Abstract
Mycoplasma bovis is a global problem for the cattle industry due to its high infection rates and associated morbidity, although its pathophysiology is poorly understood. In this study, the M. bovis transcriptome and proteome were analyzed to further investigate the biology of clinical isolates of M. bovis. A differential analysis of M. bovis, a clinical isolate (NX114), and an international type strain (PG45) at the logarithmic stage of growth, was carried out using prokaryotic transcriptome and 4D-label-free quantitative non-labeled proteomics. Transcriptomics and proteomics identified 193 DEGs and 158 DEPs, respectively, with significant differences in 49 proteins/34 transcriptomic CDS post-translational protein sequences (15 jointly up-regulated and 21 jointly down-regulated). GO comments indicate membrane, cytoplasmic and ribosome proteins were important components of the total proteins of M. bovis NX114 clinical isolate. KEGG enrichment revealed that M. bovis NX114 is mainly associated with energy metabolism, the biosynthesis of secondary metabolites, and the ABC transporters system. In addition, we annotated a novel adhesion protein that may be closely related to M. bovis infection. Triosephosphate isomerase (TpiA) and Pyruvate kinase (Pyk) genes may be the key enzymes that regulate the growth and maintenance of M. bovis and are involved in the pathogenic process as virulence factors. The results of the study revealed the biology of different isolates of M. bovis and may provide research ideas for the pathogenic mechanism of M. bovis.
1. Introduction
Mycoplasma bovis is a pathogenic microorganism that causes a wide range of diseases in cattle, including keratoconjunctivitis, mastitis, endometritis, otitis media, arthritis, pneumonia, and respiratory syndromes, and it has caused serious economic losses to the global cattle industry [1,2,3]. The lack of a cell wall on the surface of mycoplasmas and its resistance to beta-lactams and other antimicrobial compounds limit the effectiveness of antibiotic therapy [4]. The emergence of antibiotic resistance has also hampered the treatment of the disease, leading to the emergence of more resistant M. bovis isolates, especially to fluoroquinolones, doxycycline, and enrofloxacin antibiotics [5]. There is also a lack of effective vaccines to prevent M. bovis infections, so prevention and control of the disease are currently extremely difficult.
Proteins are important carriers of cellular functions and life activities and are the final form of expression of genetic information within microorganisms. The surface of mycoplasmas lacks cell wall structures, and M. bovis lacks tip structures for adhesion to host cells, like those specialized for Mycoplasma pneumoniae and Mycoplasma genitalium [6]. Therefore, it is known that M. bovis membrane proteins and membrane-associated proteins are crucial for interaction with host cells. It can play a role in the process of infecting the host by participating in bacterial adhesion, virulence-associated factors, mediators of cell signaling, effectors that modulate host immunity, and immunogens [7,8]. Thus, M. bovis proteins play a role in inducing host pathogenicity and immune response throughout the infection process. In addition, numerous studies have analyzed the secreted proteins of various mycoplasmas by untagged proteomics, isotope-tagged relative and absolute quantification (iTRAQTM), and liquid chromatography–tandem mass spectrometry (LC-MS/MS) [9,10,11,12]. Meanwhile, transcriptomics was used to identify important phosphatases involved in the glycolytic pathway in M. bovis [13]. Proteomics was used to comparatively analyze differentially secreted proteins of strongly virulent and attenuated strains of M. bovis, and advances have been made in pathogenic mechanisms [14].
Transcriptomes and proteomes are closely related to both upstream and downstream genomics. Merging transcriptome and proteome analyses will help to obtain a panoramic view of M. bovis in terms of gene expression and regulation. In this study, we analyzed the homologous and unique proteins between M. bovis clinical isolate NX114 (pneumonia isolate) and international standard strain PG45 (mastitis isolate) using a combination of prokaryotic transcriptomics and 4D-label-free quantitative proteomics. The goal is to further investigate the M. bovis proteome’s virulence, resistance, or immunological properties in order to elucidate its pathogenesis or identify effective drug or vaccine candidates.
2. Materials and Methods
2.1. Bacterial Strains and Media
M. bovis clinical strain NX114 (GenBank accession no. CP135997) was obtained from diseased Holstein cows in China, while M. bovis international type strain PG45 was generously contributed by Professor Aizhen Guo of Huazhong Agricultural University in China. The two strains were preserved in PPLO broth with 40% glycerol at −80 °C in a laboratory before being cultured in PPLO [15] broth before use.
2.2. Transmission Electron Microscopy
Samples were first treated with a 3% glutaraldehyde solution, then postfixed with 1% osmium tetroxide. They were then dehydrated using a series of acetone solutions, infiltrated with Epox 812 for an extended period, and finally embedded. The semithin sections were stained with methylene blue, while the ultrathin sections were cut using a diamond knife and stained with uranyl acetate and lead citrate. The sections were analyzed using a JEM-1400-FLASH Transmission Electron Microscope.
2.3. Transcriptomics Analysis of M. Bovis
The Trizol Reagent (Invitrogen Life Technologies, Thermo Scientific, Bremen, Germany) was used to separate the total RNA. The determination of quality and integrity was conducted using a NanoDrop spectrophotometer (Thermo Scientific, Bremen, Germany) and a Bioanalyzer 2100 system (Agilent, Palp Alto, CA, USA). The Ribo-Zero rRNA Removal Kit (Illumina, San Diego, CA, USA) is used for mRNA sequencing. The first cDNA strand was synthesized using random oligonucleotides and SuperScript III. Subsequently, the synthesis of the second-strand cDNA was carried out utilizing DNA Polymerase I and RNase H. The remaining overhangs were transformed into blunt ends using exonuclease/polymerase activities, and the enzymes were then eliminated. Following the adenylation of the 3′ ends of the DNA fragments, the Illumina PE adapter oligonucleotides were ligated in order to facilitate hybridization. In order to choose cDNA fragments that are precisely 300 base pairs long, the library fragments were purified using the AMPure XP technology (Beckman Coulter, Beverly, CA, USA). DNA fragments including ligated adaptor molecules on both ends were specifically amplified using the Illumina PCR Primer Cocktail in a 15-cycle PCR reaction. The products underwent purification using the AMPure XP system and were quantified using the Agilent high sensitivity DNA test on a Bioanalyzer 2100 system (Agilent). The sequencing library was then analyzed using a NextSeq 500 platform (Illumina) by Shanghai Personal Biotechnology Cp., Ltd., Shanghai, China.
2.4. Label-Free Quantitative Proteomics Analysis of M. Bovis
Protein was extracted from M. bovis samples using SDT lysis buffer (4% SDS, 100 mMDTT, and 100 mM Tris-HCl at pH 8). The samples were cooked for 5 min, then ultrasonically sonicated and boiled for another 5 min. Cellular debris was removed by centrifugation at 16,000 g for 15 min. The supernatant was collected and measured using a BCA Protein Assay Kit (Bio-Rad, Hercules, CA, USA). The FASP technique, as reported by Wisniewski, Zougman et al. [16], was used to digest 200 μg of protein each sample. To prevent reduced cysteine, the detergent, DTT, and IAA were added to UA buffer. Finally, the protein solution was digested with trypsin (Promega) at a 50:1 ratio overnight at 37 °C. The peptide was obtained by centrifugation at 16,000× g for 15 min. The peptide was desalted with C18 StageTip for further LC-MS analysis. The Nanodrop One gadget measured peptide concentrations using OD280.
The LC-MS/MS analysis was conducted using a Q Exactive Plus mass spectrometer, which was connected to an Easy 1200 nLC system from Thermo Fisher Scientific. The peptide was first placed onto a trap column (100 μm × 20 mm, 5 μm, C18, Dr. Maisch GmbH, Ammerbuch, Germany) using buffer A (0.1% formic acid in water). The EASY-nLC system (Thermo Fisher Scientific, Bremen, Germany) was used to perform reverse-phase high-performance liquid chromatography (RP-HPLC) separation. A self-packed column (75 μm × 150 mm; 3 μm ReproSil-Pur C18 beads, 120 Å, Dr. Maisch GmbH, Ammerbuch, Germany) was employed at a flow rate of 300 nL/min. The mobile phase A used in RP-HPLC consisted of water containing 0.1% formic acid, whereas the mobile phase B consisted of 95% acetonitrile containing 0.1% formic acid. The peptides were separated over a period of 120 min using a gradual increase in the concentration of buffer B. The acquisition of MS data was performed using a data-dependent top20 approach, which dynamically selects the most abundant precursor ions from the survey scan (300–1800 m/z) for HCD fragmentation. The instrument was operated with the peptide recognition mode on. An internal standard with a lock mass of 445.120025 Da was used for mass calibration. The complete MS scans were obtained with a resolution of 70,000 at m/z 200 and 17,500 at m/z 200 for the MS scan. The maximum injection time for MS was set to 50 ms, and for MS/MS, it was also set to 50 ms. The collision energy was adjusted to a normalized value of 27, and the isolation window was set at 1.6 T. The length of the dynamic exclusion was 60 s.
The MS data were analyzed using MaxQuant 1.6.0.16 software. MS data were searched against the Uniprot-Mycoplasmopsis bovis (Mycoplasma bovis) (28903)-6273-20221207. Trypsin was chosen as a digestive enzyme. For database search, the maximum two missed cleavage sites and a mass tolerance of 4.5 ppm for precursor ions and 20 ppm for fragment ions were specified. The carbamidomethylation of cysteines was considered a fixed modification, while the acetylation of the protein’s N-terminal and methionine oxidation were characterized as variable modifications for database searching. The database search results were filtered and exported with <1% false discovery rate (FDR) at the pep-tide-spectrum-matched and protein levels, respectively. MaxQuant was used for label-free quantification, using the intensity determination and normalization algorithms published before. We computed the “LFQ intensity” of each protein in various samples as the best estimate, meeting all pairwise peptide comparisons, and found that this LFQ intensity was almost on the same scale as the summed peptide intensities. The quantitative protein ratios were weighted and adjusted using Maxquant’s median ratio. Proteins with a fold change ≥ 1.5-fold and a p-value < 0.05 were classified as substantially differentially expressed.
2.5. Bioinformatics Analysis
Bioinformatics data were analyzed using Perseus, Microsoft Excel, and R statistical computer tools. The pheatmap package, based on the open-source statistical language R25, was used to conduct hierarchical clustering analysis, using Euclidean distance as the distance metric and the complete approach as the agglomeration technique. To annotate the sequences, information was gathered from Uni-ProtKB/Swiss-Prot, the Kyoto Encyclopedia of Genes and Genomes (KEGG), and Gene Ontology. Fisher’s exact test was used for GO and KEGG enrichment analyses, along with FDR correction for multiple testing. GO terms were divided into three categories: biological processes (BPs), molecular functions (MFs), and cellular components (CCs). The enriched GO and Kegg pathways were statistically significant (p < 0.05). Protein–protein interaction (PPI) networks were also built using the STRING database and the Cytoscape software v.3.9.1.
2.6. Combined Proteomic and Transcriptomic Analysis of M. Bovis
The combined analysis of proteomics and transcriptomics includes statistics of common differential proteins or genes between proteomics and transcriptomics, a comparison between proteomics and transcriptomics, an expression cluster analysis (Cluster_Heatmap) and a differential multiple correlation analysis (FC_correlation). KEGG analysis (Total_KEGG) is also included. All analyses in this project were performed using R 4.2.2.
2.7. qRT-PCR Analyses
TRIzol reagent was used to isolate total RNA from the M. bovis samples. The RevertAid First Strand cDNA Synthesis Kit was used to make cDNA samples. qRT-PCR was performed using the FastStart Universal SYBR Green reagent and analyzed with the StepOnePlus RealTime PCR Systems (Applied Biosystems, Waltham, MA, USA). Gene expression was measured using the comparative threshold cycle technique (2−ΔΔCt) with GAPDH. The primer sequences utilized are shown in Table S1.
3. Results
3.1. Transmission Electron Microscopy of M. Bovis
Transmission electron microscopy of the organelle structures of M. bovis NX114 and PG45 revealed a large number of ribosomes and soluble proteins in the cytoplasm of both strains (Figure 1).
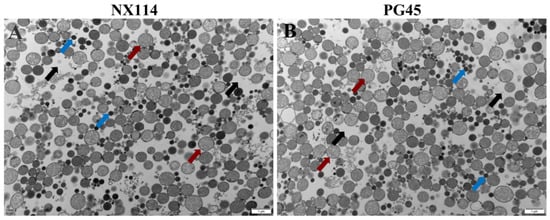
Figure 1.
Organelles of M. bovis NX114 vs. PG45 transmission electron microscopy (12,000×). (A) M. bovis NX114. (B) M. bovis PG45. (black arrow: cytoplasm; blue arrow: soluble protein; red arrow: ribosome).
3.2. Analysis of Prokaryotic Transcripts of M. Bovis
We compared the gene expression profiles of the M. bovis NX114 and PG45 groups using the Illumina platform to explore the gene profile of clinical isolate NX114. We then constructed and sequenced a total of six cDNA libraries, three from the NX114 group and three from the PG45 group. The total raw reads for the NX114 group were 15655472, 14150886, and 15689858, and the total raw reads for the PG45 group were 15509064, 15504262, and 15464574, respectively. After quality filtering (e.g., Q20, Q30, and the error rate), the clean reads were 14701712, 13306834, and 14770858 for the NX114 group, and 14770858, 14647256, and 14551518 for the PG45 group.
The RNA-seq of M. bovis NX114 and PG45 produced a total of 718 unigenes, and using BLAST software v.2.10.1 to annotate individual unigenes into 10 databases, a total of 718 unigenes (100%) were annotated in NR, 688 (95.82%) were annotated in eggNOG, 560 (77.99%) were annotated in eggNOG_Category, 453 (63.09%) were annotated in Swissprot, 419 (58.36%) were annotated in KEGG, 262 (36.49%) were annotated in Pathway, 530 (73.82%) were annotated by GO in Medium, 98 (13.65%) were annotated in by VFDB, 162 (22.56%) were annotated in by PHI, and 34 (4.74%) were annotated in by CARD (Figure 2A). FPKM density values showed gene expression profiles in M. bovis NX114 and PG45 (Figure 2B), and PCA analysis showed significant differences in the transcriptomes (Figure 2C). Based on the thresholds (|log2FoldChange| > 1 and p-value < 0.05), 193 DEGs were identified, of which 106 were up-regulated and 87 were down-regulated (Table S2 and Figure 2D). Specific databases showed that a total of 26 genes in the two M. bovis strains were simultaneously annotated in the CARH, VFDB, and PHI databases; 38 genes were found in both the VFDB and PHI, as well as 2 genes in both the CARH and PHI (Figure 3A). Among them, six unique genes (NX114:0, PG45:6) were annotated in the VFDB database and four unique genes (NX114:1, PG45:3) were annotated in the PHI database (Figure 3B). In addition, a total of 10 DEGs (3 up-regulated, 7 down-regulated) of M. bovis NX114 vs. PG45 were annotated in the CARH database, 26 DEGs (11 up-regulated, 15 down-regulated) were annotated in the VFDB database, and 42 DEGs (14 up-regulated, 28 down-regulated) were annotated in the PHI database, with a total of 5 DEGs (1 up-regulated, 4 down-regulated) being annotated in three databases at the same time (Figure 3C,D).
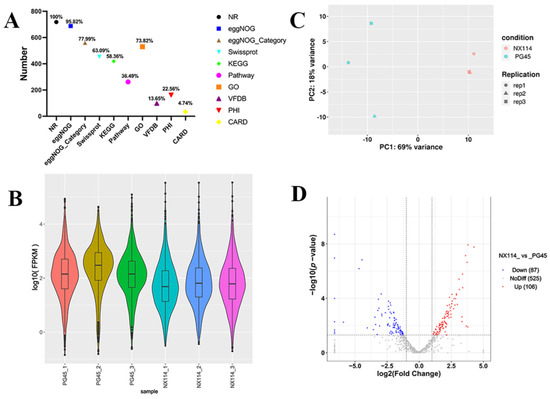
Figure 2.
The general pattern of RNA-seq data and the identification of DEGs for M. bovis NX114 vs. PG45. (A) Genomic gene annotation information. (B) FPKM density distribution. (C) PCA analysis. (D) Volcano map of DEGs.
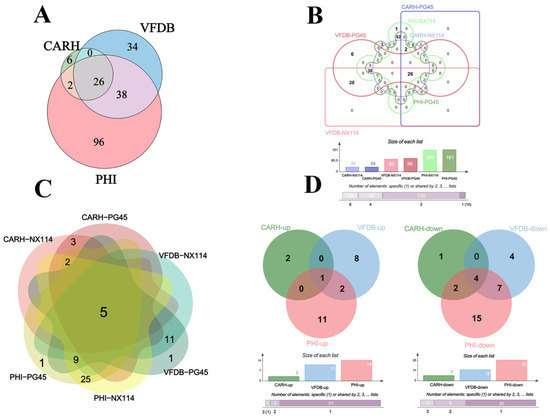
Figure 3.
The annotation of the mRNAs of M. bovis NX114 vs. PG45. (A) Equal scale area Venn diagrams of the annotation information of the two strains of M. bovis together at CARH, VFDB and PHI. (B) Interactive Venn plots of the annotation information of the two strains of M. bovis together at CARH, VFDB and PHI. (C) The annotation information of DEGs of M. bovis NX114 vs. PG45. (D) Up- and down-regulated annotation information for DEGs of M. bovis NX114 vs. PG45.
We performed GO analysis on 193 DEGs genes to better understand the underlying biological processes of M. bovis. We assigned the 193 DEGs to 135 GO categories (up-regulated DEGs accounted for 71 and down-regulated DEGs accounted for 64) (Table S3). The GO enrichment analysis of DEGs showed that most of the highest enriched terms were MF and BP. The MF category mainly included catalytic activity (GO:0003824), carbohydrate derivative binding (GO:0097367), catalytic activity acting on RNA (GO:0140098) and ion binding (GO:0043167), and for the BP category, cellular component biogenesis (GO:0044085), small molecule biosynthetic process (GO:0044283) and phosphate-containing compound metabolic process (GO:0006796) were the top three enriched GO terms (Figure S1). The KEGG pathway annotation and enrichment analysis revealed the significant assignment of DEGs to 41 metabolic pathways (Figure 4A and Figure S2), with major enrichment in the glycolysis/gluconeogenesis (ko00010), purine metabolism (ko00230), beta-lactamase resistance (ko01501), citrate cycle (TCA cycle) (ko00020), nicotine and nicotinamide metabolism (ko00760), and RNA degradation (ko03018) pathways (Figure 4B). The protein–protein interaction analysis network was composed of 22 nodes and 120 edges, with nodes representing M. bovis proteins and edges representing connections. The proteins MBOVPG45_RS01305, MBOVPG45_RS01460, MBOVPG45_RS01385, and MBOVPG45_RS01465 are the core proteins of the network (Figure 4C).
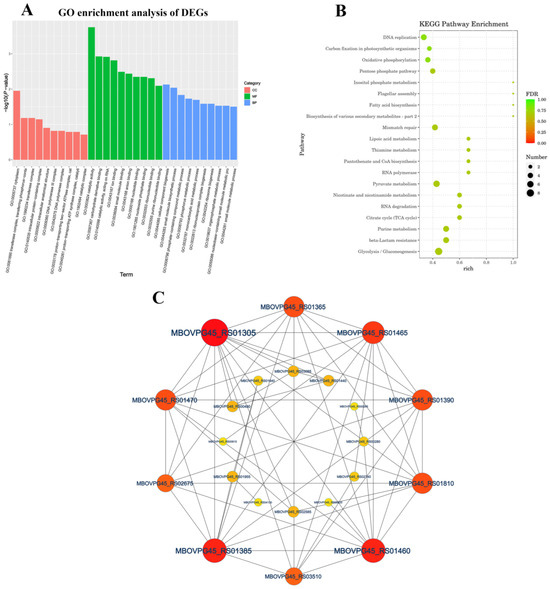
Figure 4.
Functional categorization of DEGs of M. bovis NX114 vs. PG45. (A) GO enrichment analysis of DEGs. (B) KEGG enrichment analysis of DEGs. (C) PPi network analysis of DEGs.
3.3. Four-Dimensional Label-Free Quantitative Proteomics Analysis of M. Bovis
Four-dimensional label-free quantitative proteomics comparatively analyzed the protein characterization of M. bovis. The SDS-PAGE analysis revealed that the extracted M. bovis proteins had wide ranges of molecular weights. There were also notable differences in the protein bands between M. bovis strains NX114 and PG45, which allowed for further mass spectrometry analysis (Figure S3). Mass spectrometry analysis identified a total of 11,035 unique peptides and 908 proteins, with the peptide lengths primarily distributed in the range of 7 to 15 (Figure S4). Venn diagrams revealed 885 overlapping proteins among the 908 identified (Figure 5A). The PCA analysis showed that the NX114 and PG45 groups were separate, which means that there are important differences between the M. bovis groups (Figure 5B). Cluster analysis revealed similar protein abundance profiles within the same group of M. bovis, with significant differences observed between groups (Figure 5C). Differentially expressed proteins (DEPs) were screened for 58 up-regulated and 100 down-regulated proteins based on thresholds (FC > 1.5 or <1/1.5 and p-value < 0.05) (Table S4 and Figure S5).
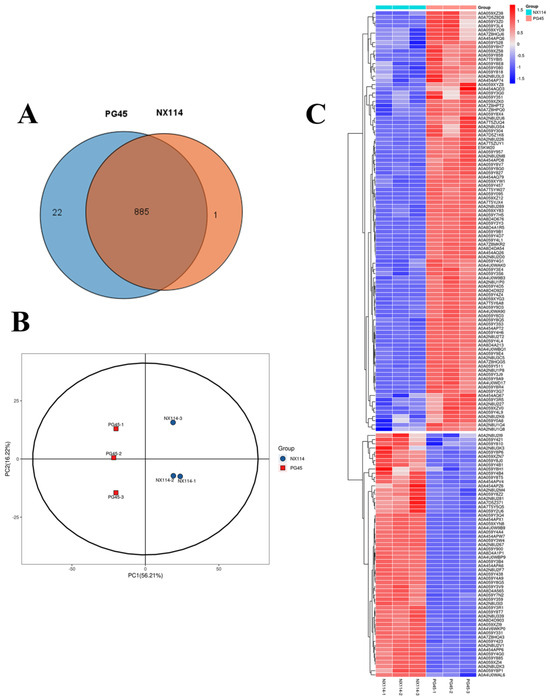
Figure 5.
The proteomic analysis of M. bovis NX114 vs. PG45. (A) A Venn diagram of the proteome. (B) A PCA analysis of proteomic profiles. (C) A cluster analysis of DEPs of M. bovis.
The GO annotations of 158 DEPs revealed 93 functional terms at level 2, including 35 biological processes (BPs), 17 cellular components (CCs), and 42 molecular functions (MFs) (Figure 6A). The GO functional enrichment analysis of DEPs showed that the 20 most significantly enriched GOs were mainly MFs and BPs, mainly N-methyltransferase activity (GO:0008170), pseudouridine synthase activity (GO:0009982), DNA binding (GO:0003677), macromolecule modification (GO:0043412), RNA modification (GO:0009451) and pseudouridine synthesis (GO:0001522) (Table S5 and Figure S6). In addition, the KEGG pathway revealed 15 KEGG pathway components out of 158 DEPs (Table S6), in which up-regulated DEPs were significantly enriched, mainly in metabolic pathways (mbv01100) and glycolysis/gluconeogenesis (mbv00010) (p < 0.05). Down-regulated DEPs were significantly enriched, mainly in homologous recombination (mbv03440) (Figure 6B).
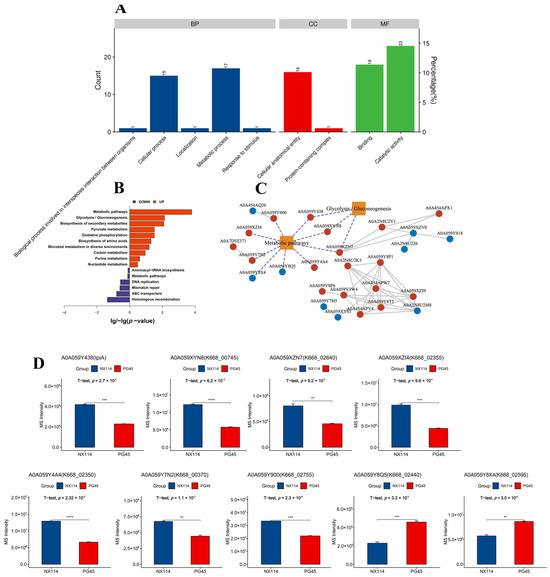
Figure 6.
Functional categorization of DEPs of M. bovis NX114 vs. PG45. (A) GO annotation of DEPs. (B) KEGG enrichment analysis of DEPs. (C) Network analysis of significantly enriched pathways (p < 0.05) interacting with significantly different proteins, red vertices indicate up-regulated proteins, blue vertices indicate down-regulated proteins, and dotted lines represent key coding proteins. (D) Abundance analysis of key DEPs. ** p < 0.05, *** p < 0.01 and **** p < 0.001.
The Protein Functional Interaction Network (PFIN) analysis was performed to construct significantly enriched pathways (p < 0.05) with significant differences in protein interactions. Metabolic pathways and glycolysis/gluconeogenesis (p < 0.05) were the core metabolic pathways (Figure 6C). The up-regulated DEPs AOA059Y438 (p = 2.7 × 10−4), AOA059XYN8 (p = 6.2 × 10−5), AOA059XZN7 (p = 9.2 × 10−3), AOA059XZl4 (p = 9.6 × 10−4), AOA059Y4A4 (p = 2.32 × 10−5), AOA059Y7N2 (p = 1.1 × 10−3) and AOA059Y900 (p = 2.3 × 10−4). The down-regulated DEPs AOA059Y8Q5 (p = 3.3 × 10−4) and AOA059Y8X4(p = 3.0 × 10−3) are key points that may be influencing metabolic or signaling pathways throughout the bovine mycoplasma system (Figure 6D).
By analyzing the subcellular localization of M. bovis proteins annotated and counted by cellular components (CCs) in the GO database, a total of 170 (152 shared, 18 unique) proteins of the two strains of M. bovis were localized to the membrane (NX114:64, PG45:68), cytoplasm (NX114:42, PG45:48) and ribosome (NX114:49, PG45:51) organelles (Figure 7). In addition, a total of 10 DEPs of M. bovis NX114 vs. PG45 were localized to the membrane (6 up-regulated, 4 down-regulated) and 4 DEPs were localized to the cytoplasm (1 up-regulated, 3 down-regulated) (Table 1).
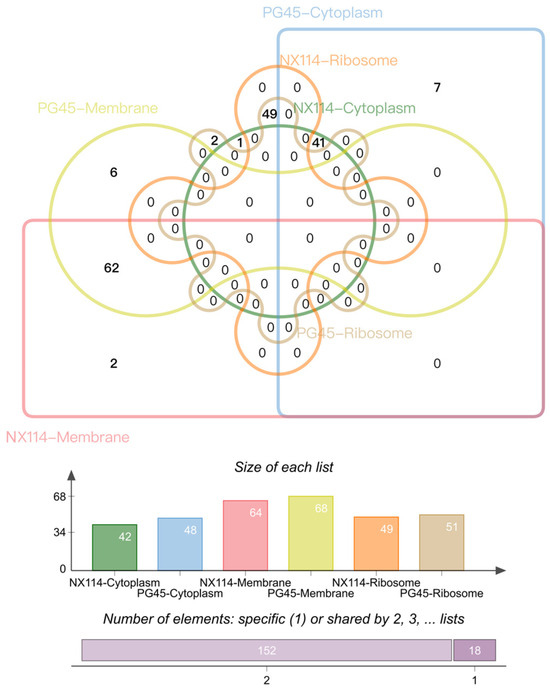
Figure 7.
Results of CC subcellular localization of M. bovis NX114 vs. PG45 histones.

Table 1.
The binding energy of compounds and core targets.
3.4. Correlation Analysis of the Transcriptome and Proteome of M. Bovis
We further analyzed the correlation between the M. bovis NX114 and PG45 transcriptomes and proteomes, due to the inconsistency between the protein level and the transcription level. We extracted a total of 718 sequences from the identified transcripts and compared them with 905 identified protein sequences for sequence similarity. The results revealed that 631 (87.88%) CDS post-translational protein sequences of the transcripts aligned with a total of 870 (96.13%) proteins. A nine-quadrant association analysis revealed shared protein correlations (R = 0.44) with the same and opposite trend in shared gene/protein differential folds (Figure 8A). For genes that were significantly different in both proteomes and transcriptomes, we found a total of 49 protein/34 transcriptome CDS post-translational protein sequences, with the 15 DEGs and DEPs that were concurrently up-regulated in expression and the 21 DEGs and DEPs that were concurrently down-regulated being the most critical (Table 2, Figure 8B and Figure 9A).
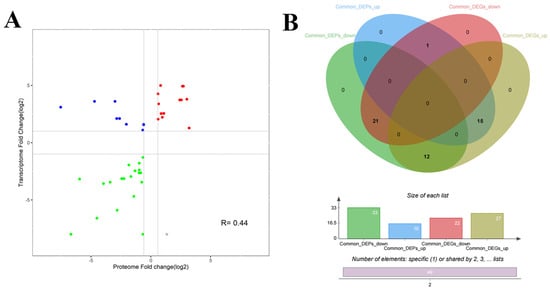
Figure 8.
Statistical analysis of DGPs and DEGs of M. bovis (A) Common differential gene/protein differential fold correlation analysis. (B) Venn diagrams of DGPs and DEGs.
Table 2.
DGPs and DEGs shared by M. bovis NX114 vs. PG45.
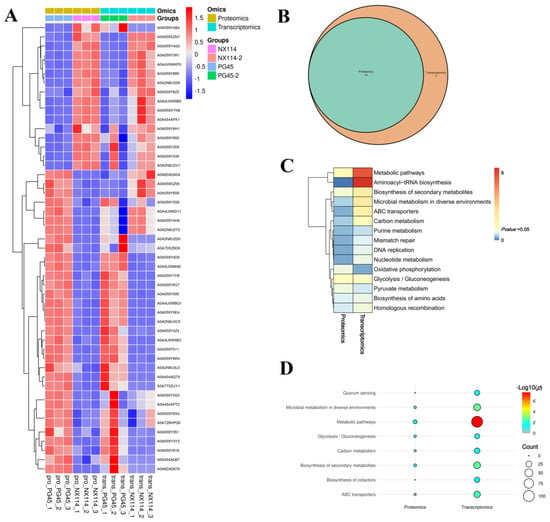
Figure 9.
Joint analysis of M. bovis DGPs and DEGs. (A) Shared significant difference gene/protein clustering analysis. (B) KEGG comparison Venn diagram. (C) pvalue_heatmap. (D) Significant KEGG comparison bubble plot.
We redrew the KEGG pathway annotation using the corresponding protein sequences of the transcriptome because the KEGG analysis of the transcriptome did not align with the chosen proteome reference. The shared KEGG analyses of the respective enrichment results revealed a total of 15 identical pathways (Figure 9B,C). The analysis of the proteome or transcriptome revealed eight significant pathways: metabolic pathways (mbv01100), glycolysis/gluconeogenesis (mbv00010), carbon metabolism (mbv01200), quorum sensing (mbv02024), biosynthesis of cofactors (mbv01240), microbial metabolism in diverse environments (mbv01120), biosynthesis of secondary metabolites (mbv01110), and ABC transporters (mbv02010) (Figure 9D). The energy metabolism pathways enriched the up-regulated DGPs TpiA and Pky among the six co-significant pathways (Figure 10)
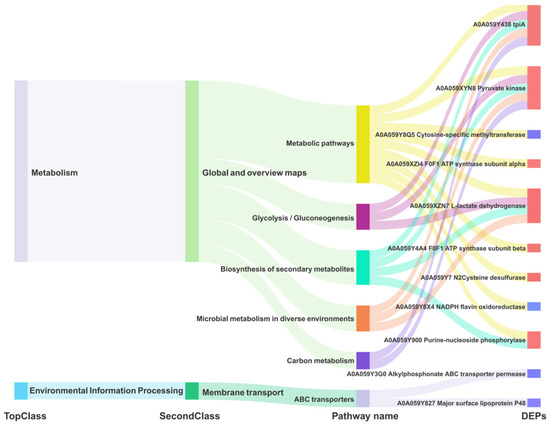
Figure 10.
Dynamic Mulberry diagram of the common significant KEGG pathway of M. bovis DGPs and DEGs.
3.5. qRT-PCR Verification
We selected some of the shared DEGs for qRT-PCR validation analysis, and calculated the expression of the corresponding genes in each group (2−ΔΔCt) using stably expressed GAPDH as an internal reference. The genes MBOVPG45_RS04010 (vspK), MBOVPG45_RS01760 (Lipoprotein), MBOVPG45_RS03700 (Lipoprotein), MBOVPG45_RS00040 (NADH-dependent flavin oxidoreductase), MBOVPG45_RS01765 (Lipoprotein), MBOVPG45_RS01755 (Lipoprotein), MBOVPG45_RS03705 (Glycerol ABC transporter, permease component), MBOVPG45_RS03865 (Ribosomal RNA small subunit methyltransferase I), MBOVPG45_RS03710 (Glycerol ABC transporter permease), MBOVPG45_RS01400 (HAD family hydrolase) and MBOVPG45_RS02835 (Lipoprotein) were down-regulated, and the genes MBOVPG45_RS01555 (Membrane lipoprotein P81), MBOVPG45_RS02985 (Lipoprotein_10 domain-containing protein) and MBOVPG45_RS00775 (Pyruvate kinase) were up-regulated (Figure 11). Although the degree of difference in gene expression varied between RNA-seq and qRT-PCR data, the DEGs, except MBOVPG45_RS01400 and MBOVPG45_RS02835, showed consistent expression trends, demonstrating the validity of the results of prokaryotic transcriptome and 4D-label-free quantitative proteomics analyses (Figure 11A).
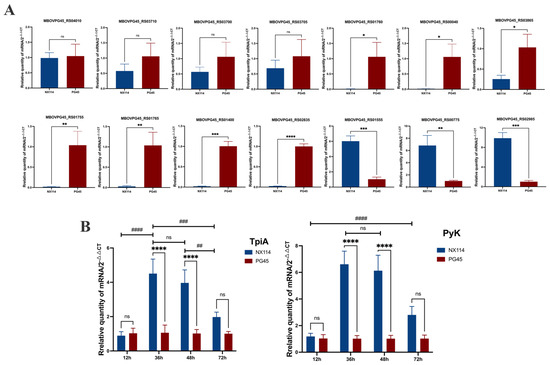
Figure 11.
The validation of differentially expressed gene expression levels in M. bovis NX114 vs. PG45. (A) The validation of RNA-seq data for randomly selected genes by real-time PCR. * p < 0.05, ** p < 0.01, *** p < 0.001 and **** p < 0.0001 ns (no significance) p > 0.05. (B) qRT-PCR was used to detect the relative mRNA expression of TpiA and PyK genes at different growth stages. ## p < 0.01, ### p < 0.001 and #### p < 0.0001, ns (no significance) p > 0.05.
In addition, the mRNA expression of TpiA and Pyk genes was not different between M. bovis NX114 vs. PG45 at the end of the delayed phase (12 h), but was significantly up-regulated at the end of the logarithmic phase (36 h) and at the persistence of the stabilized phase (48 h). The mRNA expression of TpiA and Pyk genes was not different during the decline phase (72 h) (Figure 11B).
4. Discussion
Transcriptomics and proteomics are two important tools for obtaining gene expression quantities. Proteomics provides data on the nature of the final gene products (proteins), complements comparative genomics and transcriptomics, and has a significant impact on microbiology in that it is able to generate proteomic profiles that can provide a detailed understanding of bacterial gene expression under specific conditions [17]. The biological functions of the transcription–protein dual-omics exhibit a clear backward and forward relationship, yet they are subject to influence. So, this study uses a combined transcription–protein dual-omics approach to look at the rules and nature of M. bovis’ life activities at different omics levels. It also shows how the two play a part in controlling each other and how the biology of different isolates fits together as a whole.
4.1. Important Components of Total Proteins of M. Bovis’ Clinical Strain
M. bovis has a size between that of bacteria and viruses, and does not have a nucleus and organelles such as mitochondria and Golgi apparatus, so its growth and metabolism need to obtain nutrients from the outside world [18]. We could clearly observe the cell membrane structure, the relatively low electron density of lipids comprising the intermediate layer, and the large number of ribosomes visible in both cytoplasms, as observed by transmission electron microscopy (Figure 1). The subcellular localization of this study showed that the membrane, cytoplasmic, and ribosomal proteins of the M. bovis NX114 clinical isolate are important components of total proteins. Among them, a total of 10 DEPs of M. bovis NX114 vs. PG45 were localized to the membrane, and 4 DEPs were localized to the cytoplasm (Table 1). Because there is no cell wall, M. bovis cell membranes can more fully fuse with host cells and exchange intracellular components [19]. This suggests that membrane proteins and cytoplasmic proteins play an important role in the interaction between M. bovis and its host. These key proteins serve a variety of biological functions, including acting as virulence factors, the invasion of the host, adhesion, signaling, nutrient uptake, immune system regulation, and toxic metabolite release [19].
4.2. Analysis of M. Bovis Adhesion Protein
Adhesins are key proteins in M. bovis-infected host cells that play important roles in pathogenic processes, including pathogen infection, cell invasion, colonization, immune escape, and virulence factors [20,21]. More than 10 species of M. bovis adhesins are known, which contain a variety of membrane surface proteins, some with immunogenic and conserved functions [22,23,24]. In addition, Plg, FN, HS and APLP2 proteins are now known to bind to M. bovis adhesins [20]. In this study, the M. bovis NX114 clinical isolate was found to contain six known adhesion proteins, three of which were differential proteins (Table S4). In addition, a potential M. bovis adhesion protein (A0A059XYW1) was annotated in this study (Table S4), and it remains to be verified whether it is involved in adhesion during M. bovis infection and promotes interactions with host–cell tight junctions.
4.3. Potential Virulence-Related Proteins Overrepresented in M. Bovis NX114 Whole Cell Proteins
To identify potential virulence-associated genes in M. bovis NX114, we used VFDB analysis to show that a total of 98 genes were associated with virulence in the two M. bovis strains, including 26 DEGs (11 up-regulated and 15 down-regulated) (Figure 3). Several studies have shown that P48, an important membrane protein associated with M. bovis virulence, can induce host–cell apoptosis through an endoplasmic reticulum stress-dependent signaling pathway [25]. This study found that P48 expression was downregulated. In addition, the nucleotide sequence of the M. bovis PG45 genome is known to have virulence-associated variable surface lipoproteins (Vsps), including VspE, VspI, VspO, VspJ, VspN, VspK, VspL, VspM, VspB, VspG, VspA, VspH, and VspF [26]. In this study, all 13 Vsps were jointly identified from two M. bovis strains, in which HYD67_03760, VspA, VspK, VspO, and VspJ were significantly down-regulated in DEPs and VspJ was lacking in M. bovis NX114, which indicated that the (Vsp) gene cluster differed less from M. bovis PG45 but differed more from M. bovis HB0801, which contained less than half of the Vsps [26]. The results of this study provide an important reference for further identification of potential virulence-related proteins in M. bovis.
4.4. Trend in Common DGPs and DEGs, and Pathways at All Levels of M. Bovis
The combined transcriptome–proteome analysis of M. bovis revealed a significant difference in 49 proteins out of 34 transcriptome CDS post-translational protein sequences, of which 15 were jointly up-regulated and 21 were jointly down-regulated. These proteins primarily included lipoproteins, chaperones, ABC transporters, structural domain proteins, ribosomal proteins, and key enzymes involved in glycolysis. The functional annotation and enrichment analyses of DEGs and DEPs revealed that DEPs were mainly enriched in metabolic pathways, glycolysis/gluconeogenesis, carbon metabolism, cofactor biosynthesis, microbial metabolism, cofactor biosynthesis in diverse environments, biosynthesis of secondary metabolites, and ABC transporters.
M. bovis has a limited biosynthetic capacity and requires nutrients from host cells to complete metabolism, as well as to evade host immune responses and damage cells [27]. The combined KEGG analysis revealed the potential for these proteins to exert their own virulence by influencing various metabolic pathways, thereby acquiring nutrient elements and producing M. bovis metabolites after a complex series of metabolisms in the host–cell. On the other hand, mycoplasma depends on the host for many nutrients and, thus, requires a robust transport system to transport nutrients [27]. Three types of transport systems have been identified in mycoplasma, including the ATP-binding cassette transporter (ABC), the phosphoenolpyruvate-dependent phosphotransferase system (PTS), and facile diffusion. Among them, the ABC transporter system is the main pathway for the exchange of substances between mycoplasma and the external environment [28,29]. In addition, it has been suggested that the substrate-bound ATP-dependent ABC transporter system involved in nucleoside uptake may be one of the surrogate transporters [28]. In the present study, it was shown that the ABC transporter system mediates micronutrient uptake in M. bovis, and it was found that the membrane protein P48 may be involved in the metabolism of M. bovis through the ABC transporter system, which can be utilized to carry out studies on M. bovis in this manner.
4.5. Key Enzymes Regulating Energy Metabolism and Growth in M. Bovis
Most mycoplasmas are unable to carry out a complete tricarboxylic acid cycle, so the glycolytic pathway serves as an important energy source for mycoplasmas, and it is of great significance to carry out research on enzymes related to the mycoplasma glycolytic pathway [30]. Triosephosphate isomerase (Tpi) and pyruvate kinase (Pyk) are two key enzymes in the glycolytic process carried out by mycoplasmas [30,31]. Tpi catalyzes the reversible conversion between dihydroxyacetone phosphate and D-type glyceraldehyde-3-phosphate, and Pyk is able to change phosphoenolpyruvate and ADP to pyruvate and ATP, making it one of the major rate-limiting enzymes in glycolysis [32]. In addition, pyruvate products produced by Pyk synthesis can regulate a variety of metabolic pathways. On the other hand, researchers have discovered that Tpi [31](encoded by the TpiA gene) and PK [33](encoded by the Pyk gene) of MG, along with numerous glycolytic enzymes in other mycoplasmas [34], are situated in the cell membrane and cytoplasm. These enzymes can play a role in adhesion to the host–cell, thereby exerting their harmful effects. In this study, TpiA was localized in the cytoplasm in M. bovis (Table 1), and TpiA (p < 0.001) and Pyk (p < 0.001) were significantly upregulated in M. bovis NX114 vs. PG45 (Table 2). Since most mycoplasmas lack genes related to the tricarboxylic acid cycle, the glycolytic pathway may be important for their energy acquisition. In this study, TpiA and Pyk were predominantly enriched in the energy metabolism (glycolysis/gluconeogenesis) and metabolic pathways (Figure 10), suggesting that the two enzymes regulate the metabolism of M. bovis and may be the source of the energy that provides for its survival and growth (reproduction, replication). The two strains of M. bovis in logarithmic phases showed different size morphologies, which presumably may be related to the activities of the two enzymes. Second, Tpi and PyK were enriched in the carbon metabolism pathway (Figure 10), and it was hypothesized that the two enzymes might be precursors of the carbon source of M. bovis. Because these two enzymes are less studied in M. bovis, they may provide research ideas for its pathogenic mechanism.
5. Conclusions
The study examined the biology of clinical isolates of M. bovis using transcriptomic and proteomic data. It identified 193 DEGs and 158 DEPs, and identified 49 proteins/34 transcriptomic CDS post-translational protein sequences. Membrane, cytoplasmic, and ribosome proteins were found to be significant components. M. bovis NX114 was associated with energy metabolism, secondary metabolite biosynthesis, and the ABC transport system. A new adhesion protein was annotated, and TpiA and Pyk genes were identified as key enzymes regulating M. bovis growth and maintenance. This study reveals the genes related to the regulation of M. bovis growth and provides research ideas for the pathogenic mechanism of M. bovis.
Supplementary Materials
The following supporting information can be downloaded at: (https://doi.org/10.6084/m9.figshare.26348401.v1; accessed on 15 July 2024) and (https://doi.org/10.6084/m9.figshare.26348452.v1; accessed on 15 July 2024). Figure S1: Bubble plot of GO enrichment analysis of DEGs; Figure S2: Bar graph of KEGG Pathway enrichment results for DEGs; Figure S3: SDS-PAGE results of protein from the clinical isolate and standard isolate of Mycoplasma bovis; Figure S4: Peptide length profile; Figure S5: Bar graph of DEPs number statistics; Figure S6: Term enrichment bubble map of DEPs (top10). Table S1: Primers associated with qRT-PCR validation of DEGs compared between Mycoplasma bovis NX114 VS PG45; Table S2: DEGs of Mycoplasma bovis NX114 vs PG45; Table S3: Database annotation of DEGs in Mycoplasma bovis NX114 vs PG45; Table S4: DEPs of Mycoplasma bovis NX114 vs PG45; Table S5: GO annotation of DEPs in Mycoplasma bovis NX114 vs PG45; Table S6: KEGG annotation of DEPs in Mycoplasma bovis NX114 vs PG45.
Author Contributions
Data Curation, F.Y., M.Y. and F.L.; Formal Analysis, F.Y. and M.Y.; Methodology, F.Y. and F.L.; Writing—Original Draft, F.Y.; Investigation, M.Y.; Funding Acquisition, Y.Q., Y.G. and S.H.; Conceptualization, Y.G.; Project Administration, Y.Q., Y.G. and S.H.; Supervision, S.H.; Writing—Review and Editing, S.H. All authors have read and agreed to the published version of the manuscript.
Funding
The Key R&D project of the Ningxia Hui Autonomous Region (2022BBF03024), (2022BBF03025); the Natural Fund Program of Ningxia Hui Autonomous Region (2023AAC03890), (2022AAC05045); the Tongxin County Science and Technology Research and Development Program (2024KJJH010).
Institutional Review Board Statement
The authors confirm that all methods were performed in accordance with relevant guidelines and regulations. The experimental procedure was approved by the Ethics Committee of Ningxia University (protocol NO.NXU-2022-097), approval date: 8 September 2022.
Informed Consent Statement
We obtained informed consent for this study from the farm owner.
Data Availability Statement
Data will be made available on request.
Acknowledgments
Thanks to all institutions that provided the funding.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Suwanruengsri, M.; Uemura, R.; Kanda, T.; Fuke, N.; Nueangphuet, P.; Pornthummawat, A.; Yasuda, M.; Hirai, T.; Yamaguchi, R. Production of granulomas in Mycoplasma bovis infection associated with meningitis-meningoencephalitis, endocarditis, and pneumonia in cattle. J. Vet. Diagn. Investig. 2022, 34, 68–76. [Google Scholar] [CrossRef] [PubMed]
- Gelgie, A.E.; Desai, S.E.; Gelalcha, B.D.; Kerro Dego, O. Mycoplasma bovis mastitis in dairy cattle. Front. Vet. Sci. 2024, 11, 1322267. [Google Scholar] [CrossRef] [PubMed]
- Becker, C.A.M.; Ambroset, C.; Huleux, A.; Vialatte, A.; Colin, A.; Tricot, A.; Arcangioli, M.A.; Tardy, F. Monitoring Mycoplasma bovis Diversity and Antimicrobial Susceptibility in Calf Feedlots Undergoing a Respiratory Disease Outbreak. Pathogens 2020, 9, 593. [Google Scholar] [CrossRef] [PubMed]
- Okella, H.; Tonooka, K.; Okello, E. A Systematic Review of the Recent Techniques Commonly Used in the Diagnosis of Mycoplasma bovis in Dairy Cattle. Pathogens 2023, 12, 1178. [Google Scholar] [CrossRef]
- Ammar, A.M.; Abd El-Hamid, M.I.; Mohamed, Y.H.; Mohamed, H.M.; Al-Khalifah, D.H.M.; Hozzein, W.N.; Selim, S.; El-Neshwy, W.M.; El-Malt, R.M.S. Prevalence and Antimicrobial Susceptibility of Bovine Mycoplasma Species in Egypt. Biology 2022, 11, 1083. [Google Scholar] [CrossRef]
- van der Merwe, J.; Prysliak, T.; Perez-Casal, J. Invasion of bovine peripheral blood mononuclear cells and erythrocytes by Mycoplasma bovis. Infect. Immun. 2010, 78, 4570–4578. [Google Scholar] [CrossRef]
- Nishi, K.; Gondaira, S.; Fujiki, J.; Katagata, M.; Sawada, C.; Eguchi, A.; Iwasaki, T.; Iwano, H.; Higuchi, H. Invasion of Mycoplasma bovis into bovine synovial cells utilizing the clathrin-dependent endocytosis pathway. Vet. Microbiol. 2021, 253, 108956. [Google Scholar] [CrossRef]
- Zhao, H.; Zhang, Y.; Wang, Z.; Liu, M.; Wang, P.; Wu, W.; Peng, C. MBOVPG45_0375 Encodes an IgG-Binding Protein and MBOVPG45_0376 Encodes an IgG-Cleaving Protein in Mycoplasma bovis. Front. Vet. Sci. 2021, 8, 644224. [Google Scholar] [CrossRef]
- Rebollo Couto, M.S.; Klein, C.S.; Voss-Rech, D.; Terenzi, H. Extracellular Proteins of Mycoplasma synoviae. ISRN Vet. Sci. 2012, 2012, 802308. [Google Scholar] [CrossRef]
- Paes, J.A.; Lorenzatto, K.R.; de Moraes, S.N.; Moura, H.; Barr, J.R.; Ferreira, H.B. Secretomes of Mycoplasma hyopneumoniae and Mycoplasma flocculare reveal differences associated to pathogenesis. J. Proteom. 2017, 154, 69–77. [Google Scholar] [CrossRef]
- Zubair, M.; Muhamed, S.A.; Khan, F.A.; Zhao, G.; Menghwar, H.; Faisal, M.; Zhang, H.; Zhu, X.; Rasheed, M.A.; Chen, Y.; et al. Identification of 60 secreted proteins for Mycoplasma bovis with secretome assay. Microb. Pathog. 2020, 143, 104135. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Fang, L.; Liu, W.; Song, T.; Zhao, F.; Zhang, R.; Wang, D.; Xiao, S. Quantitative Proteomic Analyses of a Pathogenic Strain and Its Highly Passaged Attenuated Strain of Mycoplasma hyopneumoniae. Biomed. Res. Int. 2019, 2019, 4165735. [Google Scholar]
- Zhang, H.; Zhang, Y.; Lu, D.; Chen, X.; Chen, Y.; Hu, C.; Guo, A. MbovP0725, a secreted serine/threonine phosphatase, inhibits the host inflammatory response and affects metabolism in Mycoplasma bovis. mSystems 2024, 9, e0089123. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Hu, G.; Lu, D.; Zhao, G.; Zhang, Y.; Zubair, M.; Chen, Y.; Hu, C.; Chen, X.; Chen, J.; et al. Comparative Secretome Analyses of Mycoplasma bovis Virulent and Attenuated Strains Revealed MbovP0145 as a Promising Diagnostic Biomarker. Front. Vet. Sci. 2021, 8, 666769. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Han, X.; Chen, Y.; Mustafa, R.; Qi, J.; Chen, X.; Hu, C.; Chen, H.; Guo, A. Attenuated Mycoplasma bovis strains provide protection against virulent infection in calves. Vaccine 2014, 32, 3107–3114. [Google Scholar] [CrossRef] [PubMed]
- Wiśniewski, J.R.; Zougman, A.; Nagaraj, N.; Mann, M. Universal sample preparation method for proteome analysis. Nat. Methods 2009, 6, 359–362. [Google Scholar] [CrossRef]
- Vranakis, I.; Goniotakis, I.; Psaroulaki, A.; Sandalakis, V.; Tselentis, Y.; Gevaert, K.; Tsiotis, G. Proteome studies of bacterial antibiotic resistance mechanisms. J. Proteom. 2014, 97, 88–99. [Google Scholar] [CrossRef]
- Citti, C.; Dordet-Frisoni, E.; Nouvel, L.X.; Kuo, C.H.; Baranowski, E. Horizontal Gene Transfers in Mycoplasmas (Mollicutes). Curr. Issues Mol. Biol. 2018, 29, 3–22. [Google Scholar] [CrossRef]
- Bürki, S.; Frey, J.; Pilo, P. Virulence, persistence and dissemination of Mycoplasma bovis. Vet. Microbiol. 2015, 179, 15–22. [Google Scholar] [CrossRef]
- Xu, Q.Y.; Pan, Q.; Wu, Q.; Xin, J.Q. Mycoplasma Bovis adhesins and their target proteins. Front. Immunol. 2022, 13, 1016641. [Google Scholar] [CrossRef]
- Calcutt, M.J.; Lysnyansky, I.; Sachse, K.; Fox, L.K.; Nicholas, R.A.J.; Ayling, R.D. Gap analysis of Mycoplasma bovis disease, diagnosis and control: An aid to identify future development requirements. Transbound. Emerg. Dis. 2018, 65 (Suppl. S1), 91–109. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Huang, J.; Zhu, H.; Guo, Y.; Khan, F.A.; Menghwar, H.; Zhao, G.; Guo, A. P27 (MBOV_RS03440) is a novel fibronectin binding adhesin of Mycoplasma bovis. Int. J. Med. Microbiol. 2018, 308, 848–857. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Dong, Y.; Baranowski, E.; Li, X.; Zhao, G.; Hao, Z.; Zhang, H.; Chen, Y.; Hu, C.; Chen, H.; et al. Mbov_0503 Encodes a Novel Cytoadhesin that Facilitates Mycoplasma bovis Interaction with Tight Junctions. Microorganisms 2020, 8, 164. [Google Scholar] [CrossRef] [PubMed]
- Adamu, J.Y.; Mitiku, F.; Hartley, C.A.; Sansom, F.M.; Marenda, M.S.; Markham, P.F.; Browning, G.F.; Tivendale, K.A. Mycoplasma bovis mbfN Encodes a Novel LRR Lipoprotein That Undergoes Proteolytic Processing and Binds Host Extracellular Matrix Components. J. Bacteriol. 2020, 203, e00154-20. [Google Scholar] [CrossRef]
- Wu, X.; Zhang, S.; Long, C.; An, Z.; Xing, X.; Wen, F.; Bao, S. Mycoplasmas bovis P48 induces apoptosis in EBL cells via an endoplasmic reticulum stress-dependent signaling pathway. Vet. Microbiol. 2021, 255, 109013. [Google Scholar] [CrossRef]
- Qi, J.; Guo, A.; Cui, P.; Chen, Y.; Mustafa, R.; Ba, X.; Hu, C.; Bai, Z.; Chen, X.; Shi, L.; et al. Comparative geno-plasticity analysis of Mycoplasma bovis HB0801 (Chinese isolate). PLoS ONE 2012, 7, e38239. [Google Scholar] [CrossRef]
- McGowin, C.L.; Totten, P.A. The Unique Microbiology and Molecular Pathogenesis of Mycoplasma genitalium. J. Infect. Dis. 2017, 216 (Suppl. S2), S382–S388. [Google Scholar] [CrossRef]
- Masukagami, Y.; Nijagal, B.; Mahdizadeh, S.; Tseng, C.W.; Dayalan, S.; Tivendale, K.A.; Markham, P.F.; Browning, G.F.; Sansom, F.M. A combined metabolomic and bioinformatic approach to investigate the function of transport proteins of the important pathogen Mycoplasma bovis. Vet. Microbiol. 2019, 234, 8–16. [Google Scholar] [CrossRef]
- Yang, F.; Yang, M.; Si, D.; Sun, J.; Liu, F.; Qi, Y.; He, S.; Guo, Y. UHPLC/MS-Based Untargeted Metabolomics Reveals Metabolic Characteristics of Clinical Strain of Mycoplasma bovis. Microorganisms 2023, 11, 2602. [Google Scholar] [CrossRef]
- Gründel, A.; Jacobs, E.; Dumke, R. Interactions of surface-displayed glycolytic enzymes of Mycoplasma pneumoniae with components of the human extracellular matrix. Int. J. Med. Microbiol. 2016, 306, 675–685. [Google Scholar] [CrossRef]
- Bao, S.; Chen, D.; Yu, S.; Chen, H.; Tan, L.; Hu, M.; Qiu, X.; Song, C.; Ding, C. Characterization of triosephosphate isomerase from Mycoplasma gallisepticum. FEMS Microbiol. Lett. 2015, 362, fnv140. [Google Scholar] [CrossRef] [PubMed]
- Mathur, D.; Malik, G.; Garg, L.C. Biochemical and functional characterization of triosephosphate isomerase from Mycobacterium tuberculosis H37Rv. FEMS Microbiol. Lett. 2006, 263, 229–235. [Google Scholar] [CrossRef] [PubMed]
- He, S.; Qi, J.; Yu, S.; Yin, Y.; Tan, L.; Bao, S.; Qiu, X.; Wang, X.; Fei, R.; Ding, C. Expression and immunological characteristics of the surface-localized pyruvate kinase in Mycoplasma gallisepticum. Microb. Pathog. 2015, 89, 161–168. [Google Scholar] [CrossRef] [PubMed]
- Qi, J.; Zhang, F.; Wang, Y.; Liu, T.; Tan, L.; Wang, S.; Tian, M.; Li, T.; Wang, X.; Ding, C.; et al. Characterization of Mycoplasma gallisepticum pyruvate dehydrogenase alpha and beta subunits and their roles in cytoadherence. PLoS ONE 2018, 13, e0208745. [Google Scholar] [CrossRef]











Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).