1. Introduction
Cellulose is the most widely distributed and abundant carbohydrate polymer in nature [
1]. It is composed of D-glucose linked by
β-1,4-glycosidic bonds. Natural cellulose forms high crystallinity through intermolecular and intramolecular hydrogen bonds, leading to high resistance to enzymatic hydrolysis [
2]. The crystallinity depends on the proportions of crystalline and amorphous areas in the cellulose materials [
3]. Cellulose is hydrolyzed by cellulases, which can be divided into three types according to their catalytic modes [
4]: (i) endo-
β-1,4-glucanase (EC 3.2.1.4); (ii) exoglucanase, including 1,4-
β-D-glucan glucanohydrolases (glucanohydrolase) (EC 3.2.1.74) and cellobiohydrolase (EC 3.2.1.91); and (iii)
β-glucosidases (EC 3.2.1.21) [
3,
5]. Endoglucanase randomly cuts at the internal amorphous sites of the cellulosic polysaccharide chain to produce oligosaccharides of different lengths. After that, new ends form. In contrast, exoglucanase functions at the reducing or nonreducing ends of the cellulosic polysaccharide chain, and then releases glucose (using glucanohydrolase) or cellobiose (using cellobiohydrolase) as the main products.
β-glucosidase hydrolyzes soluble cellodextrin and cellobiose to glucose [
2,
6]. All three types can exist and function inside or outside cells [
7]. In total, there are 26 known hydrolase families that exhibit cellulolytic activity: endoglucanase (GH5, GH6, GH7, GH8, GH9, GH10, GH12, GH44, GH45, GH48, GH51, GH74, GH124, GH131, and GH148),
β-glucosidase (GH1, GH2, GH3, GH30, GH39, and GH116), cellulase/cellulprotamine phosphorylase (GH94), and hydrolyzing cellulose monooxygenase (AA9, AA10, AA15, and AA16) [
8]. These enzyme families are key to predicting the functional genes of a cellulolytic microbe.
Most studies of cellulolytic bacteria have focused on mesophilic or thermophilic representatives in compost. Typically, these microorganisms exhibit optimum cellulase activity at room temperature or at high temperatures. Mayende et al. obtained samples from lucerne and a garden compost heap (50 °C). The samples LM01 and LM04 showed the highest cellulase activity at 60 °C, while samples LM02 and LM03 exhibited the highest cellulase activity at 70 °C, indicating thermophilic cellulase activity [
9]. Legodi et al. isolated ten cellulose-degrading strains of bacteria from decomposed banana agro-waste and decomposed
Strelitzia alba plant, which had high enzyme activity at 30–40 °C [
10]. Generally, low-temperature-resistant and cellulose-degrading microorganisms can maintain considerable activity [
11]. In particular, low-temperature-resistant microorganisms can expedite the process of composting [
12]. Such an example is that the gene M6A encoding a novel cold-adapted endoglucanase was cloned from
Microbacterium kitamiense Sa12, a strain isolated from a wasteland in Saga, Qinghai-Tibetan Plateau. Compared with an optimum temperature of 35 °C, the relative activity of the enzyme was 30–40% at 0–5 °C [
13].
The genus
Microbacterium was established based on the type species
Microbacterium lacticum, which was isolated in a study using lactic acid bacteria [
14]. Later, the genus
Aureobacterium, classified in 1983 [
15], was re-emended into the genus
Microbacterium [
16]. As of November 2023, the genus
Microbacterium comprises a total of 160 validly published species (
https://lpsn.dsmz.de/genus/microbacterium, accessed on 23 November 2023), and these species are Gram-stain-positive and rod-shaped, but are not able to form spore [
17]. Members of the genus
Microbacterium have been isolated from a diverse array of environments, including soil, sediment [
18], activated sludge [
19], insects [
20], fresh water [
21], marine environments [
22], plants [
23], and human clinical specimens [
24]. At present, it has been reported that there is a significant correlation between
Microbacterium and hemicellulose digestion in the intestinal tract of locusts [
25].
In this study, we present a novel species of the genus Microbacterium and describe the phenotypic and genotypic properties of this new strain with the ability of degrading cellulose under a wide range of temperatures and releasing reducing sugar at low temperatures. Furthermore, we elucidated the genetic basis for metabolic capability based on a whole-genome sequence.
2. Materials and Methods
2.1. Isolation and Culture Conditions
Strain QXD-8T was isolated from the soil sample of an experimental field belonging to the Institute of Geographic Sciences and Natural Resources Research, the Chinese Academy of Sciences, Heilongjiang Province, China. The soil was collected at the site with a longitude of 123°97′184″ E, a latitude of 47°34′539″ N, and an altitude of 146 m.
The CAM medium, with sodium carboxymethylcellulose as carbon source, was used to isolate the strain. CAM medium components include (per liter): 10 g of sodium carboxymethylcellulose, 0.2 g of MgSO4·7H2O, 1.2 g of NaNO3, 3 g of K2HPO4, 6 g of NH4Cl, 0.05 g of CaCl2, 0.01 g of MnSO4·7H2O, and 0.01 g of ZnSO4·7H20. The pH of the medium was adjusted to approximate 7.0 with a 1 M NaOH solution. Approximate 5 g soil samples were added to the CAM liquid medium and shake-flask cultivation was carried out for 7 days at 15 °C. The enriched culture samples were diluted and spread on CAM plates with 2% (w/v) agar and cultured at 15 °C for 2 weeks until single colonies formed. The colonies were sub-cultured on CAM agar plates using plate streaking to obtain pure cultures. The isolated strain QXD-8T was stored in 20% glycerol at −80 °C.
2.2. 16S rRNA Phylogeny
The 16S rRNA gene of strain QXD-8
T was obtained by PCR amplification using 27F (5′-AGAGTTTGATCCTGGCTCA-3′) and 1492R (5′-AGAGTTTGATCCTGGCTCAG-3′) universal primers [
26]. The sequence was uploaded into the EZBioCloud webserver (
https://www.ezbiocloud.net, accessed on 15 October 2022) to find out closely related species. The 16S rRNA gene sequences of the type strains of 34 closely related species were downloaded from EZBioCloud. Multiple sequence alignments were performed using the CLUSTAL W program [
27], and phylogenetic trees were constructed based on the 16S rRNA gene in MEGA X (version 10.2 GUI for Windows) [
28] software using maximum likelihood (ML) [
29], neighbor-joining (NJ) [
30], and minimum evolution (ME) methods. Bootstrap analysis was used with 1000 resamples to evaluate the tree topology [
31].
2.3. Genome Features
The draft genome was sequenced using the Nova PE150 platform using MAGIGENE technologies (Guangdong Province), PR China. The raw reads resulting from the genome of strain QXD-8
T were checked with metaWRAP (version 1.2.2) [
32]. Filtered reads were assembled with the metaWrap-Assembly module and the quality of the assembly was quantified with QUAST (version 5.0.2) [
33]. Genome completeness and contamination were estimated with CheckM software (version 1.0.12) [
34]. The genome sequence of strain QXD-8
T was initially annotated using PGAP (
https://www.ncbi.nlm.nih.gov/genome/annotation_prok/, accessed on 1 July 2023). The G + C content was also calculated from the whole-genome sequence.
2.4. Phylogenomic Analysis and Sequence Identity
Strain QXD-8
T was identified as a
Microbacterium by the GTDB-tk [
38]. A total of 285
Microbacterium genomes were screened from the Genome Taxonomy Database (GTDB,
https://gtdb.ecogenomic.org/, accessed on 10 April 2023). All of these genomes are representative genomes in the GTDB. The genome of
Agreia bicolorate VKM Ac-2052 was selected as an outer clade. At the same time, the GTDB-tk was used to align QXD-8
T, with the above genome based on 120 conserved proteins. IQTREE was used to reconstruct phylogenetic trees [
39]. The phylogenetic tree was visualized with the iTOL website (
https://itol.embl.de/, accessed on 15 October 2023) [
40]. The genomes of the seven species most closely related to strain QXD-8
T were selected from the phylogenetic tree for further analyses. The average nucleotide identity (ANI) and average amino acid identity (AAI) were calculated using ANI/AAI-Matreix in the Kostas lab Tools webserver (
http://enve-omics.ce.gatech.edu/, accessed on 5 April 2023). The digital DNA-DNA hybridization (dDDH) values of strain QXD-8
T and the other seven strains were obtained by submitting sequence information on a genome-to-genome distance calculator (GGDC) website (
https://ggdc.dsmz.de/ggdc.php, accessed on 5 April 2023).
2.5. Physiology, Chemotaxonomy, and Morphology
The tryptic soy broth (TSB) medium (produced by Solarbio Life Sciences in Beijing, China) was utilized to analyze the physiological characteristics of strain QXD-8T. The TSB medium, with different NaCl concentrations (0–7%, at intervals of 1%, w/v), was used to test the optimum salinity range for growth purposes. The pH values supporting growth were tested between 5 and 9.5 (at intervals of 0.5 pH unit) and different buffers were used to maintain stable pH values: 1,4-piperazinediethanesulfonic acid (PIPES for pH 5.5–6.7), phosphate-buffered saline (PBS for pH 6.5–8), and N-cyclohexyl taurine (CHES for pH 8.6–10.0). The temperature growth range was determined at 10, 15, 20, 25, 28, 30, 37, and 45 °C. OD600 values were measured with a spectrophotometer (TN5000, produced by YOKE INSTRUMENT in Shanghai, China). The analysis of variance was performed using a one-way ANOVA in GraphPad. The activity of catalase was measured by picking a small number of cells into 3% (v/v) H2O2. Gram staining was performed using crystal violet and safranin strains to observe the cell color under a light microscope. The biochemical characteristics of the novel strain and the reference strains were tested using API 20NE and API50 CH kits in conjunction with API 50CHB medium kits (produced by Biomerieux, Shanghai, China), in line with the manufacturer’s instructions. The strain was cultured in a TSB medium at 28 °C for two days. Cell morphology was observed using scanning electron microscopy (SEM, Regulus 8100, produced by HITACHI, Tokyo, Japan), and the motility pattern was observed using light microscopy. At the same time, the type species Microbacterium lacticum DSM 20427T were used for parallel experiments.
2.6. Cellulose Degradation
The sodium carboxymethylcellulose hydrolytic activity of strain QXD-8
T was identified using the Congo red staining method [
41]. Firstly, 10 µL of bacterial suspension was added to the CAM medium. After culturing at 15 °C and 28 °C for 5 days, the size of the bacterial circle (S) was measured. The medium was stained by adding 5 mL of 0.1% Congo red staining solution for 10 min, and then 5 mL of 1 M NaCl was added for 10 min to decolorize the dye. The diameters of the hydrolysis circle (H) and colony (S) were measured. Finally, the EI value (
) was calculated to quantify its hydrolysis ability.
In addition, we used the CAM-M medium by replacing sodium carboxymethyl cellulose in the CAM medium with cellulose powder and combining it with the TSB medium at a ratio of 8:2. Strain QXD-8T was cultivated at 28 °C. After 0 h, 8 h, 24 h, 32 h, 40 h, 48 h, and 72 h of culture, 1 mL of bacterial solution was taken after centrifugation, and the reducing sugar content of the supernatant was determined using a cellulase assay kit (produced by OKA, Beijing, China) based on the dinitrosalicylic acid (DNS) method.
The CAM-C medium was used to determine the growth of strain QXD-8T by replacing sodium carboxymethyl cellulose in the CAM medium with cellobiose and combining it with TSB medium at a ratio of 8:2. The multifunctional microplate reader (EnSpire 2300, produced by FUJITSU, Kawasaki, Japan) was used to determine the absorbance value of strain QXD-8T at 600 nm at different temperatures to determine the growth of the strain. The analysis of variance was performed using one-way ANOVA in GraphPad.
3. Results
3.1. General Features
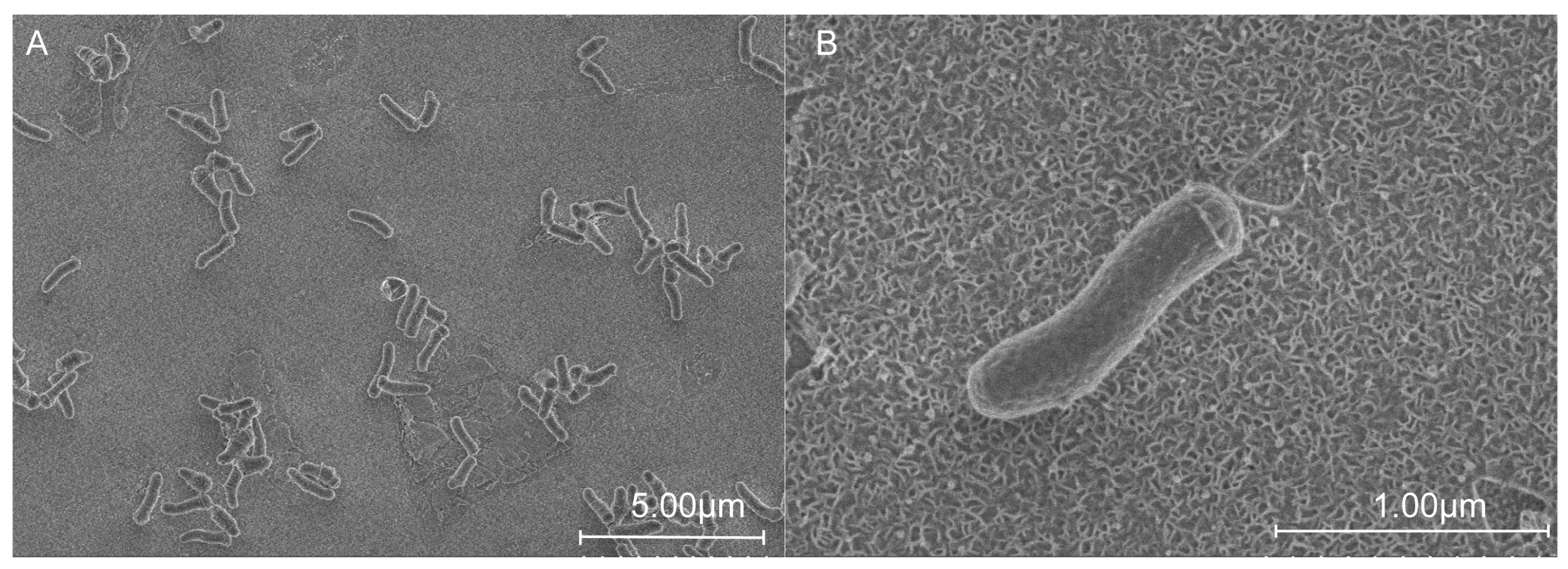
The cells of strain QXD-8
T were rod-shaped, and varied from 0.5 to 0.7 μm in width and from 1.0 to 2.2 µm in length (
Figure 1). After culturing strain QXD-8
T with the TSA medium for 2 days, colonies (with a diameter of about 1 mm) formed and they were round, raised, light yellow, and moist. Light microscopy showed that the strain was not motile. Strain QXD-8
T was a Gram-stain-positive bacterium.
The draft genome of strain QXD-8T was sequenced using next-generation genome sequencing technology, and was deposited at the NCBI (GCA_031085295.1). The size of the genome was 4.163 Mb. The genomic DNA G + C content was 70.1 mol%. Strain QXD-8T had 1 16S rRNA gene, 1 23S rRNA gene, 1 5S rRNA gene, 47 tRNA genes, and 1 tmRNA (transfer messenger RNA) gene. A total of 4110 coding sequences (CDSs) were predicted to exist in the genome.
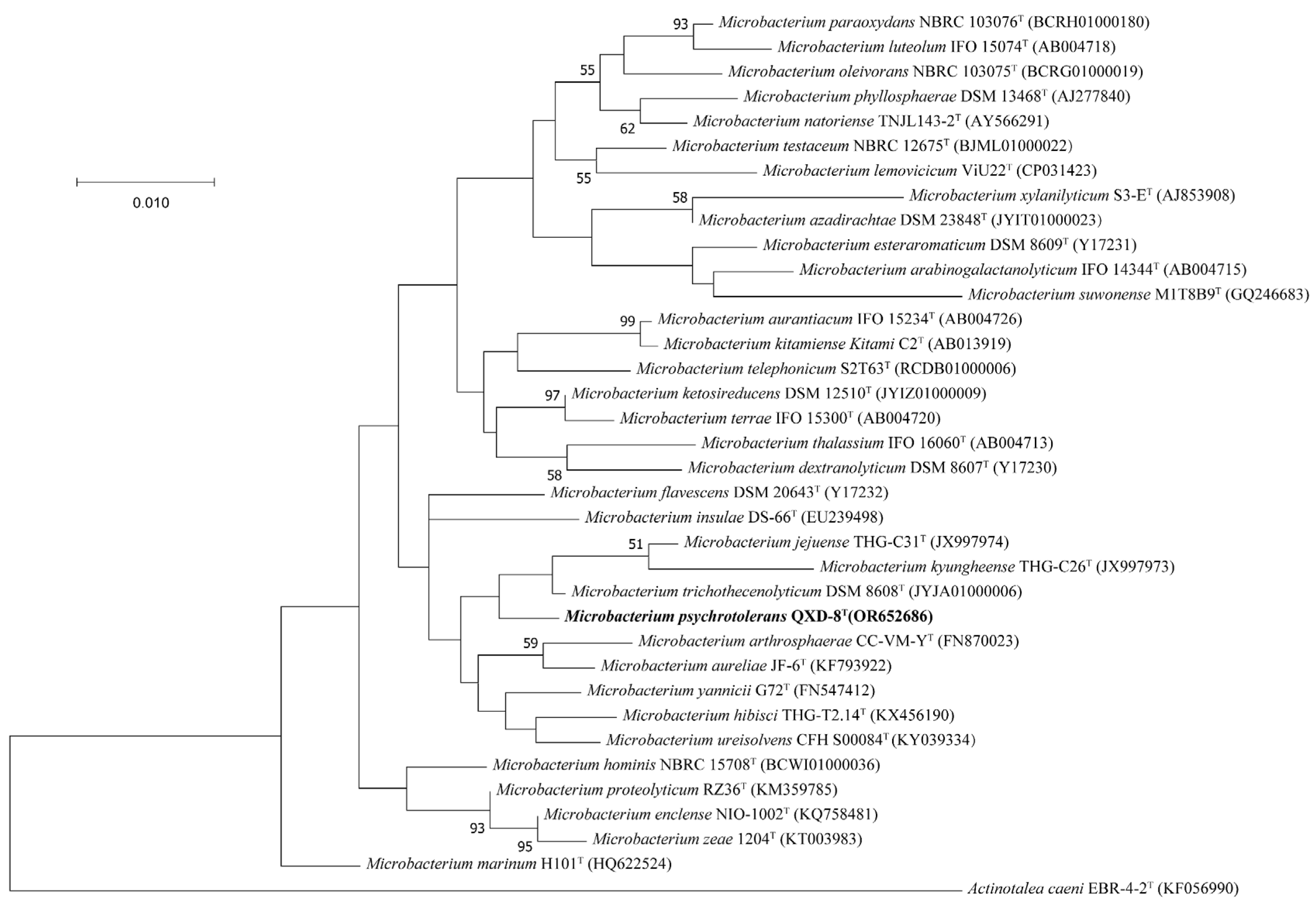
3.2. Phylogeny and Taxonomy
The partial 16S rRNA gene sequence (1401 bp) of strain QXD-8
T was obtained by PCR amplification, while the whole-length 16S rRNA gene sequence (1520 bp) of strain QXD-8
T was obtained from the genome-wide annotation information (described below), and the accession number of the latter is Q9R08_19460 at the NCBI. The two sequences showed 100% identity. The similarity analysis of the whole-length 16S rRNA gene sequence on EZBioCloud website showed that strain QXD-8
T had high similarities of 99.10%, 98.82%, and 98.82% with
Microbacterium trichothecenolyticum DSM 8608
T,
Microbacterium ketosireducens DSM 12510
T, and
Microbacterium yannicii G72
T, respectively. The similarities between QXD-8
T and other species were lower than 98.7%. Therefore, strain QXD-8
T could be considered a member of the genus
Microbacterium. Phylogenetic trees of 16S rRNA gene sequences were constructed with ML (
Figure 2), NJ (
Figure S1), and ME methods (
Figure S2), revealing that QXD-8
T clusters with
Microbacterium trichothecenolyticum DSM 8608
T,
Microbacterium jejuense THG-C31
T, and
Microbacterium kyungheense THG-C26
T in the genus
Microbacterium, and forms a separate clade with bootstrap supports of less than 50%, suggesting low credibility. A similar situation was also reported in the phylogenetic analyses of other species [
20].
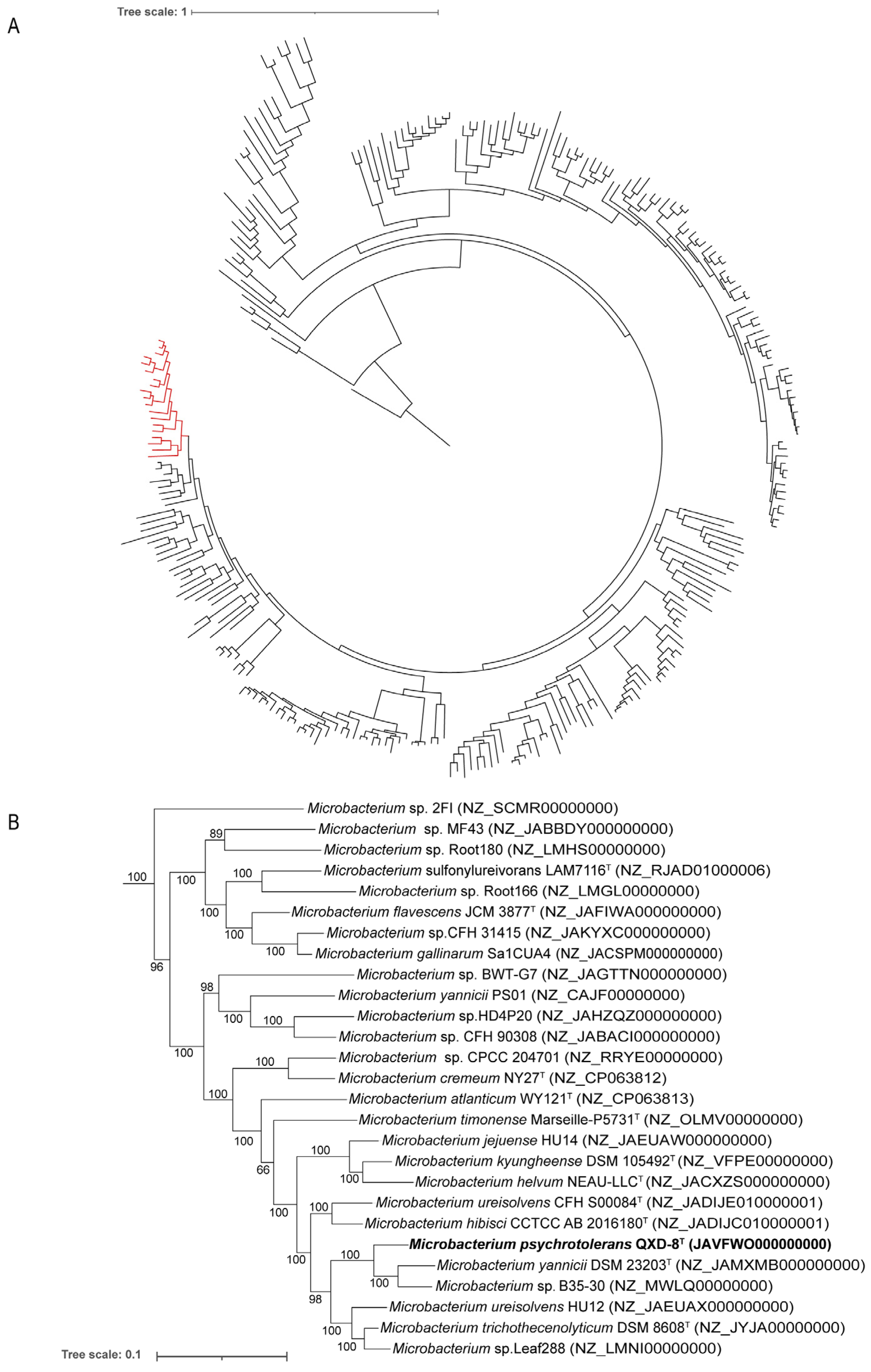
To clearly identify the phylogenetic placement of strain QXD-8
T, a phylogenomic tree was then constructed based on 120 conserved proteins. The genomes of 285 species in the genus
Microbacterium were included from the GTDB. Specifically, strain QXD-8
T was closely related to
Microbacterium yannicii G72
T and
Microbacterium sp. B35-30 with almost 100% bootstrap support (
Figure 3). Evidently, the strain represents a new taxon within the genus
Microbacterium.
In order to confirm the representativeness of strain QXD-8
T as a new species within the genus
Microbacterium, similarity analyses at the genome level were performed. The seven validly published species that shared a close relationship with QXD-8
T in the phylogenetic analyses, i.e.,
Microbacterium lacticum DSM 20427
T,
Microbacterium yannicii G72
T,
Microbacterium ureisolvens CFH S00084
T,
Microbacterium trichothecenolyticum DSM 8608
T,
Microbacterium jejuense THG-C31
T,
Microbacterium hibisci THG-T2.14
T, and
Microbacterium kyungheense THG-C26
T, were selected as references. The ANI, AAI, and dDDH values between QXD-8
T and these reference strains were 80–88%, 69–89%, and 19.1–54.5%, respectively (
Table 1). All these percentages were lower than the thresholds for the species boundaries of ANI 95–96% [
42], AAI 95–96% [
43], and dDDH 70% [
44], respectively. In particular, it exhibited the highest similarities with
Microbacterium yannicii G72
T. Consequently, strain QXD-8
T was found to be genotypically distinct from any previously described species and should be assigned to a different species.
Taken together, strain QXD-8T could, in principle, be considered a novel species within the genus Microbacterium based on the evidence obtained from the phylogenetic and phylogenomic analyses, as well as the ANI, AAI, and dDDH results.
3.3. Physiology and Chemotaxonomy
To describe the characteristics of strain QXD-8
T as a type of new species, the complete phenotypic characterization of this strain was carried out and compared with the seven closely related species in the genus
Microbacterium (
Table 2).
Generally, the results showed that strain QXD-8
T was able to grow in the NaCl concentration range of 0% to 5%, and grew slowly with an increase in salinity (
Figure 4A). Then, a significant difference in strain growth under different salt concentrations was indicated by variance analysis (
Figure S3). The pH range for growth was 5.0 to 9.5 (
Figure 4B). One-way ANOVA exhibited that there was no significant difference in the growth of strains with different pH levels (
Figure S4). The optimum NaCl concentration and pH were 0% and 7.0, respectively. However, it was interesting to find that the strain exhibited similar growth at 15 °C and 20 °C, and its optimum growth temperature was 28 °C. One-way ANOVA also supported this result, demonstrating no significant difference in growth at 15 °C and 20 °C (
Figure 4C and
Figure S5).
Bubble formation was observed in 3% hydrogen peroxide with a small number of cells, indicating positive peroxidase activity. Other enzyme activities, physiological features, and carbon assimilation results of strain QXD-8T were detected using the API 20NE kit, in line with the manufacturer’s instructions. The results showed that strain QXD-8T was positive for oxidase, nitrate reduction, aesculin hydrolysis, gelatin hydrolysis, β-galactosidase, and N-acetylglucosamine hydrolysis. H2S production, urease, glucose acidification, indole production, and arginine dihydrolase were negative. Malate, citrate, glucose, arabinose, mannose, and mannitol were utilized as a carbon source. The results of the API 50CH kit showed that acid was produced from L-arabinose, D-xylose, methyl-β-D-pyranoxyloside, D-galactose, D-glucose, D-fructose, D-mannose, mannitol, N-acetylglucosamine, arbutin, aesculin iron citrate, salicin, cellobiose, maltose, lactose, sucrose, trehalose, melezitose, raffinose, glycogen, xylitol, gentiobiose, and D-toulon sugar.
According to the results above, strain QXD-8
T and related bacteria had a similar carbon source affinity, in terms of an ability to produce acids (
Table 2). Compared with the closest related species
Microbacterium yannicii G72
T, strain QXD-8
T was able to reduce nitrate and used D-lactose and glycogen to produce acid, which were its specific characteristics. In summary, these results provided additional evidence that strain QXD-8
T belongs to
Microbacterium and represents a novel species.
3.4. Characteristics of Cellulose Decomposition at Low Temperatures
In previous studies, we found that strain QXD-8
T was still able to reproduce at low temperatures. Therefore, we performed Congo red staining on strain QXD-8
T on the CAM plate to determine whether it could degrade cellulose under low temperatures. The strain was incubated at 28 °C and 15 °C, respectively. It was observed that transparent hydrolysis zones formed from the first day at the two temperatures (
Figure 5A,D). Through quantitative analysis, it was found that under the condition of 28 °C, the EI values (the ratio of hydrolysis zone diameter to colony diameter) were 3.29 ± 0.02, 3.33 ± 0.02, and 3.43 ± 0.02 on the first, second, and third days, respectively. Under the condition of 15 °C, the EI values were 1.69 ± 0.02, 1.69 ± 0.02, and 1.71 ± 0.02 on the first, second, and third days, respectively (
Figure 5), which were almost half of those at 28 °C. In other words, strain QXD-8
T still kept respectable cellulase activity at low temperatures.
In order to identify the ability of strain QXD-8
T to hydrolyze the cellulose, we then used the CAM-M medium (cellulose used as a carbon source) to cultivate the strain and measure the concentration of extracellular reducing sugar. After adding the inoculum, the content of extracellular reducing sugar in the culture medium was measured at the beginning (0 h) as a control. The results showed that under the condition of 15 °C, the extracellular reducing sugar content gradually increased and reached the highest concentration at 32 h. The concentration of reducing sugar was 442.8939 μg/mL, and then gradually decreased to the initial content. However, it was unexpected that the content of reducing sugar was not increased under the condition of 28 °C (
Figure 6). The results of one-way ANOVA showed that there were significant differences in some time measurements at 15 °C, while there were no significant differences between the groups at 28 °C (
Figures S6 and S7). We speculate that cellulose hydrolysis requires extracellular enzyme activity, and the produced reducing sugars ultimately enter the culture broth, but they can be produced simultaneously through hydrolysis and metabolized by the strain at 28 °C. At low temperatures, due to the slower growth rate, its metabolic activity was significantly weakened. Therefore, reducing sugars can be observed to accumulate first and gradually consume.
Subsequently, in order to dissect the availability of the reducing sugar from the degradation of cellulose using strain QXD-8
T, we measured the growth of the strain in the CAM-C medium with cellobiose supplemented as a carbon source and without the addition of cellobiose as a control (
Figure 7). The results showed that at 28 °C, the group with the addition of cellobiose entered the logarithmic growth phase within 8 h and reached the stationary phase within 24 h, and the final biomass content expressed as OD
600 could reach about 1.3. However, the growth rate of the strain without cellobiose was lower than that of the strain with cellobiose, and the OD
600 at the 24th hour of cultivation (at the stationary phase) only reached around 0.75. On the contrary, under the condition of 15 °C, the addition of cellobiose had no obvious effect on the growth of the strain before 48 h. However, after 48 h, the group with added cellobiose entered the logarithmic growth phase and reached the stationary phase within 72 h, indicating that the strain should start to use cellobiose to promote the growth at about 48 h. Overall, strain QXD-8
T could decompose cellulose into reducing sugar. It is worth mentioning that the reducing sugar was accumulated under low-temperature conditions.
3.5. Genetic Basis of Cellulose Decomposition
In order to understand the cellulose degradation of strain QXD-8
T, we predicted the genes that encode the enzymes with cellulolytic activity and then reconstructed the pathway through the functional annotation and subcellular localization analyses of the genes in the genome of strain QXD-8
T. The gene products used for the enzyme families GH5, GH6, and GH10 with endoglucanase activity were annotated (
Table 3). The gene Q9R08_14620, assigned as GH5, was not predicted to have signal peptides and transmembrane regions. This protein might play a role in cytoplasm. GH6 exhibited both endoglucanase and cellobiohydrolase activity, and two encoding genes were annotated. The product of gene Q9R08_10050 contained a signal peptide of Tat/SPI, while that of gene Q9R08_14230 contained a signal peptide of Sec/SPI. However, they did not have transmembrane regions and were presumed to function extracellularly. The product of gene Q9R08_11975 was assigned as GH10, and it contained a signal peptide of Sec/SPI type and also had no transmembrane region. It could be an extracellular enzyme. Regarding
β-glucosidase, the gene of the enzyme families GH1, GH2, GH3, GH39, and GH116 were annotated in this strain (
Table 3). Most of them were not predicted to harbor the signal peptide and the transmembrane region. Therefore, they were presumed to play a role in the cell. However, two genes of the GH3 family were predicted to have signal peptides. The product of Q9R08_14550 had a signal peptide of Sec/SPI, while that of Q9R08_06855 had a signal peptide of Sec/SPII (
Table 3). Based on the genome sequence, we reconstructed the degradation pathway of cellulose in strain QXD-8
T. In short, cellulose was found to be cleaved extracellularly by the endoglucanase of the GH6 and GH10 enzyme family to form oligosaccharide chains with reducing and non-reducing ends. A part of the oligosaccharide chains entered the cell and was hydrolyzed by the endoglucanases of the GH5 enzyme family to cellobiose, cellotriose, or cellotetrasose, which were then hydrolyzed to glucose by
β-glucosidase, and the other part was hydrolyzed to cellobiose or glucose by
β-glucosidase out of the cell, and cellobiose will continue to be hydrolyzed to glucose. Cellulose was eventually hydrolyzed to glucose or oligosaccharides by the combined action of different enzyme families [
50] (
Figure 8). The physiological features of cellulose degradation and reducing sugar accumulation supported the metabolism pathway predicted from the genome sequence.
4. Discussion
In this study, we isolated a strain QXD-8
T that represents a novel species in the genus
Microbacterium and is capable of degrading cellulose to produce reducing sugar at low temperatures [
23,
45,
46,
47,
48,
49]. Phylogenetic analysis, genomic similarity, chemical classification, and physiological comparison supported the notion that the QXD-8
T could be considered the type strain of a new species of the genus
Microbacterium (
Figure 1 and
Figure 2,
Figures S1 and S2, and
Table 2). In the phylogenetic analysis based on the 16S rRNA gene, the topological structure was not highly confident because of low bootstrap values (<50%). In fact, it was also reported to exist in the species of the same genus [
17,
18,
20,
22,
23,
45,
47,
48,
49], possibly because the 16S rRNA gene was so conserved that it was not able to dissect the phylogenetic relationship of some species in the genus
Microbacterium. With the development of whole-genome sequencing, the phylogenetic relationship could be accurately estimated using the phylogenomic tree based on the concatenated sequences of single-copied conserved proteins, e.g., using the GTDB [
51]. In addition, the other sets were also widely adopted, such as UBCG2, another tool used for extracting bacterial core genes and inferring phylogenetic tree [
52]. During the experiment, it was found that strain QXD-8
T could still grow well at 15 °C, showing the resistance to low temperatures. In previous studies on the close relatives of the species, it was reported that
Microbacterium yannicii G72
T was also isolated from the middle- and high-latitude areas and could also grow under low temperatures [
23]. In addition,
Microbacterium jejuense THG-C31
T [
47],
Microbacterium suwonense M1T8B9
T [
48],
Microbacterium hibisci THG-T2.14
T [
49], and
Microbacterium kyungheense THG-C26
T [
47] exhibited similar features. In other words, low temperature tolerance may be a common characteristic of strain QXD-8
T and closely related species.
The cellulose breakdown capacity of the strain was demonstrated and the metabolism pathway was deduced based on the reconstruction of the genome sequence. Through genome annotation, we found that the enzyme family is related to the cellulose degradation pathway of the strain, and we explained how these enzyme genes function through the prediction of transmembrane regions and signal peptides. Admittedly, the predicted genes need further biochemical and genetic evidence. The reports of related bacteria do not emphasize their advantages for cellulose degradation, but the genome annotation results at least show that
Microbacterium yannicii G72
T,
Microbacterium jejuense THG-C31
T,
Microbacterium suwonense M1T8B9
T, and
Microbacterium hibisci THG-T2.14
T also harbored the genes of GH1, GH2, GH3, GH6, and GH10, as well as other enzyme families (
Table S2). Moreover, in the results of the API20NE kit, most of the carbon assimilation results of closely related bacteria were positive for ferric citrate, which also indicated that it had
β-glucosidase activity. Therefore, cellulase activity may be a common feature in
Microbacterium. The genus
Microbacterium could also be a potential microbial resource for cellulose degradation (
Table 2). It has been reported in the literature that the GH48 enzyme family is involved in the degradation of cellulose crystalline regions [
53], and we did not annotate the encoding gene from the enzyme family in strain QXD-8
T. It is possible that exoglucanase can only recognize the amorphous region in the cellulose chain for cutting and interact with
β-glucosidase to generate glucose, but the crystalline regions in cellulose may not be degraded in strain QXD-8
T.
In this study, reducing sugars could have accumulated in the extracellular environments under low temperatures, and the addition of cellobiose could have promoted the growth of cells. We speculated that the strain could not quickly transport reducing sugars into the intracellular space for growth under low-temperature conditions, although cellulase and transporter activity rates were reported to be inhibited by low temperatures [
11,
54]. The notion of whether the extracellular reducing sugars were accumulated or not depends on the cellulase and transporter activity rates at low temperatures. In this study, our results could be explained by speculating that the activity of the transporter apparently decreased more than that of cellulase (
Figure 6). The accumulated reducing sugars could also be used by other microorganisms or plants. At the same time, in some previous reports, some plant rhizosphere growth-promoting bacteria (PGPR) promote plant growth by dissolving phosphorus [
55], dissolving potassium, and fixing nitrogen, but in these cases, reducing sugars such as glucose are needed to provide energy. In addition, cellulose nitrogen-fixing bacteria (CNFB) can accelerate composting and effectively reduce the volume of compost in straw residue composting treatment [
56]. Delving into such microorganisms has a positive impact on our knowledge of the sustainable disposal of agricultural waste.
5. Conclusions
In this study, we isolated a new strain from soil, which was designated QXD-8T. Using polyphasic taxonomic methods, including phenotypic, physiological, phylogenetic, and biochemical analyses, strain QXD-8T was identified as a member of the genus Microbacterium. Phylogenetic analysis based on 16S rRNA gene sequences and 120 conserved proteins from the whole genome revealed that strain QXD-8T formed a separate clade in the genus Microbacterium. Genomic relatedness, assessed by ANI, AAI, and dDDH values, further confirmed that QXD-8T differed from its phylogenetically closest species. Overall, these results support the idea that QXD-8T represents a new member of the genus Microbacterium.
The Microbacterium sp. QXD-8T was able to grow on the CAM plate with sodium carboxymethyl cellulose as a carbon source at 15 °C and form a transparent hydrolysis circle after Congo red staining. In the liquid medium, it effectively degraded cellulose and produced reducing sugars. Functional annotation revealed the presence of encoding genes for the GH5, GH6, and GH10 enzyme families with endoglucanase activity, as well as the GH1, GH3, GH39, and GH116 enzyme families with β-glucosidase activity, suggesting their genetic basis for extracellular cellulose degradation. This study expands the diversity of psychrotolerant cellulolytic bacteria and provides a potential microbial resource for straw returning in high-latitude areas with low temperatures.
6. Description of Microbacterium psychrotolerans sp. nov.
Microbacterium psychrotolerans (psy.chro.to’le.rans. Gr. masc. adj. psychros, cold; L. pres. part. tolerans, tolerating; N.L. part. adj. psychrotolerans, cold-tolerating) cells are Gram-stain-positive, non-motile, catalase-positive, oxidase-positive, and rod-shaped, with a width of 0.5–0.7 µm, a length of 1.0–2.2 µm, an optimum growth condition of 28 °C, and a pH of 7.0 with 0% (w/v) NaCl. Colonies growing on TSA agar were light yellow, round, raised, and moist. In the API 20NE system, cells are positive for the hydrolysis of aesculin, nitrate reduction, and gelatin hydrolysis. Available carbon sources are malate, citrate, glucose, arabinose, mannose, and mannitol, and all other tests are negative. In the API 50CH test, acid is produced from L-arabinose, D-xylose, methyl-β-D-pyranoxyloside, D-galactose, D-glucose, D-fructose, D-mannose, mannitol, N-acetylglucosamine, arbutin, aesculin iron citrate, salicin, cellobiose, maltose, lactose, sucrose, trehalose, melezitose, raffinose, glycogen, xylitol, gentiobiose, and D-toulon sugar. Cells are positive for alkaline phosphatase, esterase (C4), esterase lipase (C8), lipase (C14), leucine arylamidase, valine arylamidase, trypsin hydrolysis, acid phosphatase, naphthol-AS-BI-phosphohydrolase, α-galactosidase, β-galactosidase, α-glucosidase, N-acetyl-β-glucosaminidase, and α-fucosidase, but negative for chymotrypsin hydrolysis, β-glucuronidase, glucosidase, and α-mannosidase.
The type strain, QXD-8T (=CGMCC 1.60112), with a G + C content value of 70.1 mol%, was isolated from the soil sample in Heilongjiang Province, China.
The GenBank accession number for the 16S rRNA gene sequences of Microbacterium psychrotolerans QXD-8T is OR652686 and that for the complete genome is JAVFWO000000000.
Supplementary Materials
The following supporting information can be downloaded at:
https://www.mdpi.com/article/10.3390/microorganisms12020303/s1. Figure S1: Neighbor-joining (NJ) phylogenetic tree based on 16S rRNA gene sequences; Figure S2: Minimum evolution (ME) phylogenetic tree based on 16S rRNA gene sequences; Figure S3: One-way ANOVA analysis of
Figure 4A; Figure S4: One-way ANOVA analysis of
Figure 4B; Figure S5: One-way ANOVA analysis of
Figure 4C; Figure S6: One-way ANOVA analysis of the extracellular reducing sugar content of strain QXD-8
T at 28 °C; Figure S7: One-way ANOVA analysis of the extracellular reducing sugar content of strain QXD-8
T at 15 °C; Figure S8: One-way ANOVA analysis for the group without cellobiose added at 15 °C; Figure S9: One-way ANOVA analysis for the group with cellobiose added at 15 °C in
Figure 7; Figure S10: One-way ANOVA analysis for the group without cellobiose added at 28 °C in
Figure 7; Figure S11: One-way ANOVA analysis for the group with cellobiose added at 28 °C in
Figure 7. Table S1: Prediction of signal peptide and transmembrane regions of cellulose hydrolysis-related proteins in strain QXD-8
T; Table S2: Cellulase prediction in the type strain of different
Microbacterium species.
Author Contributions
Conceptualization, H.X. and D.Z.; methodology, D.Z. and P.A.; validation, P.A. and C.Y.; formal analysis, P.A. and D.Z.; resources, P.A.; data curation, P.A.; writing—original draft preparation, P.A.; writing—review and editing, H.X., W.L. and D.Z.; visualization, P.A.; supervision, H.X.; project administration, D.Z.; funding acquisition, H.X. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Strategic Priority Research Program of the Chinese Academy of Sciences (grant no. XDA28030401).
Data Availability Statement
The 16S rRNA gene sequence for the isolate QXD-8T has been uploaded to NCBI with the accession number of OR652686. The whole genome shotgun project has been deposited at NCBI with the accession number of JAVFWO000000000 (submission ID SUB13746405, BioSample ID SAMN36886429, and BioProject ID PRJNA1003280).
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Brunecky, R.; Subramanian, V.; Yarbrough, J.M.; Donohoe, B.S.; Vinzant, T.B.; Vanderwall, T.A.; Knott, B.C.; Chaudhari, Y.B.; Bomble, Y.J.; Himmel, M.E.; et al. Synthetic fungal multifunctional cellulases for enhanced biomass conversion. Green Chem. 2019, 22, 478–489. [Google Scholar] [CrossRef]
- Béguin, P.; Aubert, J.P. The biological degradation of cellulose. FEMS Microbiol. Rev. 1994, 13, 25–58. [Google Scholar] [CrossRef] [PubMed]
- Lynd, L.R.; Weimer, P.J.; van Zyl, W.H.; Pretorius, I.S. Microbial cellulose utilization: Fundamentals and biotechnology. Microbiol. Mol. Biol. Rev. 2002, 66, 506–577. [Google Scholar] [CrossRef] [PubMed]
- Henrissat, B.; Teeri, T.T.; Warren, R.A.J. A scheme for designating enzymes that hydrolyse the polysaccharides in the cell walls of plants. FEBS Lett. 1998, 425, 352–354. [Google Scholar] [CrossRef] [PubMed]
- Bayer, E.A.; Chanzy, H.; Lamed, R.; Shoham, Y. Cellulose, cellulases and cellulosomes. Curr. Opin. Struct. Biol. 1998, 8, 548–557. [Google Scholar] [CrossRef] [PubMed]
- Bhat, M.K.; Bhat, S. Cellulose degrading enzymes and their potential industrial applications. Biotechnol. Adv. 1997, 15, 583–620. [Google Scholar] [CrossRef]
- Lee, H.-J.; Kang, T.-G.; Kim, Y.-W.; Lee, H.-S.; Kim, S.-K. Functional expression and extracellular secretion of Clostridium thermocellum Cel48S cellulase in Escherichia coli via the signal recognition particle-dependent translocation pathway. Enzym. Microb. Technol. 2021, 151, 109918. [Google Scholar] [CrossRef] [PubMed]
- Elcheninov, A.G.; Ugolkov, Y.A.; Elizarov, I.M.; Klyukina, A.A.; Kublanov, I.V.; Sorokin, D.Y. Cellulose metabolism in halo(natrono)archaea: A comparative genomics study. Front. Microbiol. 2023, 14, 1112247. [Google Scholar] [CrossRef]
- Mayende, L.; Wilhelmi, B.; Pletschke, B. Cellulases (CMCases) and polyphenol oxidases from thermophilic Bacillus spp. isolated from compost. Soil Biol. Biochem. 2006, 38, 2963–2966. [Google Scholar] [CrossRef]
- Legodi, L.M.; La Grange, D.; van Rensburg, E.L.J.; Ncube, I. Isolation of cellulose degrading fungi from decaying banana pseudostem and Strelitzia alba. Enzym. Res. 2019, 2019, 1390890. [Google Scholar] [CrossRef]
- Steiner, E.; Margesin, R. Production and partial characterization of a crude cold-active cellulase (CMCase) from Bacillus mycoides AR20-61 isolated from an Alpine forest site. Ann. Microbiol. 2020, 70, 67. [Google Scholar] [CrossRef]
- Li, D.; Feng, L.; Liu, K.; Cheng, Y.; Hou, N.; Li, C. Optimization of cold-active CMCase production by psychrotrophic Sphingomonas sp. FLX-7 from the cold region of China. Cellulose 2016, 23, 1335–1347. [Google Scholar] [CrossRef]
- Lin, L.; Qin, N.; Guan, L. A novel cold-adapted endoglucanase (M6A) from Microbacterium kitamiense S12 Isolated from Qinghai-Tibetan Plateau. Biotechnol. Bioprocess Eng. 2019, 24, 544–551. [Google Scholar] [CrossRef]
- Orla-Jensen, S. The Lactic Acid Bacteria; Høst & Sons: Copenhagen, Denmark, 1919. [Google Scholar]
- Collins, M.D.; Jones, D.; Keddie, R.M.; Kroppenstedt, R.M.; Schleifer, K.H. Classification of some coryneform bacteria in a new genus Aureobacterium. Syst. Appl. Microbiol. 1983, 4, 236–252. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, M.; Hatano, K. Union of the genera Microbacterium Orla-Jensen and Aureobacterium Collins et al. in a redefined genus Microbacterium. Int. J. Syst. Bacteriol. 1998, 48 Pt 3, 739–747. [Google Scholar] [CrossRef] [PubMed]
- Fidalgo, C.; Riesco, R.; Henriques, I.; Trujillo, M.E.; Alves, A. Microbacterium diaminobutyricum sp. nov., isolated from Halimione portulacoides, which contains diaminobutyric acid in its cell wall, and emended description of the genus Microbacterium. Int. J. Syst. Evol. Microbiol. 2016, 66, 4492–4500. [Google Scholar] [CrossRef]
- Mawlankar, R.R.; Mual, P.; Sonalkar, V.V.; Thorat, M.N.; Verma, A.; Srinivasan, K.; Dastager, S.G. Microbacterium enclense sp. nov., isolated from sediment sample. Int. J. Syst. Evol. Microbiol. 2015, 65, 2064–2070. [Google Scholar] [CrossRef]
- Malik, A.; Sakamoto, M.; Ono, T.; Kakii, K. Coaggregation between Acinetobacter johnsonii S35 and Microbacterium esteraromaticum strains isolated from sewage activated sludge. J. Biosci. Bioeng. 2003, 96, 10–15. [Google Scholar] [CrossRef]
- Lee, S.D.; Kim, I.S. Microbacterium tenebrionis sp. nov. and Microbacterium allomyrinae sp. nov., isolated from larvae of Tenebrio molitor L. and Allomyrina dichotoma, respectively. Int. J. Syst. Evol. Microbiol. 2023, 73, 005729. [Google Scholar] [CrossRef]
- Nayarisseri, A.; Khandelwal, R.; Singh, S.K. Identification and characterization of lipopeptide biosurfactant producing Microbacterium sp isolated from Brackish River Water. Curr. Top Med. Chem. 2020, 20, 2221–2234. [Google Scholar] [CrossRef]
- Kageyama, A.; Takahashi, Y.; Matsuo, Y.; Kasai, H.; Shizuri, Y.; Ōmura, S. Microbacterium sediminicola sp. nov. and Microbacterium marinilacus sp. nov., isolated from marine environments. Int. J. Syst. Evol. Microbiol. 2007, 57, 2355–2359. [Google Scholar] [CrossRef] [PubMed]
- Karojet, S.; Kunz, S.; van Dongen, J.T. Microbacterium yannicii sp. nov., isolated from Arabidopsis thaliana roots. Int. J. Syst. Evol. Microbiol. 2012, 62, 822–826. [Google Scholar] [CrossRef]
- Gneiding, K.; Frodl, R.; Funke, G. Identities of Microbacterium spp. encountered in human clinical specimens. J. Clin. Microbiol. 2008, 46, 3646–3652. [Google Scholar] [CrossRef] [PubMed]
- Bai, J.; Ling, Y.; Li, W.J.; Wang, L.; Xue, X.B.; Gao, Y.Y.; Li, F.F.; Li, X.J. Analysis of intestinal microbial diversity of four species of grasshoppers and determination of cellulose digestibility. Insects 2022, 13, 432. [Google Scholar] [CrossRef]
- Ventura, M.; Elli, M.; Reniero, R.; Zink, R. Molecular microbial analysis of Bifidobacterium isolates from different environments by the species-specific amplified ribosomal DNA restriction analysis (ARDRA). FEMS Microbiol. Ecol. 2001, 36, 113–121. [Google Scholar] [CrossRef] [PubMed]
- Thompson, J.D.; Gibson, T.J.; Plewniak, F.; Jeanmougin, F.; Higgins, D.G. The CLUSTAL_X windows interface: Flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 1997, 25, 4876–4882. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Felsenstein, J. Evolutionary trees from DNA sequences: A maximum likelihood approach. J. Mol. Evol. 1981, 17, 368–376. [Google Scholar] [CrossRef]
- Saitou, N.; Nei, M. The neighbor-joining method: A new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 1987, 4, 406–425. [Google Scholar]
- Felsenstein, J. Confidence-limits on phylogenies—An approach using the bootstrap. Evolution 1985, 39, 783–791. [Google Scholar] [CrossRef]
- Uritskiy, G.V.; DiRuggiero, J.; Taylor, J. MetaWRAP—A flexible pipeline for genome-resolved metagenomic data analysis. Microbiome 2018, 6, 158. [Google Scholar] [CrossRef] [PubMed]
- Gurevich, A.; Saveliev, V.; Vyahhi, N.; Tesler, G. QUAST: Quality assessment tool for genome assemblies. Bioinformatics 2013, 29, 1072–1075. [Google Scholar] [CrossRef]
- Parks, D.H.; Imelfort, M.; Skennerton, C.T.; Hugenholtz, P.; Tyson, G.W. CheckM: Assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 2015, 25, 1043–1055. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.; Ge, Q.; Yan, Y.; Zhang, X.; Huang, L.; Yin, Y. dbCAN3: Automated carbohydrate-active enzyme and substrate annotation. Nucleic Acids Res. 2023, 51, W115–W121. [Google Scholar] [CrossRef]
- Almagro Armenteros, J.J.; Tsirigos, K.D.; Sønderby, C.K.; Petersen, T.N.; Winther, O.; Brunak, S.; von Heijne, G.; Nielsen, H. SignalP 5.0 improves signal peptide predictions using deep neural networks. Nat. Biotechnol. 2019, 37, 420–423. [Google Scholar] [CrossRef] [PubMed]
- Hallgren, J.; Tsirigos, K.D.; Pedersen, M.D.; Armenteros, J.J.A.; Marcatili, P.; Nielsen, H.; Krogh, A.; Winther, O. DeepTMHMM predicts alpha and beta transmembrane proteins using deep neural networks. J. BioRxiv 2022, 2022.04.08.487609. [Google Scholar] [CrossRef]
- Chaumeil, P.A.; Mussig, A.J.; Hugenholtz, P.; Parks, D.H. GTDB-Tk: A toolkit to classify genomes with the Genome Taxonomy Database. Bioinformatics 2019, 36, 1925–1927. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef]
- Letunic, I.; Bork, P. Interactive Tree of Life (iTOL) v5: An online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 2021, 49, W293–W296. [Google Scholar] [CrossRef]
- Florencio, C.; Couri, S.; Farinas, C.S. Correlation between agar plate screening and solid-state fermentation for the prediction of cellulase production by Trichoderma strains. Enzym. Res. 2012, 2012, 793708. [Google Scholar] [CrossRef]
- Jain, C.; Rodriguez-R, L.M.; Phillippy, A.M.; Konstantinidis, K.T.; Aluru, S. High throughput ANI analysis of 90K prokaryotic genomes reveals clear species boundaries. Nat. Commun. 2018, 9, 5114. [Google Scholar] [CrossRef] [PubMed]
- Konstantinidis, K.T.; Tiedje, J.M. Towards a genome-based taxonomy for prokaryotes. J. Bacteriol. 2005, 187, 6258–6264. [Google Scholar] [CrossRef] [PubMed]
- Wayne, L.G.; Moore, W.E.C.; Stackebrandt, E.; Kandler, O.; Colwell, R.R.; Krichevsky, M.I.; Truper, H.G.; Murray, R.G.E.; Grimont, P.A.D.; Brenner, D.J.; et al. Report of the ad hoc committee on reconciliation of approaches to bacterial systematics. Int. J. Syst. Evol. Microbiol. 1987, 37, 463–464. [Google Scholar] [CrossRef]
- Cheng, L.J.; Ming, H.; Zhao, Z.L.; Ji, W.L.; Zhang, L.Y.; Li, L.Y.; Meng, X.L.; Li, M.; Niu, M.M.; Nie, G.X. Microbacterium ureisolvens sp. nov., isolated from a Yellow River sample. Int. J. Syst. Evol. Microbiol. 2019, 69, 560–566. [Google Scholar] [CrossRef]
- Yokota, A.; Takeuchi, M.; Sakane, T.; Weiss, N. Proposal of six new species in the genus Aureobacterium and transfer of Flavobacterium esteraromaticum Omelianski to the genus Aureobacterium as Aureobacterium esteraromaticum comb. nov. Int. J. Syst. Bacteriol. 1993, 43, 555–564. [Google Scholar] [CrossRef][Green Version]
- Kook, M.; Son, H.M.; Yi, T.H. Microbacterium kyungheense sp. nov. and Microbacterium jejuense sp. nov., isolated from salty soil. Int. J. Syst. Evol. Microbiol. 2014, 64, 2267–2273. [Google Scholar] [CrossRef] [PubMed]
- Anandham, R.; Tamura, T.; Hamada, M.; Weon, H.Y.; Kim, S.J.; Kim, Y.S.; Suzuki, K.; Kwon, S.W. Microbacterium suwonense sp. nov., isolated from cow dung. J. Microbiol. 2011, 49, 852–856. [Google Scholar] [CrossRef]
- Yan, Z.F.; Lin, P.; Won, K.H.; Yang, J.E.; Li, C.T.; Kook, M.; Wang, Q.J.; Yi, T.H. Microbacterium hibisci sp. nov., isolated from rhizosphere of mugunghwa (Hibiscus syriacus L.). Int. J. Syst. Evol. Microbiol. 2017, 67, 3564–3569. [Google Scholar] [CrossRef]
- Singh, R.; Pal, D.B.; Alkhanani, M.F.; Almalki, A.H.; Areeshi, M.Y.; Haque, S.; Srivastava, N. Prospects of soil microbiome application for lignocellulosic biomass degradation: An overview. Sci. Total Environ. 2022, 838, 155966. [Google Scholar] [CrossRef]
- Parks, D.H.; Chuvochina, M.; Rinke, C.; Mussig, A.J.; Chaumeil, P.-A.; Hugenholtz, P. GTDB: An ongoing census of bacterial and archaeal diversity through a phylogenetically consistent, rank normalized and complete genome-based taxonomy. Nucleic Acids Res. 2022, 50, D785–D794. [Google Scholar] [CrossRef]
- Kim, J.; Na, S.-I.; Kim, D.; Chun, J. UBCG2: Up-to-date bacterial core genes and pipeline for phylogenomic analysis. J. Microbiol. 2021, 59, 609–615. [Google Scholar] [CrossRef] [PubMed]
- Wilson, D.B. Microbial diversity of cellulose hydrolysis. Curr. Opin. Microbiol. 2011, 14, 259–263. [Google Scholar] [CrossRef] [PubMed]
- Collins, T.; Margesin, R. Psychrophilic lifestyles: Mechanisms of adaptation and biotechnological tools. Appl. Microbiol. Biotechnol. 2019, 103, 2857–2871. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Chang, J.; Yin, Q.; Wang, P.; Lu, M.; Wang, X.; Dang, X. Effect of the combined physical and chemical treatments with microbial fermentation on corn straw degradation. Bioresour. Technol. 2013, 148, 361–365. [Google Scholar] [CrossRef]
- Harindintwali, J.D.; Zhou, J.; Yu, X. Lignocellulosic crop residue composting by cellulolytic nitrogen-fixing bacteria: A novel tool for environmental sustainability. Sci. Total Environ. 2020, 715, 136912. [Google Scholar] [CrossRef]
Figure 1.
Morphological observation of strain QXD-8T using scanning electron microscopy. The cells were cultured in TSB at 28 °C for 2 days. Magnification levels of ×6.0k (A) and ×45.0k (B).
Figure 1.
Morphological observation of strain QXD-8T using scanning electron microscopy. The cells were cultured in TSB at 28 °C for 2 days. Magnification levels of ×6.0k (A) and ×45.0k (B).
Figure 2.
Maximum likelihood phylogenetic tree based on 16S rRNA gene sequences. Bootstrap values (%) were based on 1000 replicates and shown with more than 50% bootstrap support. The sequence of Actinotalea caeni EBR-4-2T was used as the outgroup. Bar, 0.010 substitution per nucleotide position. The name of the strain obtained in this study is bold.
Figure 2.
Maximum likelihood phylogenetic tree based on 16S rRNA gene sequences. Bootstrap values (%) were based on 1000 replicates and shown with more than 50% bootstrap support. The sequence of Actinotalea caeni EBR-4-2T was used as the outgroup. Bar, 0.010 substitution per nucleotide position. The name of the strain obtained in this study is bold.
Figure 3.
Maximum likelihood phylogenetic tree based on 120 conserved proteins in the genome. (A) The phylogenomic tree was composed of 285 Microbacterium genomes. Bar, 1 substitution per amino acid position. (B) The red part in subfigure A to show the details of the branch including QXD-8T. Bar, 0.1 substitution per amino acid position. Bootstrap values (%) were based on 1000 replicates, and more than 50% bootstrap support was exhibited. The name of the strain obtained in this study is bold.
Figure 3.
Maximum likelihood phylogenetic tree based on 120 conserved proteins in the genome. (A) The phylogenomic tree was composed of 285 Microbacterium genomes. Bar, 1 substitution per amino acid position. (B) The red part in subfigure A to show the details of the branch including QXD-8T. Bar, 0.1 substitution per amino acid position. Bootstrap values (%) were based on 1000 replicates, and more than 50% bootstrap support was exhibited. The name of the strain obtained in this study is bold.
Figure 4.
Optical density of the cultivation broth of strain QXD-8T growing under different conditions. The OD600 values of strains at 24 h of growth with different NaCl concentrations (A), pH values (B), and temperatures (C).
Figure 4.
Optical density of the cultivation broth of strain QXD-8T growing under different conditions. The OD600 values of strains at 24 h of growth with different NaCl concentrations (A), pH values (B), and temperatures (C).
Figure 5.
The hydrolysis circle of strain QXD-8T was stained with Congo red on the CAM plate. The cells were incubated at 15 °C for 1 day (A), 2 days (B), and 3 days (C). The cells were incubated at 28 °C for 1 day (D), 2 days (E), and 3 days (F). The graph represents the EI value (diameter of the hydrolysis zone and diameter of the colony) of the transparent hydrolysis zone of strain QXD-8T at 15 °C and 28 °C for different incubation times, and five duplicates were set for each experiment (G). All values are representative of five duplicates (****: p ≤ 0.0001).
Figure 5.
The hydrolysis circle of strain QXD-8T was stained with Congo red on the CAM plate. The cells were incubated at 15 °C for 1 day (A), 2 days (B), and 3 days (C). The cells were incubated at 28 °C for 1 day (D), 2 days (E), and 3 days (F). The graph represents the EI value (diameter of the hydrolysis zone and diameter of the colony) of the transparent hydrolysis zone of strain QXD-8T at 15 °C and 28 °C for different incubation times, and five duplicates were set for each experiment (G). All values are representative of five duplicates (****: p ≤ 0.0001).
Figure 6.
Time course of reducing sugar accumulation levels in the CAM-M medium using strain QXD-8T at 15 °C and 28 °C.
Figure 6.
Time course of reducing sugar accumulation levels in the CAM-M medium using strain QXD-8T at 15 °C and 28 °C.
Figure 7.
Time course of reducing sugar accumulation and cell growth with cellobiose. Growth curve of strain QXD-8
T. The growth with and without cellobiose was measured at 15 °C and 28 °C, respectively. The results of one-way ANOVA analyses are exhibited in
Figures S8–S11.
Figure 7.
Time course of reducing sugar accumulation and cell growth with cellobiose. Growth curve of strain QXD-8
T. The growth with and without cellobiose was measured at 15 °C and 28 °C, respectively. The results of one-way ANOVA analyses are exhibited in
Figures S8–S11.
Figure 8.
The decomposition process of cellulose using QXD-8T. The metabolism pathway was reconstructed based on the genome sequence. Note: The 15 genes encoding intracellular β-glucosidase (superscript red star) are Q9R08_02235, Q9R08_03730, Q9R08_06910, Q9R08_16030, Q9R08_16505, Q9R08_11550, Q9R08_03725, Q9R08_13945, Q9R08_14280, Q9R08_15860, Q9R08_19625, Q9R08_01910, Q9R08_09335, Q9R08_01530, and Q9R08_04245. The red question mark: the transport processes of the compounds were not annotated.
Figure 8.
The decomposition process of cellulose using QXD-8T. The metabolism pathway was reconstructed based on the genome sequence. Note: The 15 genes encoding intracellular β-glucosidase (superscript red star) are Q9R08_02235, Q9R08_03730, Q9R08_06910, Q9R08_16030, Q9R08_16505, Q9R08_11550, Q9R08_03725, Q9R08_13945, Q9R08_14280, Q9R08_15860, Q9R08_19625, Q9R08_01910, Q9R08_09335, Q9R08_01530, and Q9R08_04245. The red question mark: the transport processes of the compounds were not annotated.
Table 1.
Genome relatedness based on ANI, AAI, and dDDH values between QXD-8T and close related species of the genus Microbacterium.
Table 1.
Genome relatedness based on ANI, AAI, and dDDH values between QXD-8T and close related species of the genus Microbacterium.
| Close Related Species | ANI (%) | AAI (%) | dDDH (%) |
|---|
| Microbacterium yannicii G72T | 88 | 89 | 54.5 |
| Microbacterium trichothecenolyticum DSM 8608T | 85 | 85 | 44.8 |
| Microbacterium ureisolvens CFH S00084T | 84 | 85 | 43.3 |
| Microbacterium hibisci THG-T2.14T | 84 | 82 | 38.7 |
| Microbacterium jejuense THG-C31T | 83 | 82 | 36.7 |
| Microbacterium kyungheense THG-C26T | 83 | 81 | 34.5 |
| Microbacterium lacticum DSM 20427T | 80 | 69 | 19.1 |
Table 2.
Physiological characteristics of strain QXD-8T and eight Microbacterium species type strains with the highest similarity scores.
Table 2.
Physiological characteristics of strain QXD-8T and eight Microbacterium species type strains with the highest similarity scores.
| Characteristic | 1 * | 2 * | 3 | 4 | 5 | 6 | 7 | 8 | 9 |
|---|
| Motility | - | - | - | - | - | - | - | - | - |
| Colony colour | Y | Y | Y | Y | Y | Y | Y | Y | Y |
| Production of H2S | - | - | + | - | + | - | N/A | N/A | - |
| Temperature growth range (°C) | 15–37 | 20–37 | 04–37 | 15–37 | 25–30 | 12–35 | 10–40 | 10–40 | 12–35 |
| Optimum salinity (%) | 0 | 0 | 0 | 0 | 0 | 1–2 | 0–3 | 0–7 | 0–6 |
| Optimum pH | 7 | 7 | 7.5 | 7 | 8 | 7 | 7 | 7 | 7 |
| Catalase activity | + | + | + | + | + | + | + | - | + |
| Activity/assimilation (API 20NE): |
| Oxidase | + | + | + | - | - | + | + | - | + |
| Urease | - | - | - | + | + | - | - | + | + |
| Nitrate reduction | + | + | - | - | + | + | + | - | + |
| Gelatin hydrolysis | + | - | + | + | - | - | + | + | - |
| Aesculin iron citrate | + | + | N/A | + | N/A | + | + | N/A | N/A |
| N-Acetylglucosamine | + | + | N/A | - | N/A | + | + | N/A | + |
| Arabinose sugar | + | + | N/A | - | N/A | + | + | N/A | + |
| Acid production (API 50CH): |
| Mannitol | - | - | - | + | N/A | N/A | - | N/A | N/A |
| L-Arabinose | + | - | + | + | N/A | - | + | - | - |
| Methyl-β-D pyranoxyloside | + | - | N/A | + | N/A | - | - | N/A | + |
| D-Glucose | + | + | N/A | - | N/A | + | + | - | + |
| L-Rhamnose | - | - | N/A | + | N/A | N/A | + | + | N/A |
| myo-Inositol | - | - | N/A | - | N/A | - | + | + | - |
| Mannitol | + | + | N/A | - | N/A | + | + | - | + |
| Arbutin | w | - | N/A | - | N/A | + | - | N/A | + |
| D-Lactose | w | + | - | + | N/A | + | - | N/A | - |
| Sucrose | + | - | + | + | N/A | - | + | N/A | - |
| Trehalose | w | - | N/A | + | N/A | + | + | + | - |
| Melezitose | + | - | N/A | + | N/A | - | - | N/A | - |
| Raffinose | + | - | N/A | + | N/A | - | - | N/A | - |
| Glycogen | + | + | - | + | N/A | + | + | N/A | + |
| Xylitol | w | - | N/A | - | N/A | N/A | - | N/A | N/A |
| Gentiobiose | + | - | N/A | + | N/A | + | - | N/A | - |
Table 3.
Prediction results of signal peptide and transmembrane region of cellulose hydrolysis related genes in strain QXD-8
T (
Table S1).
Table 3.
Prediction results of signal peptide and transmembrane region of cellulose hydrolysis related genes in strain QXD-8
T (
Table S1).
| Activity Rates in Family | CAZy Family | Locus Tag
(Q9R08_X) | Signal Peptide Prediction |
|---|
| Protein Type | Cleavage Site |
|---|
| endo-β-1,4-glucanase | GH5 | 14620 | NA | - |
| GH6 | 10055 | (Tat/SPI) | 47 and 48 |
| 14230 | (Sec/SPI) | 39 and 40 |
| GH10 | 11975 | (Sec/SPI) | 29 and 30 |
| β-glucosidase | GH1 | 02235 | NA | - |
| 03730 | NA | - |
| 06910 | NA | - |
| 16030 | NA | - |
| 16505 | NA | - |
| GH2 | 11550 | NA | - |
| GH3 | 03725 | NA | - |
| 13945 | NA | - |
| 14280 | NA | - |
| 14550 | (Sec/SPI) | 43 and 44 |
| 15860 | NA | - |
| 19625 | NA | - |
| 01910 | NA | - |
| 06855 | (Sec/SPII) | 22 and 23 |
| 09335 | NA | - |
| GH39 | 01530 | NA | - |
| GH116 | 04245 | NA | - |
| Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}