
Residue K28 of Zika Virus NS5 Protein Is Implicated in Virus Replication and Antagonism of STAT2


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Sequence Alignment and Analysis of NS5 Sequences
2.2. Cells
2.3. Generation of Interferon α/β Receptor Knockout A549 Cells
2.4. Western Blotting
2.5. Generation of Viruses with Substitutions at Selected NS5 Residues by Circular Polymerase Extension Reaction
2.6. Growth Kinetics
2.7. Immunofluorescence
2.8. Statistical Analyses
3. Results
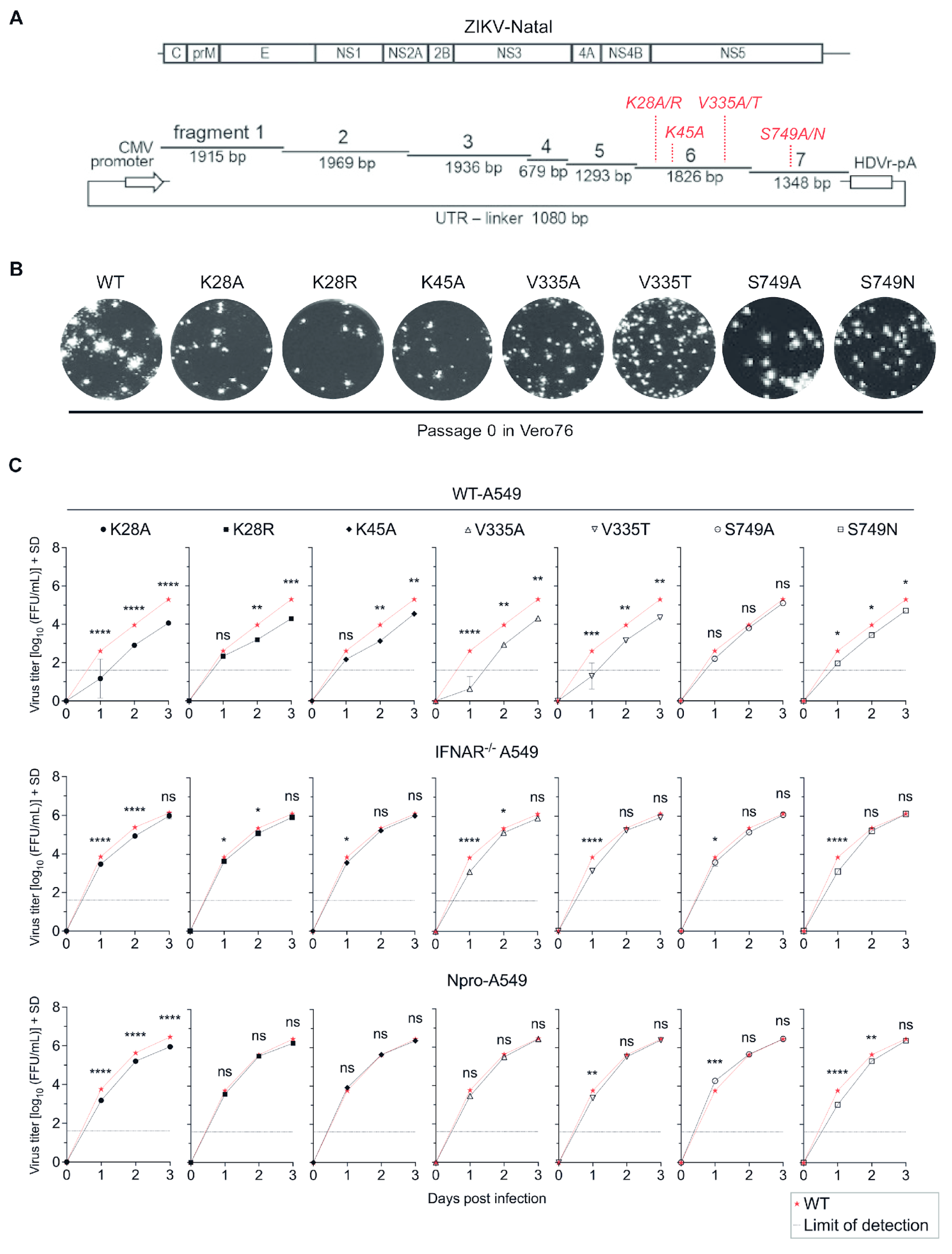
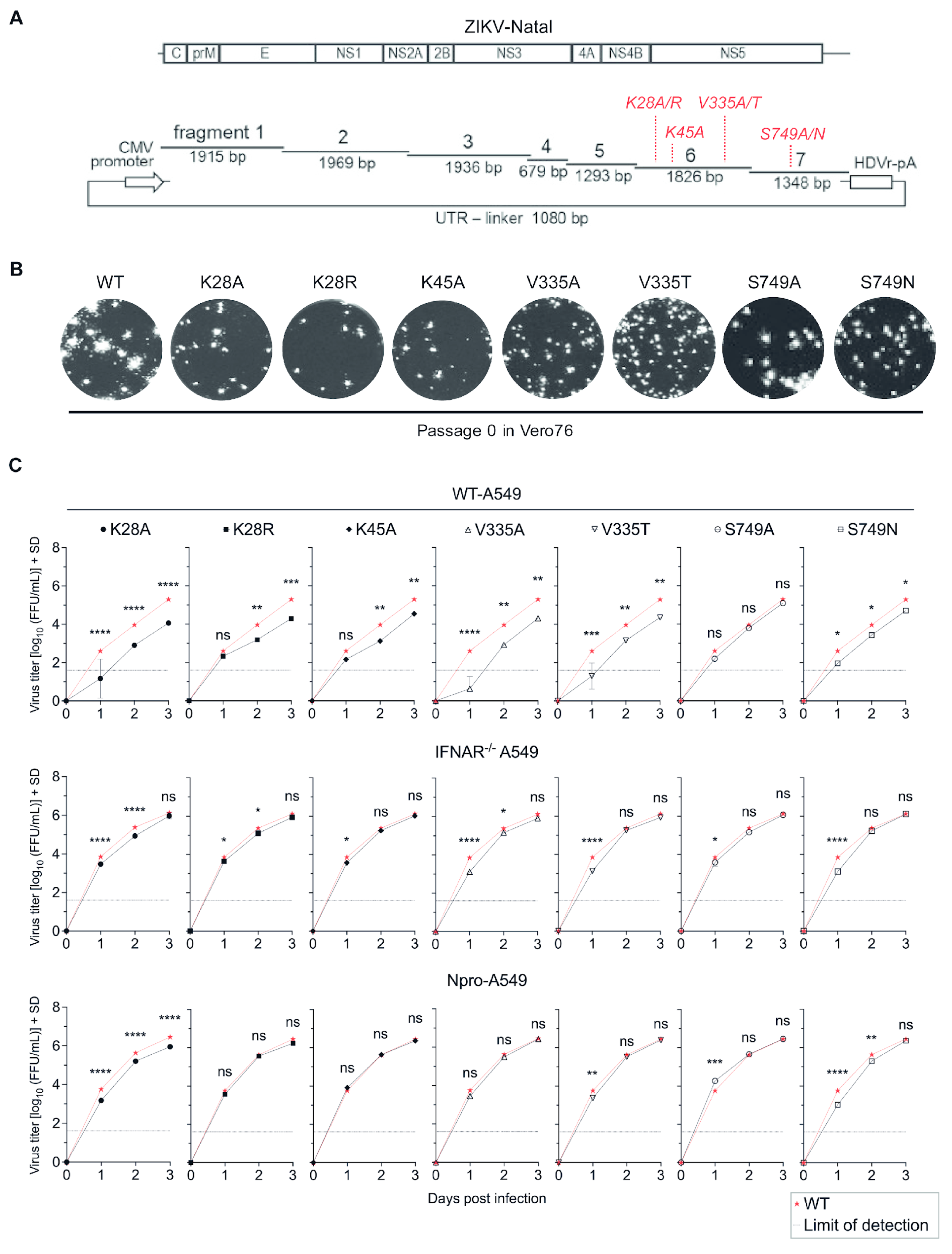
3.1. Substitutions at Residues K28, K45, V335, and S749 Attenuated Virus Replication in Type I IFN Competent Cells
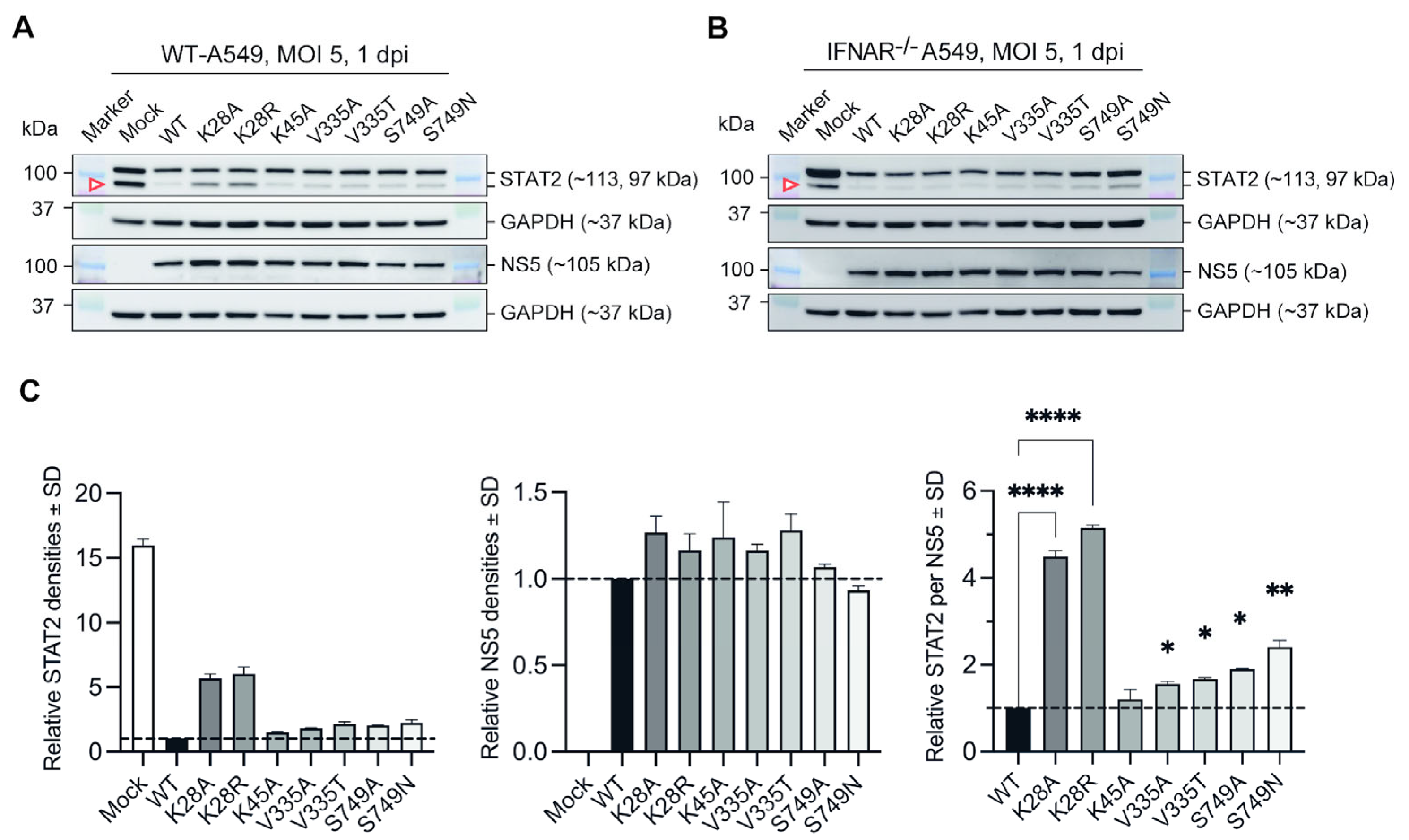
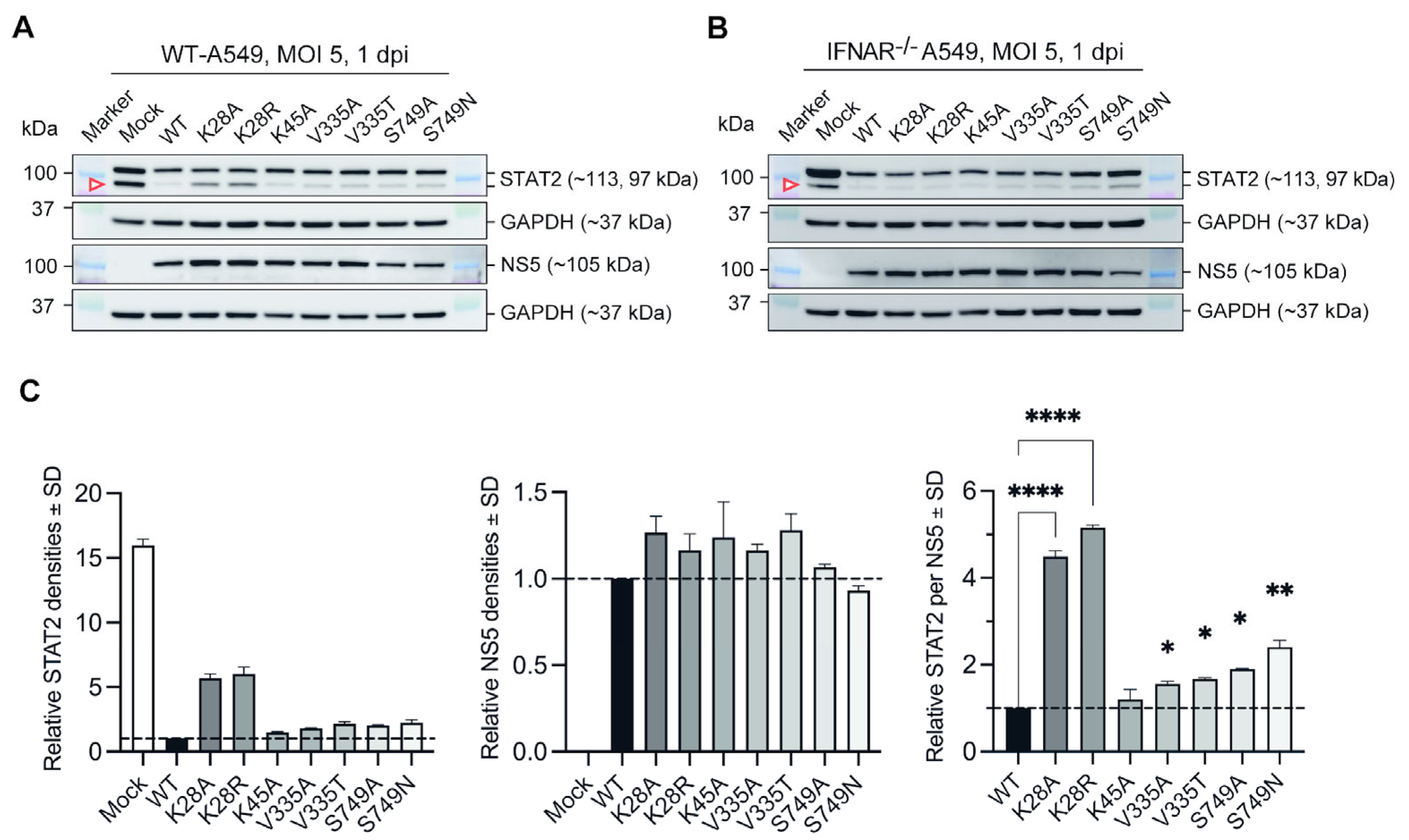
3.2. Substitutions at Selected Residues in NS5 Reduced ZIKV Efficiency to Antagonise STAT2
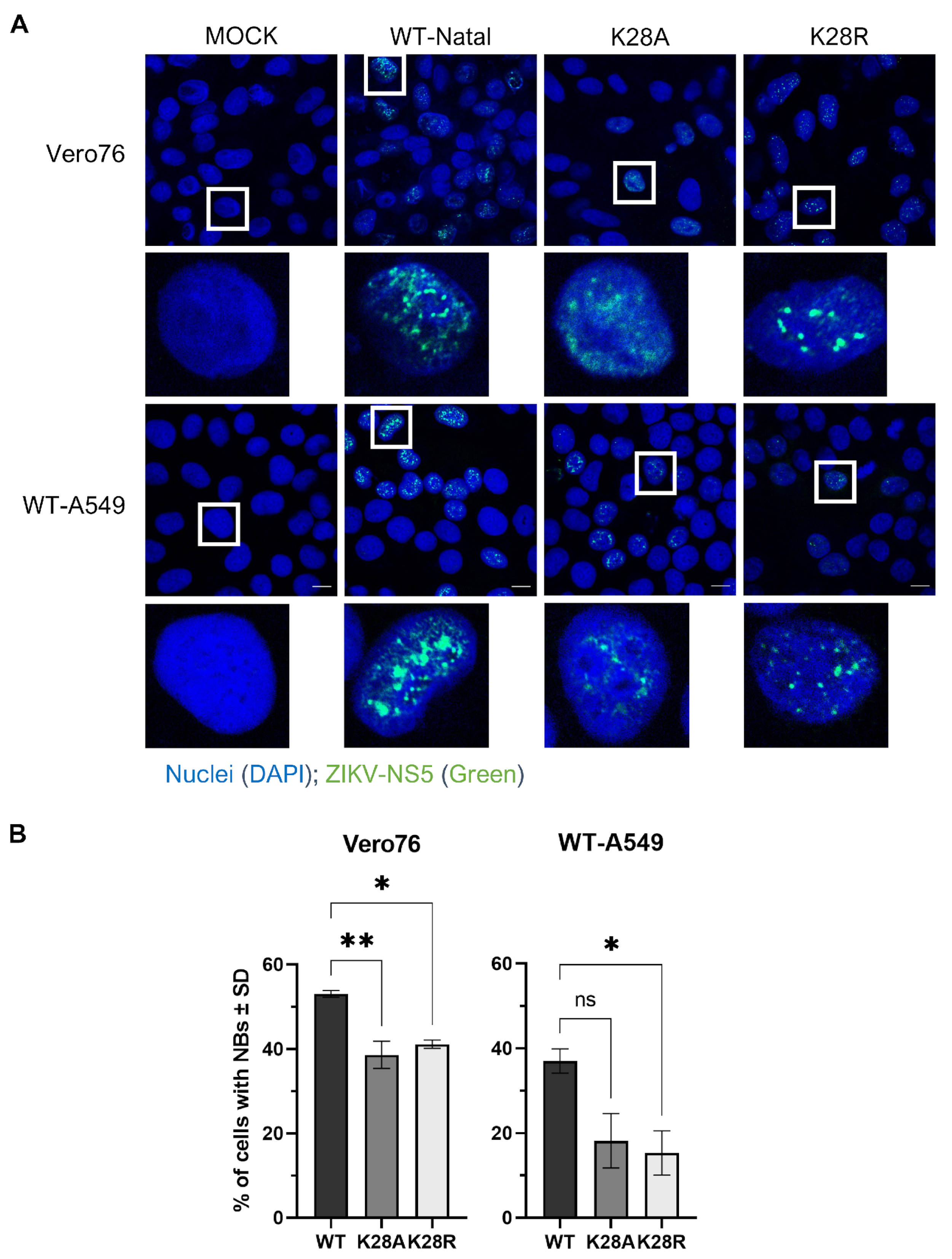
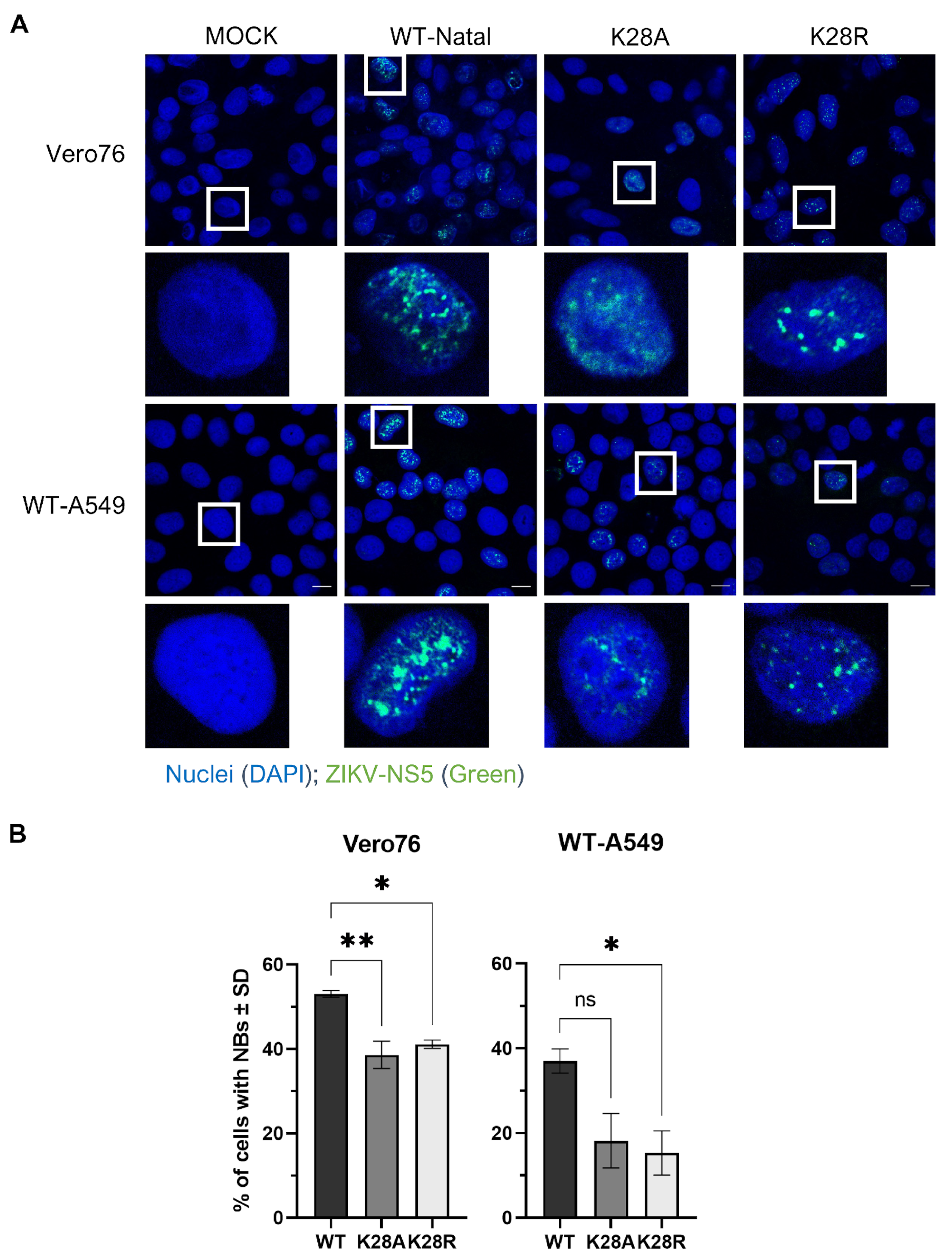
3.3. Substitutions at Residue K28 Resulted in Different NS5 Nuclear Body Phenotypes
4. Discussion
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ashour, J.; Laurent-Rolle, M.; Shi, P.Y.; Garcia-Sastre, A. NS5 of dengue virus mediates STAT2 binding and degradation. J. Virol. 2009, 83, 5408–5418. [Google Scholar] [CrossRef] [PubMed]
- Mazzon, M.; Jones, M.; Davidson, A.; Chain, B.; Jacobs, M. Dengue virus NS5 inhibits interferon-alpha signaling by blocking signal transducer and activator of transcription 2 phosphorylation. J. Infect. Dis. 2009, 200, 1261–1270. [Google Scholar] [CrossRef] [PubMed]
- Morrison, J.; Laurent-Rolle, M.; Maestre, A.M.; Rajsbaum, R.; Pisanelli, G.; Simon, V.; Mulder, L.C.; Fernandez-Sesma, A.; Garcia-Sastre, A. Dengue virus co-opts UBR4 to degrade STAT2 and antagonize type I interferon signaling. PLoS Pathog. 2013, 9, e1003265. [Google Scholar] [CrossRef] [PubMed]
- Laurent-Rolle, M.; Boer, E.F.; Lubick, K.J.; Wolfinbarger, J.B.; Carmody, A.B.; Rockx, B.; Liu, W.; Ashour, J.; Shupert, W.L.; Holbrook, M.R.; et al. The NS5 protein of the virulent West Nile virus NY99 strain is a potent antagonist of type I interferon-mediated JAK-STAT signaling. J. Virol. 2010, 84, 3503–3515. [Google Scholar] [CrossRef] [PubMed]
- Lubick, K.J.; Robertson, S.J.; McNally, K.L.; Freedman, B.A.; Rasmussen, A.L.; Taylor, R.T.; Walts, A.D.; Tsuruda, S.; Sakai, M.; Ishizuka, M.; et al. Flavivirus Antagonism of Type I Interferon Signaling Reveals Prolidase as a Regulator of IFNAR1 Surface Expression. Cell Host Microbe 2015, 18, 61–74. [Google Scholar] [CrossRef] [PubMed]
- Grant, A.; Ponia, S.S.; Tripathi, S.; Balasubramaniam, V.; Miorin, L.; Sourisseau, M.; Schwarz, M.C.; Sanchez-Seco, M.P.; Evans, M.J.; Best, S.M.; et al. Zika Virus Targets Human STAT2 to Inhibit Type I Interferon Signaling. Cell Host Microbe 2016, 19, 882–890. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Hou, S.; Airo, A.M.; Limonta, D.; Mancinelli, V.; Branton, W.; Power, C.; Hobman, T.C. Zika virus inhibits type-I interferon production and downstream signaling. EMBO Rep. 2016, 17, 1766–1775. [Google Scholar] [CrossRef]
- Roby, J.A.; Esser-Nobis, K.; Dewey-Verstelle, E.C.; Fairgrieve, M.R.; Schwerk, J.; Lu, A.Y.; Soveg, F.W.; Hemann, E.A.; Hatfield, L.D.; Keller, B.C.; et al. Flavivirus Nonstructural Protein NS5 Dysregulates HSP90 to Broadly Inhibit JAK/STAT Signaling. Cells 2020, 9, 899. [Google Scholar] [CrossRef]
- Ivashkiv, L.B.; Donlin, L.T. Regulation of type I interferon responses. Nat. Rev. Immunol. 2014, 14, 36–49. [Google Scholar] [CrossRef]
- Greenlund, A.C.; Morales, M.O.; Viviano, B.L.; Yan, H.; Krolewski, J.; Schreiber, R.D. Stat recruitment by tyrosine-phosphorylated cytokine receptors: An ordered reversible affinity-driven process. Immunity 1995, 2, 677–687. [Google Scholar] [CrossRef]
- Shuai, K.; Horvath, C.M.; Huang, L.H.; Qureshi, S.A.; Cowburn, D.; Darnell, J.E., Jr. Interferon activation of the transcription factor Stat91 involves dimerization through SH2-phosphotyrosyl peptide interactions. Cell 1994, 76, 821–828. [Google Scholar] [CrossRef] [PubMed]
- Heim, M.H.; Kerr, I.M.; Stark, G.R.; Darnell, J.E., Jr. Contribution of STAT SH2 groups to specific interferon signaling by the Jak-STAT pathway. Science 1995, 267, 1347–1349. [Google Scholar] [CrossRef] [PubMed]
- Fu, X.Y.; Kessler, D.S.; Veals, S.A.; Levy, D.E.; Darnell, J.E., Jr. ISGF3, the transcriptional activator induced by interferon alpha, consists of multiple interacting polypeptide chains. Proc. Natl. Acad. Sci. USA 1990, 87, 8555–8559. [Google Scholar] [CrossRef] [PubMed]
- Der, S.D.; Zhou, A.; Williams, B.R.; Silverman, R.H. Identification of genes differentially regulated by interferon alpha, beta, or gamma using oligonucleotide arrays. Proc. Natl. Acad. Sci. USA 1998, 95, 15623–15628. [Google Scholar] [CrossRef] [PubMed]
- Schneider, W.M.; Chevillotte, M.D.; Rice, C.M. Interferon-stimulated genes: A complex web of host defenses. Annu. Rev. Immunol. 2014, 32, 513–545. [Google Scholar] [CrossRef] [PubMed]
- Joyce, M.A.; Berry-Wynne, K.M.; Dos Santos, T.; Addison, W.R.; McFarlane, N.; Hobman, T.; Tyrrell, D.L. HCV and flaviviruses hijack cellular mechanisms for nuclear STAT2 degradation: Up-regulation of PDLIM2 suppresses the innate immune response. PLoS Pathog. 2019, 15, e1007949. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Thurmond, S.; Zhou, K.; Sanchez-Aparicio, M.T.; Fang, J.; Lu, J.; Gao, L.; Ren, W.; Cui, Y.; Veit, E.C.; et al. Structural basis for STAT2 suppression by flavivirus NS5. Nat. Struct. Mol. Biol. 2020, 27, 875–885. [Google Scholar] [CrossRef] [PubMed]
- Laurent-Rolle, M.; Morrison, J.; Rajsbaum, R.; Macleod, J.M.L.; Pisanelli, G.; Pham, A.; Ayllon, J.; Miorin, L.; Martinez, C.; tenOever, B.R.; et al. The interferon signaling antagonist function of yellow fever virus NS5 protein is activated by type I interferon. Cell Host Microbe 2014, 16, 314–327. [Google Scholar] [CrossRef]
- Dar, H.A.; Zaheer, T.; Paracha, R.Z.; Ali, A. Structural analysis and insight into Zika virus NS5 mediated interferon inhibition. Infect. Genet. Evol. 2017, 51, 143–152. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef]
- Waterhouse, A.M.; Procter, J.B.; Martin, D.M.; Clamp, M.; Barton, G.J. Jalview Version 2—A multiple sequence alignment editor and analysis workbench. Bioinformatics 2009, 25, 1189–1191. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef]
- Sanjana, N.E.; Shalem, O.; Zhang, F. Improved vectors and genome-wide libraries for CRISPR screening. Nat. Methods 2014, 11, 783–784. [Google Scholar] [CrossRef] [PubMed]
- Shalem, O.; Sanjana, N.E.; Hartenian, E.; Shi, X.; Scott, D.A.; Mikkelson, T.; Heckl, D.; Ebert, B.L.; Root, D.E.; Doench, J.G.; et al. Genome-scale CRISPR-Cas9 knockout screening in human cells. Science 2014, 343, 84–87. [Google Scholar] [CrossRef] [PubMed]
- Peng, N.Y.G.; Amarilla, A.A.; Hugo, L.E.; Modhiran, N.; Sng, J.D.J.; Slonchak, A.; Watterson, D.; Setoh, Y.X.; Khromykh, A.A. The distinguishing NS5-M114V mutation in American Zika virus isolates has negligible impacts on virus replication and transmission potential. PLoS Neglected Trop. Dis. 2022, 16, e0010426. [Google Scholar] [CrossRef] [PubMed]
- Edmonds, J.; van Grinsven, E.; Prow, N.; Bosco-Lauth, A.; Brault, A.C.; Bowen, R.A.; Hall, R.A.; Khromykh, A.A. A novel bacterium-free method for generation of flavivirus infectious DNA by circular polymerase extension reaction allows accurate recapitulation of viral heterogeneity. J. Virol. 2013, 87, 2367–2372. [Google Scholar] [CrossRef]
- Setoh, Y.X.; Prow, N.A.; Peng, N.; Hugo, L.E.; Devine, G.; Hazlewood, J.E.; Suhrbier, A.; Khromykh, A.A. De Novo Generation and Characterization of New Zika Virus Isolate Using Sequence Data from a Microcephaly Case. mSphere 2017, 2, e00190-17. [Google Scholar] [CrossRef]
- Setoh, Y.X.; Peng, N.Y.; Nakayama, E.; Amarilla, A.A.; Prow, N.A.; Suhrbier, A.; Khromykh, A.A. Fetal Brain Infection Is Not a Unique Characteristic of Brazilian Zika Viruses. Viruses 2018, 10, 541. [Google Scholar] [CrossRef]
- Amarilla, A.A.; Modhiran, N.; Setoh, Y.X.; Peng, N.Y.G.; Sng, J.D.J.; Liang, B.; McMillan, C.L.D.; Freney, M.E.; Cheung, S.T.M.; Chappell, K.J.; et al. An Optimized High-Throughput Immuno-Plaque Assay for SARS-CoV-2. Front. Microbiol. 2021, 12, 625136. [Google Scholar] [CrossRef]
- Zhao, B.; Yi, G.; Du, F.; Chuang, Y.C.; Vaughan, R.C.; Sankaran, B.; Kao, C.C.; Li, P. Structure and function of the Zika virus full-length NS5 protein. Nat. Commun. 2017, 8, 14762. [Google Scholar] [CrossRef]
- Ferrero, D.S.; Ruiz-Arroyo, V.M.; Soler, N.; Uson, I.; Guarne, A.; Verdaguer, N. Supramolecular arrangement of the full-length Zika virus NS5. PLoS Pathog. 2019, 15, e1007656. [Google Scholar] [CrossRef] [PubMed]
- Lazear, H.M.; Nice, T.J.; Diamond, M.S. Interferon-lambda: Immune Functions at Barrier Surfaces and Beyond. Immunity 2015, 43, 15–28. [Google Scholar] [CrossRef]
- McNab, F.; Mayer-Barber, K.; Sher, A.; Wack, A.; O’Garra, A. Type I interferons in infectious disease. Nat. Rev. Immunol. 2015, 15, 87–103. [Google Scholar] [CrossRef]
- Wack, A.; Terczynska-Dyla, E.; Hartmann, R. Guarding the frontiers: The biology of type III interferons. Nat. Immunol. 2015, 16, 802–809. [Google Scholar] [CrossRef] [PubMed]
- Signal Transducer and Activator of Transcription 2. UniProt. Available online: https://www.uniprot.org/uniprotkb/P52630/entry#P52630-4 (accessed on 12 April 2022).
- Sugiyama, T.; Nishio, Y.; Kishimoto, T.; Akira, S. Identification of alternative splicing form of Stat2. FEBS Lett. 1996, 381, 191–194. [Google Scholar] [CrossRef]
- Ota, T.; Suzuki, Y.; Nishikawa, T.; Otsuki, T.; Sugiyama, T.; Irie, R.; Wakamatsu, A.; Hayashi, K.; Sato, H.; Nagai, K.; et al. Complete sequencing and characterization of 21,243 full-length human cDNAs. Nat. Genet. 2004, 36, 40–45. [Google Scholar] [CrossRef] [PubMed]
- Stat2 (D9J7L) Rabbit mAb #72604. Cell Signaling Technology. Available online: https://www.cellsignal.com/products/primary-antibodies/stat2-d9j7l-rabbit-mab/72604 (accessed on 11 January 2018).
- Leung, S.; Qureshi, S.A.; Kerr, I.M.; Darnell, J.E., Jr.; Stark, G.R. Role of STAT2 in the alpha interferon signaling pathway. Mol. Cell. Biol. 1995, 15, 1312–1317. [Google Scholar] [CrossRef]
- Ng, I.H.W.; Chan, K.W.; Tan, M.J.A.; Gwee, C.P.; Smith, K.M.; Jeffress, S.J.; Saw, W.G.; Swarbrick, C.M.D.; Watanabe, S.; Jans, D.A.; et al. Zika Virus NS5 Forms Supramolecular Nuclear Bodies That Sequester Importin-alpha and Modulate the Host Immune and Pro-Inflammatory Response in Neuronal Cells. ACS Infect. Dis. 2019, 5, 932–948. [Google Scholar] [CrossRef]
- Tan, M.J.A.; Chan, K.W.K.; Ng, I.H.W.; Kong, S.Y.Z.; Gwee, C.P.; Watanabe, S.; Vasudevan, S.G. The Potential Role of the ZIKV NS5 Nuclear Spherical-Shell Structures in Cell Type-Specific Host Immune Modulation during ZIKV Infection. Cells 2019, 8, 1519. [Google Scholar] [CrossRef]
- Zhao, Z.; Tao, M.; Han, W.; Fan, Z.; Imran, M.; Cao, S.; Ye, J. Nuclear localization of Zika virus NS5 contributes to suppression of type I interferon production and response. J. Gen. Virol. 2021, 102, 001376. [Google Scholar] [CrossRef]
- Brooks, A.J.; Johansson, M.; John, A.V.; Xu, Y.; Jans, D.A.; Vasudevan, S.G. The interdomain region of dengue NS5 protein that binds to the viral helicase NS3 contains independently functional importin beta 1 and importin alpha/beta-recognized nuclear localization signals. J. Biol. Chem. 2002, 277, 36399–36407. [Google Scholar] [CrossRef]
- Pryor, M.J.; Rawlinson, S.M.; Butcher, R.E.; Barton, C.L.; Waterhouse, T.A.; Vasudevan, S.G.; Bardin, P.G.; Wright, P.J.; Jans, D.A.; Davidson, A.D. Nuclear localization of dengue virus nonstructural protein 5 through its importin alpha/beta-recognized nuclear localization sequences is integral to viral infection. Traffic 2007, 8, 795–807. [Google Scholar] [CrossRef]
- Coloma, J.; Jain, R.; Rajashankar, K.R.; Garcia-Sastre, A.; Aggarwal, A.K. Structures of NS5 Methyltransferase from Zika Virus. Cell Rep. 2016, 16, 3097–3102. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Feng, T.; Cheng, J.; Li, Y.; Yin, X.; Zeng, W.; Jin, X.; Li, Y.; Guo, F.; Jin, T. Structure of the NS5 methyltransferase from Zika virus and implications in inhibitor design. Biochem. Biophys. Res. Commun. 2017, 492, 624–630. [Google Scholar] [CrossRef] [PubMed]
- Bollati, M.; Milani, M.; Mastrangelo, E.; Ricagno, S.; Tedeschi, G.; Nonnis, S.; Decroly, E.; Selisko, B.; de Lamballerie, X.; Coutard, B.; et al. Recognition of RNA cap in the Wesselsbron virus NS5 methyltransferase domain: Implications for RNA-capping mechanisms in Flavivirus. J. Mol. Biol. 2009, 385, 140–152. [Google Scholar] [CrossRef]
- Ray, D.; Shah, A.; Tilgner, M.; Guo, Y.; Zhao, Y.; Dong, H.; Deas, T.S.; Zhou, Y.; Li, H.; Shi, P.Y. West Nile virus 5’-cap structure is formed by sequential guanine N-7 and ribose 2’-O methylations by nonstructural protein 5. J. Virol. 2006, 80, 8362–8370. [Google Scholar] [CrossRef]
- Issur, M.; Geiss, B.J.; Bougie, I.; Picard-Jean, F.; Despins, S.; Mayette, J.; Hobdey, S.E.; Bisaillon, M. The flavivirus NS5 protein is a true RNA guanylyltransferase that catalyzes a two-step reaction to form the RNA cap structure. RNA 2009, 15, 2340–2350. [Google Scholar] [CrossRef] [PubMed]
- Daffis, S.; Szretter, K.J.; Schriewer, J.; Li, J.; Youn, S.; Errett, J.; Lin, T.Y.; Schneller, S.; Zust, R.; Dong, H.; et al. 2’-O methylation of the viral mRNA cap evades host restriction by IFIT family members. Nature 2010, 468, 452–456. [Google Scholar] [CrossRef]
- Szretter, K.J.; Daniels, B.P.; Cho, H.; Gainey, M.D.; Yokoyama, W.M.; Gale, M., Jr.; Virgin, H.W.; Klein, R.S.; Sen, G.C.; Diamond, M.S. 2’-O methylation of the viral mRNA cap by West Nile virus evades ifit1-dependent and -independent mechanisms of host restriction in vivo. PLoS Pathog. 2012, 8, e1002698. [Google Scholar] [CrossRef]
- Zust, R.; Dong, H.; Li, X.F.; Chang, D.C.; Zhang, B.; Balakrishnan, T.; Toh, Y.X.; Jiang, T.; Li, S.H.; Deng, Y.Q.; et al. Rational design of a live attenuated dengue vaccine: 2’-o-methyltransferase mutants are highly attenuated and immunogenic in mice and macaques. PLoS Pathog. 2013, 9, e1003521. [Google Scholar] [CrossRef]
- Tay, M.Y.; Saw, W.G.; Zhao, Y.; Chan, K.W.; Singh, D.; Chong, Y.; Forwood, J.K.; Ooi, E.E.; Gruber, G.; Lescar, J.; et al. The C-terminal 50 amino acid residues of dengue NS3 protein are important for NS3-NS5 interaction and viral replication. J. Biol. Chem. 2015, 290, 2379–2394. [Google Scholar] [CrossRef] [PubMed]
- Zou, G.; Chen, Y.L.; Dong, H.; Lim, C.C.; Yap, L.J.; Yau, Y.H.; Shochat, S.G.; Lescar, J.; Shi, P.Y. Functional analysis of two cavities in flavivirus NS5 polymerase. J. Biol. Chem. 2011, 286, 14362–14372. [Google Scholar] [CrossRef] [PubMed]
- Johansson, M.; Brooks, A.J.; Jans, D.A.; Vasudevan, S.G. A small region of the dengue virus-encoded RNA-dependent RNA polymerase, NS5, confers interaction with both the nuclear transport receptor importin-beta and the viral helicase, NS3. J. Gen. Virol. 2001, 82, 735–745. [Google Scholar] [CrossRef] [PubMed]
- Saade, M.; Ferrero, D.S.; Blanco-Ameijeiras, J.; Gonzalez-Gobartt, E.; Flores-Mendez, M.; Ruiz-Arroyo, V.M.; Martinez-Saez, E.; Ramon, Y.C.S.; Akizu, N.; Verdaguer, N.; et al. Multimerization of Zika Virus-NS5 Causes Ciliopathy and Forces Premature Neurogenesis. Cell Stem Cell 2020, 27, 920–936.e928. [Google Scholar] [CrossRef] [PubMed]
- Conde, J.N.; Schutt, W.R.; Mladinich, M.; Sohn, S.Y.; Hearing, P.; Mackow, E.R. NS5 Sumoylation Directs Nuclear Responses That Permit Zika Virus To Persistently Infect Human Brain Microvascular Endothelial Cells. J. Virol. 2020, 94, e01086-20. [Google Scholar] [CrossRef]
- Flory, C.; Chan, K.W.K.; Bifani, A.M.; Choy, M.M.J.; Lescar, J.; Ooi, E.E.; Tautz, N.; Vasudevan, S.G. Optimal flexibility of the linker region of Zika virus NS5 methyltransferase-polymerase is critical for virus replication. Antiviral Res. 2021, 195, 105194. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Peng, N.Y.G.; Sng, J.D.J.; Setoh, Y.X.; Khromykh, A.A. Residue K28 of Zika Virus NS5 Protein Is Implicated in Virus Replication and Antagonism of STAT2. Microorganisms 2024, 12, 660. https://doi.org/10.3390/microorganisms12040660
Peng NYG, Sng JDJ, Setoh YX, Khromykh AA. Residue K28 of Zika Virus NS5 Protein Is Implicated in Virus Replication and Antagonism of STAT2. Microorganisms. 2024; 12(4):660. https://doi.org/10.3390/microorganisms12040660
Chicago/Turabian StylePeng, Nias Y. G., Julian D. J. Sng, Yin Xiang Setoh, and Alexander A. Khromykh. 2024. "Residue K28 of Zika Virus NS5 Protein Is Implicated in Virus Replication and Antagonism of STAT2" Microorganisms 12, no. 4: 660. https://doi.org/10.3390/microorganisms12040660
APA StylePeng, N. Y. G., Sng, J. D. J., Setoh, Y. X., & Khromykh, A. A. (2024). Residue K28 of Zika Virus NS5 Protein Is Implicated in Virus Replication and Antagonism of STAT2. Microorganisms, 12(4), 660. https://doi.org/10.3390/microorganisms12040660