, M.C.; Realpe, J.L.; Schneider, B.G.; Morgan, D.R.; et al. Dynamics of Helicobacter pylori infection as a determinant of progression of gastric precancerous lesions: 16-year follow-up of an eradication trial. Gut 2018, 67, 1239–1246. [Google Scholar] [CrossRef] [PubMed]
- Correa, P.; Houghton, J. Carcinogenesis of Helicobacter pylori. Gastroenterology 2007, 133, 659–672. [Google Scholar] [CrossRef] [PubMed]
- Kodaman, N.; Pazos, A.; Schneider, B.G.; Piazuelo, M.B.; Mera, R.; Sobota, R.S.; Sicinschi, L.A.; Shaffer, C.L.; Romero-Gallo, J.; de Sablet, T.; et al. Human and Helicobacter pylori coevolution shapes the risk of gastric disease. Proc. Natl. Acad. Sci. USA 2014, 111, 1455–1460. [Google Scholar] [CrossRef]
- Correa, P.; Piazuelo, M.B. Evolutionary History of the Helicobacter pylori Genome: Implications for Gastric Carcinogenesis. Gut Liver 2012, 6, 21–28. [Google Scholar] [CrossRef]
- de Sablet, T.; Piazuelo, M.B.; Shaffer, C.L.; Schneider, B.G.; Asim, M.; Chaturvedi, R.; Bravo, L.E.; Sicinschi, L.A.; Delgado, A.G.; Mera, R.M.; et al. Phylogeographic origin of Helicobacter pylori is a determinant of gastric cancer risk. Gut 2011, 60, 1189–1195. [Google Scholar] [CrossRef]
- Hooi, J.K.Y.; Lai, W.Y.; Ng, W.K.; Suen, M.M.Y.; Underwood, F.E.; Tanyingoh, D.; Malfertheiner, P.; Graham, D.Y.; Wong, V.W.S.; Wu, J.C.Y.; et al. Global Prevalence of Helicobacter pylori Infection: Systematic Review and Meta-Analysis. Gastroenterology 2017, 153, 420–429. [Google Scholar] [CrossRef] [PubMed]
- Fauzia, K.A.; Tuan, V.P. Rising resistance: Antibiotic choices for Helicobacter pylori infection. Lancet Gastroenterol. Hepatol. 2024, 9, 7–8. [Google Scholar] [CrossRef] [PubMed]
- Shanks, A.M.; El-Omar, E.M. Helicobacter pylori infection, host genetics and gastric cancer. J. Dig. Dis. 2009, 10, 157–164. [Google Scholar] [CrossRef]
- Dai, J.; Zhao, J.; Mao, L.; Hu, Y.; Lv, B. Study on the value of antibiotic-resistant gene detection in Helicobacter pylori in China. Exp. Ther. Med. 2022, 23, 228. [Google Scholar] [CrossRef]
- Balloux, F.; Brønstad Brynildsrud, O.; van Dorp, L.; Shaw, L.P.; Chen, H.; Harris, K.A.; Wang, H.; Eldholm, V. From Theory to Practice: Translating Whole-Genome Sequencing (WGS) into the Clinic. Trends Microbiol. 2018, 26, 1035–1048. [Google Scholar] [CrossRef] [PubMed]
- Cross, A.S. What is a virulence factor? Crit. Care 2008, 12, 196. [Google Scholar] [CrossRef]
- Hatakeyama, M.; Higashi, H. Helicobacter pylori CagA: A new paradigm for bacterial carcinogenesis. Cancer Sci. 2005, 96, 835–843. [Google Scholar] [CrossRef] [PubMed]
- Ansari, S.; Yamaoka, Y. Survival of Helicobacter pylori in gastric acidic territory. Helicobacter 2017, 22, e12386. [Google Scholar] [CrossRef] [PubMed]
- Yamaoka, Y.; Ojo, O.; Fujimoto, S.; Odenbreit, S.; Haas, R.; Gutierrez, O.; El-Zimaity, H.M.; Reddy, R.; Arnqvist, A.; Graham, D.Y. Helicobacter pylori outer membrane proteins and gastroduodenal disease. Gut 2006, 55, 775–781. [Google Scholar] [CrossRef]
- Backert, S.; Tegtmeyer, N.; Fischer, W. Composition, structure and function of the Helicobacter pylori cag pathogenicity island encoded type IV secretion system. Future Microbiol. 2015, 10, 955–965. [Google Scholar] [CrossRef] [PubMed]
- Backert, S.; Haas, R.; Gerhard, M.; Naumann, M. The Helicobacter pylori Type IV Secretion System Encoded by the cag Pathogenicity Island: Architecture, Function, and Signaling. Curr. Top. Microbiol. Immunol. 2017, 413, 187–220. [Google Scholar] [CrossRef] [PubMed]
- Tshibangu-Kabamba, E.; Yamaoka, Y. Helicobacter pylori infection and antibiotic resistance—From biology to clinical implications. Nat. Rev. Gastroenterol. Hepatol. 2021, 18, 613–629. [Google Scholar] [CrossRef] [PubMed]
- Doohan, D.; Fauzia, K.A.; Rathnayake, J.; Lamawansa, M.D.; Waskito, L.A.; Tuan, V.P.; Dashdorj, A.; Kabamba, E.T.; Phuc, B.H.; Ansari, S.; et al. Pepsinogen and Serum IgG Detection Is a Valuable Diagnostic Method for Helicobacter pylori Infection in a Low-Prevalence Country: A Report from Sri Lanka. Diagnostics 2021, 11, 1364. [Google Scholar] [CrossRef] [PubMed]
- Stolte, M.; Meining, A. The updated Sydney system: Classification and grading of gastritis as the basis of diagnosis and treatment. Can. J. Gastroenterol. 2001, 15, 591–598. [Google Scholar] [CrossRef]
- Dixon, M.F.; Genta, R.M.; Yardley, J.H.; Correa, P. Classification and grading of gastritis. The updated Sydney System. International Workshop on the Histopathology of Gastritis, Houston 1994. Am. J. Surg. Pathol. 1996, 20, 1161–1181. [Google Scholar] [CrossRef] [PubMed]
- Uchida, T.; Kanada, R.; Tsukamoto, Y.; Hijiya, N.; Matsuura, K.; Yano, S.; Yokoyama, S.; Kishida, T.; Kodama, M.; Murakami, K.; et al. Immunohistochemical diagnosis of the cagA-gene genotype of Helicobacter pylori with anti-East Asian CagA-specific antibody. Cancer Sci. 2007, 98, 521–528. [Google Scholar] [CrossRef]
- Wick, R.R.; Judd, L.M.; Gorrie, C.L.; Holt, K.E. Unicycler: Resolving bacterial genome assemblies from short and long sequencing reads. PLoS Comput. Biol. 2017, 13, e1005595. [Google Scholar] [CrossRef] [PubMed]
- Tanizawa, Y.; Fujisawa, T.; Nakamura, Y. DFAST: A flexible prokaryotic genome annotation pipeline for faster genome publication. Bioinformatics 2018, 34, 1037–1039. [Google Scholar] [CrossRef] [PubMed]
- Gurevich, A.; Saveliev, V.; Vyahhi, N.; Tesler, G. QUAST: Quality assessment tool for genome assemblies. Bioinformatics 2013, 29, 1072–1075. [Google Scholar] [CrossRef]
- Chen, L.; Zheng, D.; Liu, B.; Yang, J.; Jin, Q. VFDB 2016: Hierarchical and refined dataset for big data analysis—10 years on. Nucleic Acids Res. 2016, 44, D694–D697. [Google Scholar] [CrossRef] [PubMed]
- Sayers, S.; Li, L.; Ong, E.; Deng, S.; Fu, G.; Lin, Y.; Yang, B.; Zhang, S.; Fa, Z.; Zhao, B. Victors: A web-based knowledge base of virulence factors in human and animal pathogens. Nucleic Acids Res. 2019, 47, D693–D700. [Google Scholar] [CrossRef]
- Kumar, S.; Nei, M.; Dudley, J.; Tamura, K. MEGA: A biologist-centric software for evolutionary analysis of DNA and protein sequences. Brief. Bioinform. 2008, 9, 299–306. [Google Scholar] [CrossRef]
- Carattoli, A.; Hasman, H. PlasmidFinder and in silico pMLST: Identification and typing of plasmid replicons in whole-genome sequencing (WGS). In Horizontal Gene Transfer; Springer: Berlin/Heidelberg, Germany, 2020; pp. 285–294. [Google Scholar]
- Arndt, D.; Grant, J.R.; Marcu, A.; Sajed, T.; Pon, A.; Liang, Y.; Wishart, D.S. PHASTER: A better, faster version of the PHAST phage search tool. Nucleic Acids Res. 2016, 44, W16–W21. [Google Scholar] [CrossRef] [PubMed]
- Siguier, P.; Pérochon, J.; Lestrade, L.; Mahillon, J.; Chandler, M. ISfinder: The reference centre for bacterial insertion sequences. Nucleic Acids Res. 2006, 34, D32–D36. [Google Scholar] [CrossRef]
- Stothard, P.; Wishart, D.S. Circular genome visualization and exploration using CGView. Bioinformatics 2005, 21, 537–539. [Google Scholar] [CrossRef]
- Page, A.J.; Cummins, C.A.; Hunt, M.; Wong, V.K.; Reuter, S.; Holden, M.T.; Fookes, M.; Falush, D.; Keane, J.A.; Parkhill, J. Roary: Rapid large-scale prokaryote pan genome analysis. Bioinformatics 2015, 31, 3691–3693. [Google Scholar] [CrossRef] [PubMed]
- Hadfield, J.; Croucher, N.J.; Goater, R.J.; Abudahab, K.; Aanensen, D.M.; Harris, S.R. Phandango: An interactive viewer for bacterial population genomics. Bioinformatics 2018, 34, 292–293. [Google Scholar] [CrossRef]
- Yahara, K.; Furuta, Y.; Oshima, K.; Yoshida, M.; Azuma, T.; Hattori, M.; Uchiyama, I.; Kobayashi, I. Chromosome painting in silico in a bacterial species reveals fine population structure. Mol. Biol. Evol. 2013, 30, 1454–1464. [Google Scholar] [CrossRef] [PubMed]
- Petersen, A.M.; Blom, J.; Andersen, L.P.; Krogfelt, K.A. Role of strain type, AGS cells and fetal calf serum in Helicobacter pylori adhesion and invasion assays. FEMS Immunol. Med. Microbiol. 2000, 29, 59–67. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.-H.; Tsai, W.-H.; Wu, H.-Y.; Chen, C.-Y.; Yeh, W.-L.; Chen, Y.-H.; Hsu, H.-Y.; Chen, W.-W.; Chen, Y.-W.; Chang, W.-W. Probiotic Lactobacillus spp. act against Helicobacter pylori-induced inflammation. J. Clin. Med. 2019, 8, 90. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.-C.; Kuo, W.-S.; Chen, Y.-C.; Perng, C.-L.; Lin, H.-J.; Ou, Y.-H. Fragmentation of CagA reduces hummingbird phenotype induction by helicobactor pylori. PLoS ONE 2016, 11, e0150061. [Google Scholar] [CrossRef] [PubMed]
- The European Committee on Antimicrobial Susceptibility Testing. European Committee on Antimicrobial Susceptibility Testing, Breakpoint Tables for Interpretation of MICs and Zone Diameters; EUCAST: Växjö, Sweden, 2015. [Google Scholar]
- Kumar, N.; Mukhopadhyay, A.K.; Patra, R.; De, R.; Baddam, R.; Shaik, S.; Alam, J.; Tiruvayipati, S.; Ahmed, N. Next-Generation Sequencing and De Novo Assembly, Genome Organization, and Comparative Genomic Analyses of the Genomes of Two Helicobacter pylori Isolates from Duodenal Ulcer Patients in India. J. Bacteriol. 2012, 194, 5963–5964. [Google Scholar] [CrossRef] [PubMed]
- Kauser, F.; Hussain, M.A.; Ahmed, I.; Ahmad, N.; Habeeb, A.; Khan, A.A.; Ahmed, N. Comparing genomes of Helicobacter pylori strains from the high-altitude desert of Ladakh, India. J. Clin. Microbiol. 2005, 43, 1538–1545. [Google Scholar] [CrossRef] [PubMed]
- Ansari, S.; Kabamba, E.T.; Shrestha, P.K.; Aftab, H.; Myint, T.; Tshering, L.; Sharma, R.P.; Ni, N.; Aye, T.T.; Subsomwong, P.; et al. Helicobacter pylori bab characterization in clinical isolates from Bhutan, Myanmar, Nepal and Bangladesh. PLoS ONE 2017, 12, e0187225. [Google Scholar] [CrossRef] [PubMed]
- Suerbaum, S.; Smith, J.M.; Bapumia, K.; Morelli, G.; Smith, N.H.; Kunstmann, E.; Dyrek, I.; Achtman, M. Free recombination within Helicobacter pylori. Proc. Natl. Acad. Sci. USA 1998, 95, 12619–12624. [Google Scholar] [CrossRef]
- Falush, D.; Kraft, C.; Taylor, N.S.; Correa, P.; Fox, J.G.; Achtman, M.; Suerbaum, S. Recombination and mutation during long-term gastric colonization by Helicobacter pylori: Estimates of clock rates, recombination size, and minimal age. Proc. Natl. Acad. Sci. USA 2001, 98, 15056–15061. [Google Scholar] [CrossRef]
- Muñoz-Ramirez, Z.Y.; Pascoe, B.; Mendez-Tenorio, A.; Mourkas, E.; Sandoval-Motta, S.; Perez-Perez, G.; Morgan, D.R.; Dominguez, R.L.; Ortiz-Princz, D.; Cavazza, M.E. A 500-year tale of co-evolution, adaptation, and virulence: Helicobacter pylori in the Americas. ISME J. 2021, 15, 78–92. [Google Scholar] [CrossRef] [PubMed]
- Kumar, N.; Mariappan, V.; Baddam, R.; Lankapalli, A.K.; Shaik, S.; Goh, K.-L.; Loke, M.F.; Perkins, T.; Benghezal, M.; Hasnain, S.E.; et al. Comparative genomic analysis of Helicobacter pylori from Malaysia identifies three distinct lineages suggestive of differential evolution. Nucleic Acids Res. 2014, 43, 324–335. [Google Scholar] [CrossRef] [PubMed]
- Falush, D.; Wirth, T.; Linz, B.; Pritchard, J.K.; Stephens, M.; Kidd, M.; Blaser, M.J.; Graham, D.Y.; Vacher, S.; Perez-Perez, G.I.; et al. Traces of Human Migrations in Helicobacter pylori Populations. Science 2003, 299, 1582–1585. [Google Scholar] [CrossRef] [PubMed]
- Tegtmeyer, N.; Wessler, S.; Backert, S. Role of the cag-pathogenicity island encoded type IV secretion system in Helicobacter pylori pathogenesis. FEBS J. 2011, 278, 1190–1202. [Google Scholar] [CrossRef]
- Furuta, Y.; Yahara, K.; Hatakeyama, M.; Kobayashi, I. Evolution of cagA oncogene of Helicobacter pylori through recombination. PLoS ONE 2011, 6, e23499. [Google Scholar] [CrossRef] [PubMed]
- Muñoz-Ramírez, Z.Y.; Mendez-Tenorio, A.; Kato, I.; Bravo, M.M.; Rizzato, C.; Thorell, K.; Torres, R.; Aviles-Jimenez, F.; Camorlinga, M.; Canzian, F.; et al. Whole Genome Sequence and Phylogenetic Analysis Show Helicobacter pylori Strains from Latin America Have Followed a Unique Evolution Pathway. Front. Cell. Infect. Microbiol. 2017, 7, 50. [Google Scholar] [CrossRef] [PubMed]
- Petzold, K.; Olofsson, A.; Arnqvist, A.; Boren, T.; Gröbner, G.; Schleucher, J. Helicobacter pylori: How is Adhesin BabA, a Blood Group Antigen Binding Membrane Protein, Involved in Bacterial Adherence? Biophys. J. 2009, 96, 409a. [Google Scholar] [CrossRef]
- Quintana-Hayashi, M.P.; Rocha, R.; Padra, M.; Thorell, A.; Jin, C.; Karlsson, N.G.; Roxo-Rosa, M.; Oleastro, M.; Lindén, S.K. BabA-mediated adherence of pediatric ulcerogenic H. pylori strains to gastric mucins at neutral and acidic pH. Virulence 2018, 9, 1699–1717. [Google Scholar] [CrossRef]
- Gangwer, K.A.; Shaffer, C.L.; Suerbaum, S.; Lacy, D.B.; Cover, T.L.; Bordenstein, S.R. Molecular evolution of the Helicobacter pylori vacuolating toxin gene vacA. J. Bacteriol. 2010, 192, 6126–6135. [Google Scholar] [CrossRef] [PubMed]
- Palframan, S.L.; Kwok, T.; Gabriel, K. Vacuolating cytotoxin A (VacA), a key toxin for Helicobacter pylori pathogenesis. Front. Cell. Infect. Microbiol. 2012, 2, 92. [Google Scholar] [CrossRef] [PubMed]
- Gutiérrez-Escobar, A.J.; Bravo, M.M.; Acevedo, O.; Backert, S. Molecular evolution of the VacA p55 binding domain of Helicobacter pylori in mestizos from a high gastric cancer region of Colombia. PeerJ 2019, 7, e6634. [Google Scholar] [CrossRef] [PubMed]
- Hatakeyama, M. Structure and function of Helicobacter pylori CagA, the first-identified bacterial protein involved in human cancer. Proc. Jpn. Academy. Ser. B Phys. Biol. Sci. 2017, 93, 196–219. [Google Scholar] [CrossRef] [PubMed]
- Matos, J.I.; de Sousa, H.A.; Marcos-Pinto, R.; Dinis-Ribeiro, M. Helicobacter pylori CagA and VacA genotypes and gastric phenotype: A meta-analysis. Eur. J. Gastroenterol. Hepatol. 2013, 25, 1431–1441. [Google Scholar] [CrossRef] [PubMed]
- Park, J.Y.; Forman, D.; Waskito, L.A.; Yamaoka, Y.; Crabtree, J.E. Epidemiology of Helicobacter pylori and CagA-positive infections and global variations in gastric cancer. Toxins 2018, 10, 163. [Google Scholar] [CrossRef]
- Alandiyjany, M.N.; Croxall, N.J.; Grove, J.I.; Delahay, R.M. A role for the tfs3 ICE-encoded type IV secretion system in pro-inflammatory signalling by the Helicobacter pylori Ser/Thr kinase, CtkA. PLoS ONE 2017, 12, e0182144. [Google Scholar] [CrossRef] [PubMed]
- Fischer, W.; Tegtmeyer, N.; Stingl, K.; Backert, S. Four chromosomal type IV secretion systems in Helicobacter pylori: Composition, structure and function. Front. Microbiol. 2020, 11, 1592. [Google Scholar] [CrossRef] [PubMed]
- He, S.; Corneloup, A.; Guynet, C.; Lavatine, L.; Caumont-Sarcos, A.; Siguier, P.; Marty, B.; Dyda, F.; Chandler, M.; Ton Hoang, B. The IS 200/IS 605 Family and “Peel and Paste” Single-Strand Transposition Mechanism. Microbiol. Spectr. 2015, 3, 4. [Google Scholar] [CrossRef] [PubMed]
- Abadi, A.T.B.; Mobarez, A.M.; Bonten, M.J.; Wagenaar, J.A.; Kusters, J.G. Clinical relevance of the cagA, tnpA and tnpB genes in Helicobacter pylori. BMC Gastroenterol. 2014, 14, 33. [Google Scholar] [CrossRef] [PubMed]
- Jenks, P.J.; Labigne, A.; Ferrero, R.L. Exposure to Metronidazole In Vivo Readily Induces Resistance in Helicobacter pylori and Reduces the Efficacy of Eradication Therapy in Mice. Antimicrob. Agents Chemother. 1999, 43, 777–781. [Google Scholar] [CrossRef] [PubMed]
- Fauzia, K.A.; Aftab, H.; Tshibangu-Kabamba, E.; Alfaray, R.I.; Saruuljavkhlan, B.; Cimuanga-Mukanya, A.; Matsumoto, T.; Subsomwong, P.; Akada, J.; Miftahussurur, M.; et al. Mutations Related to Antibiotics Resistance in Helicobacter pylori Clinical Isolates from Bangladesh. Antibiotics 2023, 12, 279. [Google Scholar] [CrossRef] [PubMed]
- Miftahussurur, M.; Aftab, H.; Shrestha, P.K.; Sharma, R.P.; Subsomwong, P.; Waskito, L.A.; Doohan, D.; Fauzia, K.A.; Yamaoka, Y. Effective therapeutic regimens in two South Asian countries with high resistance to major Helicobacter pylori antibiotics. Antimicrob. Resist. Infect. Control 2019, 8, 40. [Google Scholar] [CrossRef] [PubMed]
- Tillekeratne, L.G.; Bodinayake, C.K.; Dabrera, T.; Nagahawatte, A.; Arachchi, W.K.; Sooriyaarachchi, A.; Stewart, K.; Watt, M.; Østbye, T.; Woods, C.W. Antibiotic overuse for acute respiratory tract infections in Sri Lanka: A qualitative study of outpatients and their physicians. BMC Fam. Pract. 2017, 18, 37. [Google Scholar] [CrossRef] [PubMed]
- Gunasekera, Y.D.; Kinnison, T.; Kottawatta, S.A.; Silva-Fletcher, A.; Kalupahana, R.S. Misconceptions of antibiotics as a potential explanation for their misuse. A survey of the general public in a rural and urban community in Sri Lanka. Antibiotics 2022, 11, 454. [Google Scholar] [CrossRef]
- Alkan, C.; Sajjadian, S.; Eichler, E.E. Limitations of next-generation genome sequence assembly. Nat. Methods 2011, 8, 61–65. [Google Scholar] [CrossRef]
- Adewale, B.A. Will long-read sequencing technologies replace short-read sequencing technologies in the next 10 years? Afr. J. Lab. Med. 2020, 9, 1340. [Google Scholar] [CrossRef] [PubMed]
- Pearman, W.S.; Freed, N.E.; Silander, O.K. Testing the advantages and disadvantages of short- and long- read eukaryotic metagenomics using simulated reads. BMC Bioinform. 2020, 21, 220. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | SLK36 | SLK40 | SLK91 | SLK231 | SLK237 | SLK260 |
|---|---|---|---|---|---|---|
| cagA | Absent | Absent | Absent | Present | Present | Absent |
| EPIYA motif | BCCC | ABC | ||||
| CagA type | western | western | ||||
| vacA | Present | Present | Present | Present | Present | Present |
| Signal (s) | s2 | s2 | s2 | s1a | s1a | s2 |
| Intermediate (i) | i2 | i2 | i2 | i1 | i1 | i2 |
| Deletion (d) | d2 | d2 | d2 | d1 | d1 | d2 |
| Middle (m) | m2 | m2 | m2 | m1a | m1a | m2 |
| C-region (c) | c2 | c2 | c2 | c1 | c1 | c2 |
| oipA | Present | Present | Present | Present | Present | Present |
| CT repeat | 8 | 7 | 10 | 6 | 6 | 7 |
| On/off status | On | Off | On | On | On | Off |
| iceA1 | Present | Absent | Present | Present | Present | Absent |
| Strain | tfs4a | tfs4b | tfs3 | cagPAI | comB | IS |
|---|---|---|---|---|---|---|
| SLK36 | P12 | None | None | None | 26695 | IS21 |
| SLK40 | None | None | None | None | 26695 | IS21 |
| SLK91 | None | Shi470 | None | None | 26695 | None |
| SLK231 | None | None | None | 26695 | 26695 | IS21, IS605 |
| SLK237 | None | None | None | 26695 | 26695 | IS21 |
| SLK260 | P12 | None | Gambia24 | None | 26695 | IS21, IS200/605 |
| Strain Name | AMX | TCN | CAM | MNZ | LEV | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| MIC | Susc | MIC | Susc | MIC | Susc | MIC | Susc | MIC | Susc | |
| SLK36 | 0.023 | S | <0.016 | S | 0.023 | S | >256 | R | <0.02 | S |
| SLK40 | 0.032 | S | 0.032 | S | 0.064 | S | 192 | R | 0.094 | S |
| SLK91 | 0.023 | S | 0.032 | S | 0.023 | S | 0.75 | S | 0.19 | S |
| SLK231 | <0.016 | S | 0.094 | S | 0.125 | S | 48 | R | 0.5 | S |
| SLK237 | 0.023 | S | 0.032 | S | 0.047 | S | 24 | R | 0.19 | S |
| SLK260 | 0.19 | S | 0.094 | S | 0.125 | S | >256 | R | >32 | R |
| Gene | SLK36 | SLK40 | SLK91 | SLK231 | SLK237 | SLK260 |
|---|---|---|---|---|---|---|
| frxA | Premature-stop codon | Premature-stop codon | Truncated locus 1–96 | - | Truncated locus 1–96 | - |
| - | N124S | - | N124S | N124S | - | |
| fur | - | - | - | P114H | - | |
| - | - | - | - | - | C150Y | |
| omp11 | - | T13A | - | - | - | - |
| rdxA | - | - | - | Premature-stop codon locus 52 | - | - |
| R16C | - | - | - | R16C | - | |
| - | K64N | - | K64N | K64N | - | |
| H97Y | H97T | - | H97T | H97T | - | |
| - | P106S | - | P106S | P106S | - | |
| - | - | - | V204I | V204I | V204I | |
| - | - | - | D205A | D205A | D205A | |
| ribF | - | Q78R | - | Q78R | Q78R | - |
| D86E | - | - | - | - | D86E | |
| - | H94N | - | - | H94N | - | |
| E109G | - | - | - | - | E109G | |
| - | Q111K | - | Q111K | Q111K | - | |
| Q242K | - | - | Q242K | - | Q242K |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fauzia, K.A.; Rathnayake, J.; Doohan, D.; Lamawansa, M.D.; Alfaray, R.I.; Batsaikhan, S.; Phuc, B.H.; Waskito, L.A.; Tuan, V.P.; Kabamba, E.T.; et al. Beyond Low Prevalence: Exploring Antibiotic Resistance and Virulence Profiles in Sri Lankan Helicobacter pylori with Comparative Genomics. Microorganisms 2025, 13, 420. https://doi.org/10.3390/microorganisms13020420
Fauzia KA, Rathnayake J, Doohan D, Lamawansa MD, Alfaray RI, Batsaikhan S, Phuc BH, Waskito LA, Tuan VP, Kabamba ET, et al. Beyond Low Prevalence: Exploring Antibiotic Resistance and Virulence Profiles in Sri Lankan Helicobacter pylori with Comparative Genomics. Microorganisms. 2025; 13(2):420. https://doi.org/10.3390/microorganisms13020420
Chicago/Turabian StyleFauzia, Kartika Afrida, Jeewantha Rathnayake, Dalla Doohan, Meegahalande Durage Lamawansa, Ricky Indra Alfaray, Saruuljavkhlan Batsaikhan, Bui Hoang Phuc, Langgeng Agung Waskito, Vo Phuoc Tuan, Evariste Tshibangu Kabamba, and et al. 2025. "Beyond Low Prevalence: Exploring Antibiotic Resistance and Virulence Profiles in Sri Lankan Helicobacter pylori with Comparative Genomics" Microorganisms 13, no. 2: 420. https://doi.org/10.3390/microorganisms13020420
APA StyleFauzia, K. A., Rathnayake, J., Doohan, D., Lamawansa, M. D., Alfaray, R. I., Batsaikhan, S., Phuc, B. H., Waskito, L. A., Tuan, V. P., Kabamba, E. T., Ansari, S., Matsumoto, T., Akada, J., Matsuhisa, T., & Yamaoka, Y. (2025). Beyond Low Prevalence: Exploring Antibiotic Resistance and Virulence Profiles in Sri Lankan Helicobacter pylori with Comparative Genomics. Microorganisms, 13(2), 420. https://doi.org/10.3390/microorganisms13020420