
Association between High Interferon-Gamma Production in Avian Tuberculin-Stimulated Blood from Mycobacterium avium subsp. paratuberculosis-Infected Cattle and Candidate Genes Implicated in Necroptosis
 , , , , and
, , , , and 
Abstract
1. Introduction
2. Materials and Methods
2.1. Animals and Disease Status
2.2. Interferon-Gamma Release Assay (IGRA)
2.3. Genotyping and Imputation to Whole-Genome Sequence (WGS)
2.4. GWAS Analysis, Variance Components, and h2 Estimation
2.5. GWAS Data Postprocessing
2.6. SNPs, Quantitative Trait Loci (QTLs), and Candidate Genes Identification
2.7. Gene Ontology (GO) and Pathway Enrichment Analysis
2.8. Estimated Breeding Values (EBVs) for IFNγ Production
2.9. Statistical Analysis
3. Results
3.1. Assessment of IFNɣ Production
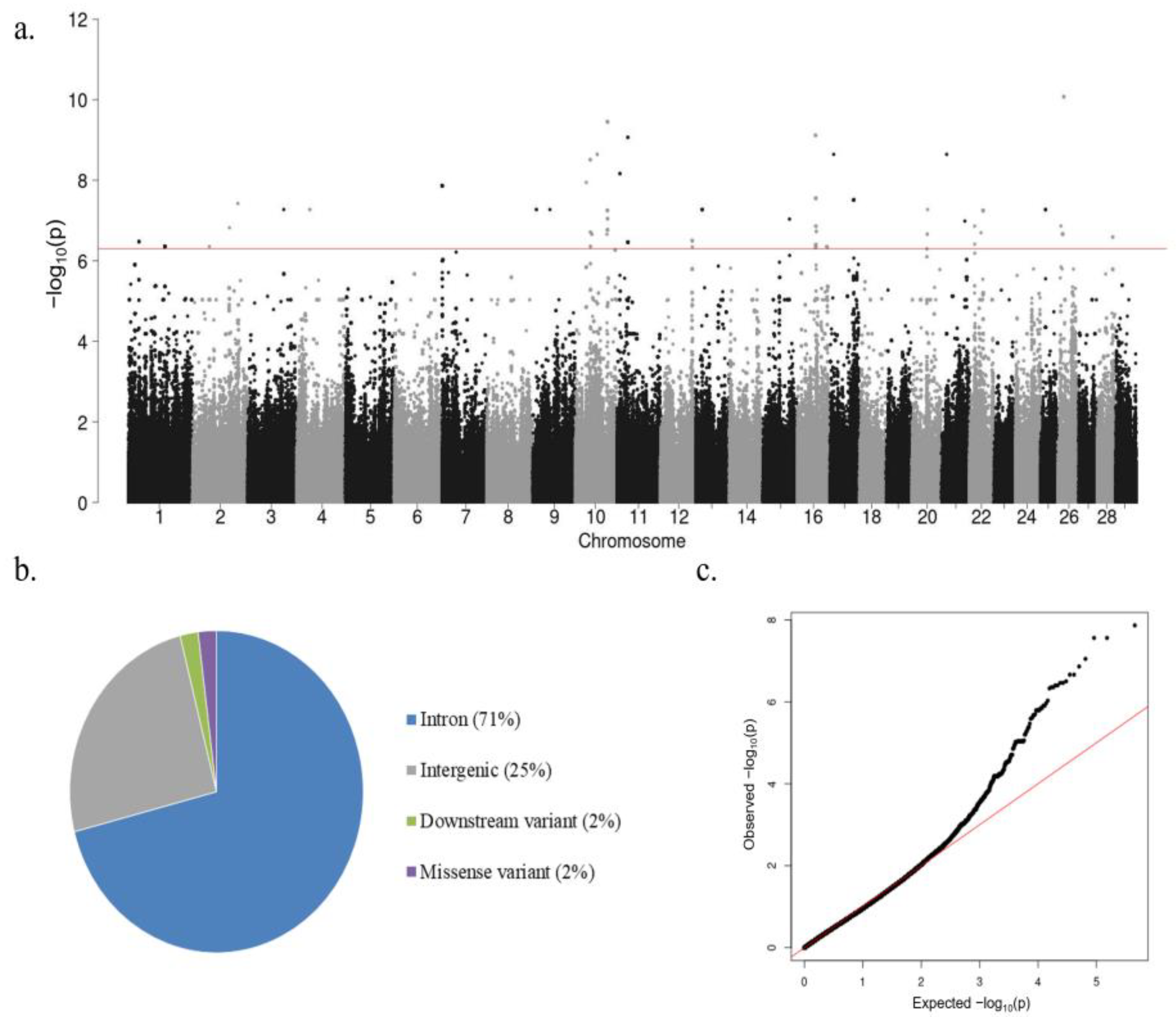
3.2. Heritability (h2) Estimate, Variance Components, and GWAS Results
3.3. SNPs, QTLs, and Candidate Genes Associated with High IFNγ Production
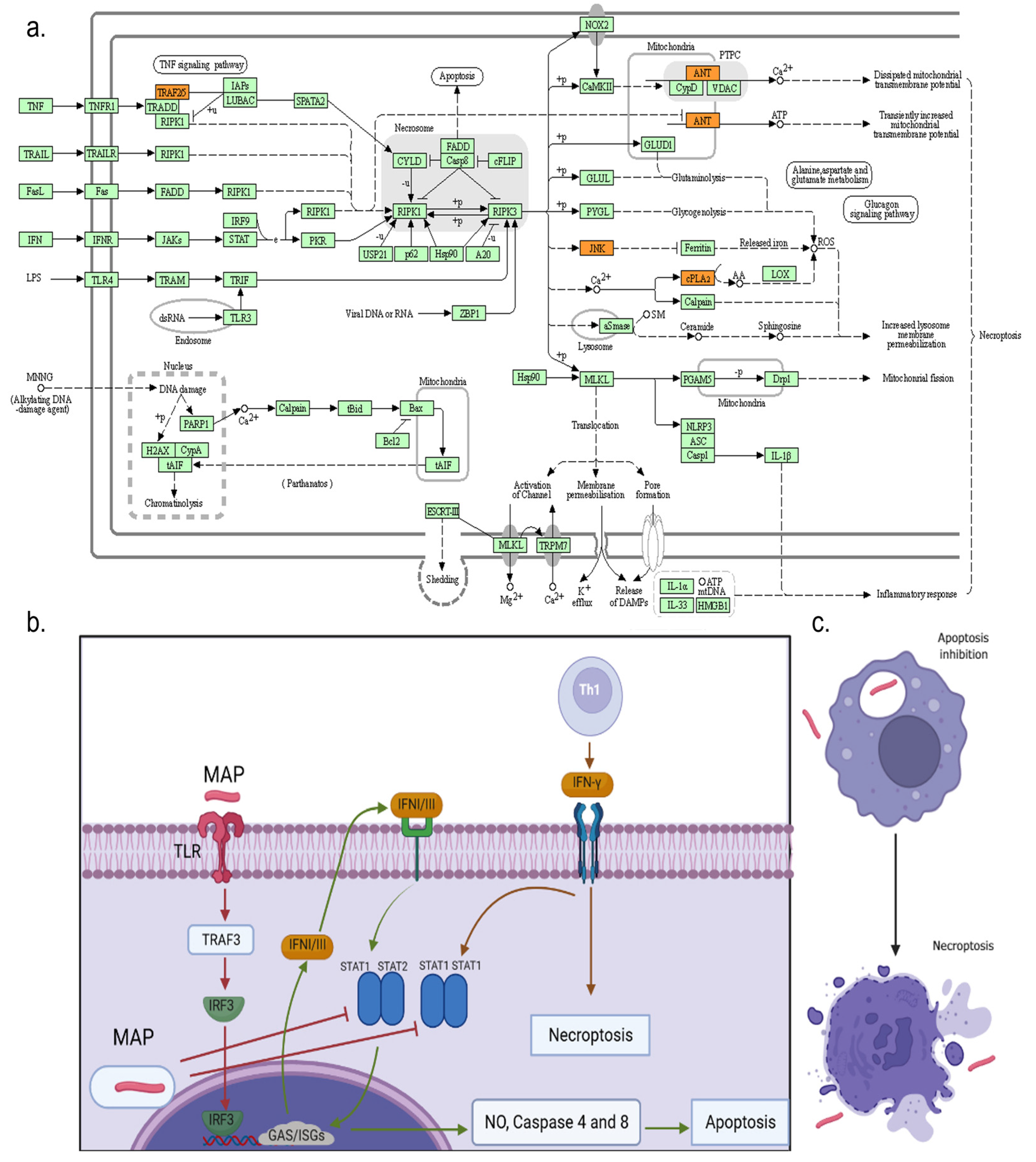
3.4. Gene Ontologies (GOs) and KEGG Pathway Enrichment Analysis
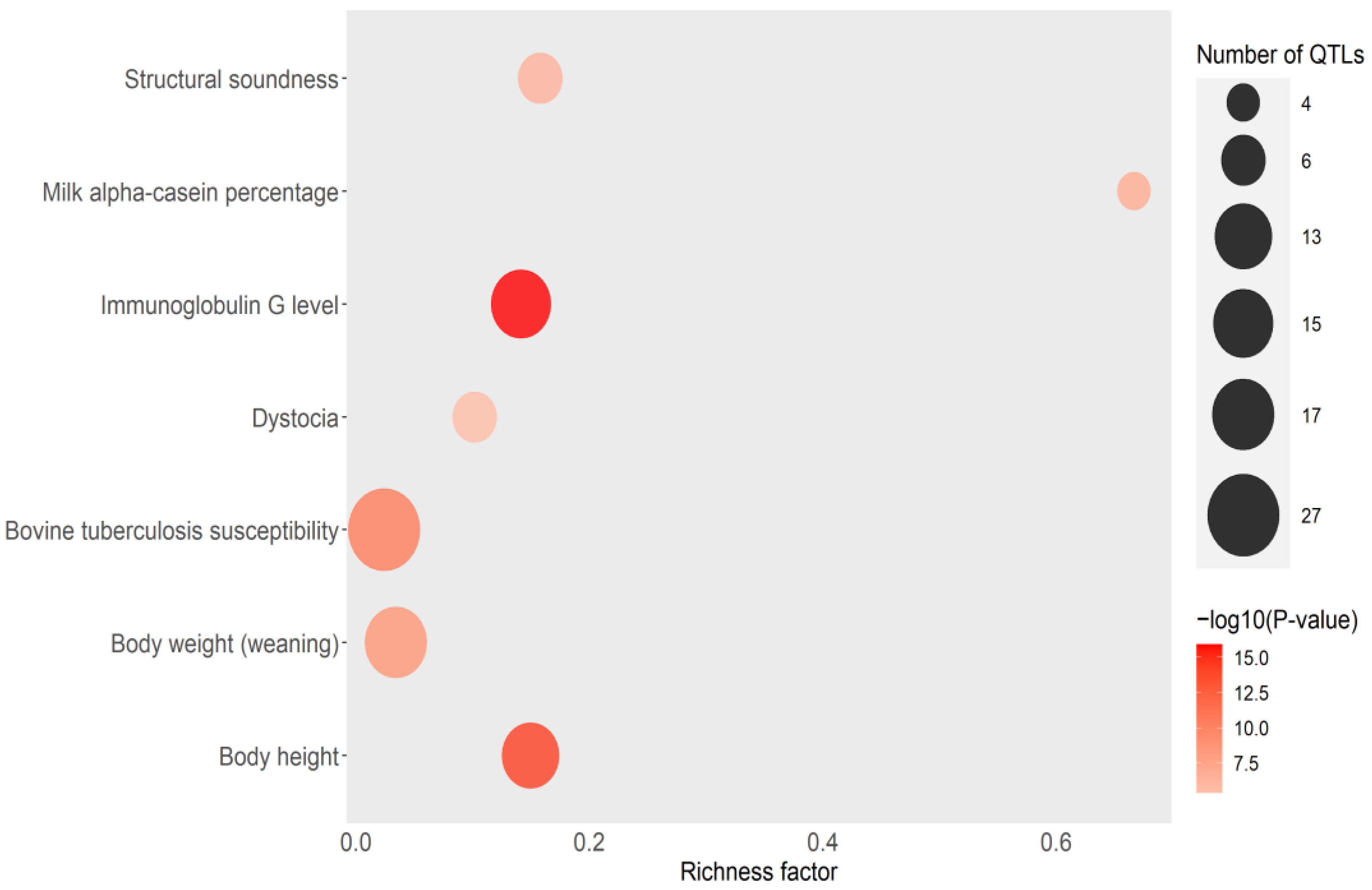
3.5. EBVs for IFNγ Production and Correlations with Other Bovine Traits
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rasmussen, P.; Barkema, H.W.; Mason, S.; Beaulieu, E.; Hall, D.C. Economic Losses Due to Johne’s Disease (Paratuberculosis) in Dairy Cattle. J. Dairy Sci. 2021, 104, 3123–3143. [Google Scholar] [CrossRef] [PubMed]
- Whittington, R.; Donat, K.; Weber, M.F.; Kelton, D.; Nielsen, S.S.; Eisenberg, S.; Arrigoni, N.; Juste, R.; Sáez, J.L.; Dhand, N.; et al. Control of Paratuberculosis: Who, Why and How. A Review of 48 Countries. BMC Vet. Res. 2019, 15, 198. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, S.S.; Toft, N. A Review of Prevalences of Paratuberculosis in Farmed Animals in Europe. Prev. Vet. Med. 2009, 88, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Feller, M.; Huwiler, K.; Stephan, R.; Altpeter, E.; Shang, A.; Furrer, H.; Pfyffer, G.E.; Jemmi, T.; Baumgartner, A.; Egger, M. Mycobacterium avium Subspecies paratuberculosis and Crohn’s Disease: A Systematic Review and Meta-Analysis. Lancet Infect. Dis. 2007, 7, 607–613. [Google Scholar] [CrossRef]
- Juste, R.A.; Elguezabal, N.; Garrido, J.M.; Pavon, A.; Geijo, M.V.; Sevilla, I.; Cabriada, J.L.; Tejada, A.; Garcia-Campos, F.; Casado, R.; et al. On the Prevalence of M. avium Subspecies paratuberculosis DNA in the Blood of Healthy Individuals and Patients with Inflammatory Bowel Disease. PLoS ONE 2008, 3, e2537. [Google Scholar] [CrossRef]
- Jeyanathan, M.; Boutros-Tadros, O.; Radhi, J.; Semret, M.; Bitton, A.; Behr, M.A. Visualization of Mycobacterium avium in Crohn’s Tissue by Oil-Immersion Microscopy. Microbes Infect. 2007, 9, 1567–1573. [Google Scholar] [CrossRef]
- Pierce, E.S. Could Mycobacterium avium Subspecies paratuberculosis Cause Crohn’s Disease, Ulcerative Colitis... and Colorectal Cancer? Infect. Agents Cancer 2018, 13, 1. [Google Scholar] [CrossRef]
- Dow, C.T. Warm, Sweetened Milk at the Twilight of Immunity—Alzheimer’s Disease—Inflammaging, Insulin Resistance, M. paratuberculosis and Immunosenescence. Front. Immunol. 2021, 12, 714179. [Google Scholar] [CrossRef] [PubMed]
- Garrido, J.M.; Vazquez, P.; Molina, E.; Plazaola, J.M.; Sevilla, I.A.; Geijo, M.V.; Alonso-Hearn, M.; Juste, R.A. Paratuberculosis Vaccination Causes Only Limited Cross-Reactivity in the Skin Test for Diagnosis of Bovine Tuberculosis. PLoS ONE 2013, 8, e80985. [Google Scholar] [CrossRef]
- Blanco-Vázquez, C.; Alonso-Hearn, M.; Juste, R.A.; Canive, M.; Iglesias, T.; Iglesias, N.; Amado, J.; Vicente, F.; Balseiro, A.; Casais, R. Detection of Latent Forms of Mycobacterium avium Subsp. paratuberculosis Infection Using Host Biomarker-Based ELISAs Greatly Improves Paratuberculosis Diagnostic Sensitivity. PLoS ONE 2020, 15, e0236336. [Google Scholar] [CrossRef]
- Sweeney, R.W. Pathogenesis of Paratuberculosis. Vet. Clin. N. Am. Food Anim. Pract. 2011, 27, 537–546. [Google Scholar] [CrossRef]
- Weiss, D.J.; Souza, C.D. Review Paper: Modulation of Mononuclear Phagocyte Function by Mycobacterium avium Subsp. paratuberculosis. Vet. Pathol. 2008, 45, 829–841. [Google Scholar] [CrossRef] [PubMed]
- Souza, C.; Davis, W.C.; Eckstein, T.M.; Sreevatsan, S.; Weiss, D.J. Mannosylated Lipoarabinomannans from Mycobacterium Avium Subsp. Paratuberculosis Alters the Inflammatory Response by Bovine Macrophages and Suppresses Killing of Mycobacterium Avium Subsp. Avium Organisms. PLoS ONE 2013, 8, e75924. [Google Scholar] [CrossRef] [PubMed]
- Kabara, E.; Coussens, P.M. Infection of Primary Bovine Macrophages with Mycobacterium avium Subspecies paratuberculosis Suppresses Host Cell Apoptosis. Front. Microbiol. 2012, 3, 215. [Google Scholar] [CrossRef] [PubMed]
- Ssekitoleko, J.; Ojok, L.; Abd El Wahed, A.; Erume, J.; Amanzada, A.; Eltayeb, E.; Eltom, K.H.; Okuni, J.B. Mycobacterium avium Subsp. paratuberculosis Virulence: A Review. Microorganisms 2021, 9, 2623. [Google Scholar] [CrossRef]
- Stabel, J. Transitions in Immune Responses to Mycobacterium paratuberculosis. Vet. Microbiol. 2000, 77, 465–473. [Google Scholar] [CrossRef]
- Coussens, P.M. Model for Immune Responses to Mycobacterium avium Subspecies paratuberculosis in Cattle. Infect. Immun. 2004, 72, 3089–3096. [Google Scholar] [CrossRef]
- Zhang, X.; Sun, Y.; He, C.; Qiu, X.; Zhou, D.; Ye, Z.; Long, Y.; Tang, T.; Su, X.; Ma, J. The Immune Characterization of Interferon-β Responses in Tuberculosis Patients. Microbiol. Immunol. 2018, 62, 281–290. [Google Scholar] [CrossRef] [PubMed]
- Canive, M.; González-Recio, O.; Fernández, A.; Vázquez, P.; Badia-Bringué, G.; Lavín, J.L.; Garrido, J.M.; Juste, R.A.; Alonso-Hearn, M. Identification of Loci Associated with Susceptibility to Mycobacterium avium Subsp. paratuberculosis Infection in Holstein Cattle Using Combinations of Diagnostic Tests and Imputed Whole-Genome Sequence Data. PLoS ONE 2021, 16, e0256091. [Google Scholar] [CrossRef]
- Kravitz, A.; Pelzer, K.; Sriranganathan, N. The Paratuberculosis Paradigm Examined: A Review of Host Genetic Resistance and Innate Immune Fitness in Mycobacterium avium Subsp. Paratuberculosis Infection. Front. Vet. Sci. 2021, 8, 721706. [Google Scholar] [CrossRef]
- Sanchez, M.P.; Tribout, T.; Fritz, S.; Guatteo, R.; Fourichon, C.; Schibler, L.; Delafosse, A.; Boichard, D. New Insights into the Genetic Resistance to Paratuberculosis in Holstein Cattle via Single-Step Genomic Evaluation. Genet. Sel. Evol. 2022, 54, 67. [Google Scholar] [CrossRef]
- Alonso-Hearn, M.; Badia-Bringué, G.; Canive, M. Genome-Wide Association Studies for the Identification of Cattle Susceptible and Resilient to Paratuberculosis. Front. Vet. Sci. 2022, 9, 935133. [Google Scholar] [CrossRef]
- Canive, M.; Badia-Bringué, G.; Vázquez, P.; González-Recio, O.; Fernández, A.; Garrido, J.M.; Juste, R.A.; Alonso-Hearn, M. Identification of Loci Associated with Pathological Outcomes in Holstein Cattle Infected with Mycobacterium avium Subsp. paratuberculosis Using Whole-Genome Sequence Data. Sci. Rep. 2021, 11, 20177. [Google Scholar] [CrossRef] [PubMed]
- Canive, M.; Badia-Bringué, G.; Vázquez, P.; Garrido, J.M.; Juste, R.A.; Fernandez, A.; González-Recio, O.; Alonso-Hearn, M. A Genome-Wide Association Study for Tolerance to Paratuberculosis Identifies Candidate Genes Involved in DNA Packaging, DNA Damage Repair, Innate Immunity, and Pathogen Persistence. Front. Immunol. 2022, 13, 820965. [Google Scholar] [CrossRef]
- Ko, D.C.; Shukla, K.P.; Fong, C.; Wasnick, M.; Brittnacher, M.J.; Wurfel, M.M.; Holden, T.D.; O’Keefe, G.E.; Van Yserloo, B.; Akey, J.M.; et al. A Genome-Wide In Vitro Bacterial-Infection Screen Reveals Human Variation in the Host Response Associated with Inflammatory Disease. Am. J. Hum. Genet. 2009, 85, 214–227. [Google Scholar] [CrossRef]
- Badia-Bringué, G.; Canive, M.; Alonso-Hearn, M. Control of Mycobacterium avium Subsp. paratuberculosis Load within Infected Bovine Monocyte-Derived Macrophages Is Associated with Host Genetics. Front. Immunol. 2023, 14, 1042638. [Google Scholar] [CrossRef]
- Frank, S.A. Chapter 8, Genetic Variability of Hosts. In Immunology and Evolution of Infectious Disease; Princeton University Press: Princeton, NJ, USA, 2002. Available online: https://www.ncbi.nlm.nih.gov/books/NBK2401/ (accessed on 13 July 2023).
- Vázquez, P.; Ruiz-Larrañaga, O.; Garrido, J.M.; Iriondo, M.; Manzano, C.; Agirre, M.; Estonba, A.; Juste, R.A. Genetic Association Analysis of Paratuberculosis Forms in Holstein-Friesian Cattle. Vet. Med. Int. 2014, 2014, 321327. [Google Scholar] [CrossRef] [PubMed]
- González, J.; Geijo, M.V.; García-Pariente, C.; Verna, A.; Corpa, J.M.; Reyes, L.E.; Ferreras, M.C.; Juste, R.A.; García Marín, J.F.; Pérez, V. Histopathological Classification of Lesions Associated with Natural Paratuberculosis Infection in Cattle. J. Comp. Pathol. 2005, 133, 184–196. [Google Scholar] [CrossRef]
- Loh, P.-R.; Palamara, P.F.; Price, A.L. Fast and Accurate Long-Range Phasing in a UK Biobank Cohort. Nat. Genet. 2016, 48, 811–816. [Google Scholar] [CrossRef] [PubMed]
- Das, S.; Forer, L.; Schönherr, S.; Sidore, C.; Locke, A.E.; Kwong, A.; Vrieze, S.I.; Chew, E.Y.; Levy, S.; McGue, M.; et al. Next-Generation Genotype Imputation Service and Methods. Nat. Genet. 2016, 48, 1284–1287. [Google Scholar] [CrossRef]
- Hayes, B.J.; Daetwyler, H.D. 1000 Bull Genomes Project to Map Simple and Complex Genetic Traits in Cattle: Applications and Outcomes. Annu. Rev. Anim. Biosci. 2019, 7, 89–102. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Lee, S.H.; Goddard, M.E.; Visscher, P.M. GCTA: A Tool for Genome-Wide Complex Trait Analysis. Am. J. Hum. Genet. 2011, 88, 76–82. [Google Scholar] [CrossRef] [PubMed]
- Burton, P.R.; Clayton, D.G.; Cardon, L.R.; Craddock, N.; Deloukas, P.; Duncanson, A.; Kwiatkowski, D.P.; McCarthy, M.I.; Ouwehand, W.H.; Samani, N.J.; et al. Genome-Wide Association Study of 14,000 Cases of Seven Common Diseases and 3,000 Shared Controls. Nature 2007, 447, 661–678. [Google Scholar] [CrossRef]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.R.; Bender, D.; Maller, J.; Sklar, P.; De Bakker, P.I.W.; Daly, M.J.; et al. PLINK: A Tool Set for Whole-Genome Association and Population-Based Linkage Analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef] [PubMed]
- Ensembl Genome Browser 109. Available online: http://www.ensembl.org (accessed on 24 October 2022).
- Animal Genome Databases. Available online: https://www.animalgenome.org (accessed on 2 December 2022).
- Fonseca, P.A.S.; Suárez-Vega, A.; Marras, G.; Cánovas, Á. GALLO: An R Package for Genomic Annotation and Integration of Multiple Data Sources in Livestock for Positional Candidate Loci. GigaScience 2020, 9, giaa149. [Google Scholar] [CrossRef]
- GWAS Catalog. Available online: https://www.ebi.ac.uk/gwas (accessed on 2 December 2022).
- GeneCards—Human Genes | Gene Database | Gene Search. Available online: https://www.genecards.org (accessed on 2 December 2022).
- InnateDB: Systems Biology of the Innate Immune Response. Available online: https://www.innatedb.com (accessed on 4 April 2023).
- R Core Team. R: A Language and Environment for Statistical Computing. Available online: https://www.r-project.org/ (accessed on 5 December 2022).
- Wu, T.; Hu, E.; Xu, S.; Chen, M.; Guo, P.; Dai, Z.; Feng, T.; Zhou, L.; Tang, W.; Zhan, L.; et al. ClusterProfiler 4.0: A Universal Enrichment Tool for Interpreting Omics Data. Innovation 2021, 2, 100141. [Google Scholar] [CrossRef]
- VanRaden, P.M.; Sullivan, P.G. International Genomic Evaluation Methods for Dairy Cattle. Genet. Sel. Evol. 2010, 42, 7. [Google Scholar] [CrossRef]
- Jamovi—Open Statistical Software for the Desktop and Cloud. Available online: https://www.jamovi.org (accessed on 25 October 2022).
- McGovern, S.P.; Purfield, D.C.; Ring, S.C.; Carthy, T.R.; Graham, D.A.; Berry, D.P. Candidate Genes Associated with the Heritable Humoral Response to Mycobacterium avium ssp. paratuberculosis in Dairy Cows Have Factors in Common with Gastrointestinal Diseases in Humans. J. Dairy Sci. 2019, 102, 4249–4263. [Google Scholar] [CrossRef]
- Kim, Y.M.; Shin, E.C. Type I and III Interferon Responses in SARS-CoV-2 Infection. Exp. Mol. Med. 2021, 53, 750–760. [Google Scholar] [CrossRef]
- Lai, G.; Liu, H.; Deng, J.; Li, K.; Zhang, C.; Zhong, X.; Xie, B. The Characteristics of Tumor Microenvironment Predict Survival and Response to Immunotherapy in Adrenocortical Carcinomas. Cells 2023, 12, 755. [Google Scholar] [CrossRef]
- Park, S.H.; Park-Min, K.H.; Chen, J.; Hu, X.; Ivashkiv, L.B. Tumor Necrosis Factor Induces GSK3 Kinase-Mediated Cross-Tolerance to Endotoxin in Macrophages. Nat. Immunol. 2011, 12, 607–615. [Google Scholar] [CrossRef]
- Wu, T.; Ikezono, T.; Angus, C.W.; Shelhamer, J.H. Characterization of the Promoter for the Human 85 KDa Cytosolic Phospholipase A2 Gene. Nucleic Acid. Res. 1994, 22, 5093–5098. [Google Scholar] [CrossRef]
- Wang, Q.S.; Shen, S.Q.; Sun, H.W.; Xing, Z.X.; Yang, H.L. Interferon-Gamma Induces Autophagy-Associated Apoptosis through Induction of CPLA2-Dependent Mitochondrial ROS Generation in Colorectal Cancer Cells. Biochem. Biophys. Res. Commun. 2018, 498, 1058–1065. [Google Scholar] [CrossRef]
- Cekay, M.J.; Roesler, S.; Frank, T.; Knuth, A.K.; Eckhardt, I.; Fulda, S. Smac Mimetics and Type II Interferon Synergistically Induce Necroptosis in Various Cancer Cell Lines. Cancer Lett. 2017, 410, 228–237. [Google Scholar] [CrossRef] [PubMed]
- Pestka, S.; Krause, C.D.; Walter, M.R. Interferons, Interferon-like Cytokines, and Their Receptors. Immunol. Rev. 2004, 202, 8–32. [Google Scholar] [CrossRef]
- Ivashkiv, L.B.; Donlin, L.T. Regulation of Type I Interferon Responses. Nat. Rev. Immunol. 2014, 14, 36–49. [Google Scholar] [CrossRef]
- González-Navajas, J.M.; Lee, J.; David, M.; Raz, E. Immunomodulatory Functions of Type I Interferons. Nat. Rev. Immunol. 2012, 12, 125–135. [Google Scholar] [CrossRef]
- Xu, G.; Wang, J.; Gao, G.F.; Liu, C.H. Insights into Battles between Mycobacterium tuberculosis and Macrophages. Protein Cell 2014, 5, 728–736. [Google Scholar] [CrossRef] [PubMed]
- Berghe, T.V.; Linkermann, A.; Jouan-Lanhouet, S.; Walczak, H.; Vandenabeele, P. Regulated Necrosis: The Expanding Network of Non-Apoptotic Cell Death Pathways. Nat. Rev. Mol. Cell Biol. 2014, 15, 135–147. [Google Scholar] [CrossRef]
- Kearney, C.J.; Cullen, S.P.; Tynan, G.A.; Henry, C.M.; Clancy, D.; Lavelle, E.C.; Martin, S.J. Necroptosis Suppresses Inflammation via Termination of TNF-or LPS-Induced Cytokine and Chemokine Production. Cell Death Differ. 2015, 22, 1313–1327. [Google Scholar] [CrossRef] [PubMed]
- Stutz, M.D.; Ojaimi, S.; Allison, C.; Preston, S.; Arandjelovic, P.; Hildebrand, J.M.; Sandow, J.J.; Webb, A.I.; Silke, J.; Alexander, W.S.; et al. Necroptotic Signaling Is Primed in Mycobacterium Tuberculosis-Infected Macrophages, but Its Pathophysiological Consequence in Disease Is Restricted. Cell Death Differ. 2018, 25, 951–965. [Google Scholar] [CrossRef] [PubMed]
- Roca, F.J.; Ramakrishnan, L. TNF Dually Mediates Resistance and Susceptibility to Mycobacteria via Mitochondrial Reactive Oxygen Species. Cell 2013, 153, 521–534. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| BTA 1 | QTL Start (bp) | QTL End (bp) | Peak p-Value of Peak SNP | Regression Coefficient (b) | Genes in QTL 2 | No. of SNPs in QTL |
|---|---|---|---|---|---|---|
| 1 | 26,283,242 | 26,283,242 | 3.34 × 10−7 | 0.526 | ROBO1 | 1 |
| 1 | 90,034,825 | 90,360,896 | 4.42 × 10−7 | 1.127 | 5S_rRNA, TBL1XR1, ENSBTAG00000054926 | 3 |
| 2 | 41,453,339 | 41,453,339 | 4.42 × 10−7 | 1.127 | 1 | |
| 2 | 90,850,109 | 90,850,109 | 1.51 × 10−7 | 0.474 | TBL1XR1, SNORD11B, SNORD11, BMPR2 | 1 |
| 2 | 111,599,011 | 111,599,011 | 3.76 × 10−8 | 0.843 | 1 | |
| 3 | 88,432,545 | 88,432,545 | 5.32 × 10−8 | 1.223 | 1 | |
| 4 | 31,901,616 | 31,901,616 | 5.32 × 10−8 | 1.223 | GPNMB, MALSU1, IGF2BP3, ENSBTAG00000054861 | 1 |
| 7 | 512,191 | 941,753 | 1.36 × 10−8 | 0.720 | 5S_rRNA, FLT4, CNOT6, GFPT2, MAPK9 | 5 |
| 9 | 9,107,205 | 9,107,205 | 5.32 × 10−8 | 1.223 | 1 | |
| 9 | 42,469,434 | 42,469,434 | 5.32 × 10−8 | 1.223 | SOBP | 1 |
| 10 | 27,643,294 | 27,643,294 | 1.13 × 10−8 | 0.854 | OR4G10, OR4F67B, OR4G18, OR4K36 | 1 |
| 10 | 37,469,311 | 37,833,767 | 3.07 × 10−9 | 0.811 | PLA2G4E, PLA2G4D, PLA2G4F, VPS39, TMEM87A, GANC, CAPN3, ZNF106, SNAP23, LRRC57, HAUS2 | 3 |
| 10 | 41,108,685 | 41,108,685 | 2.18 × 10−7 | 1.105 | 1 | |
| 10 | 54,450,209 | 54,461,425 | 2.27 × 10−9 | 1.338 | NEDD4, ENSBTAG00000031396 | 2 |
| 10 | 78,308,107 | 78,308,107 | 2.18 × 10−7 | 1.105 | 1 | |
| 10 | 79,501,139 | 79,957,849 | 3.55 × 10−10 | 0.839 | PLEKHH1, PIGH, ARG2, U6, VTI1B, RDH11, RDH12, ZFYVE26, RAD51B, ENSBTAG00000054736 | 9 |
| 11 | 7,148,731 | 7,148,731 | 6.81 × 10−9 | 1.036 | IL1RL1, IL18R1 | 1 |
| 11 | 26,763,300 | 26,794,996 | 8.58 × 10−10 | 1.058 | SLC3A1, PREPL, CAMKMT, ENSBTAG00000040564, ENSBTAG00000043226 | 2 |
| 12 | 78,134,904 | 78,134,904 | 3.16 × 10−7 | 0.536 | ITGBL1, 5S_rRNA, FGF14 | 1 |
| 12 | 78,813,208 | 78,813,208 | 4.52 × 10−7 | 0.338 | 1 | |
| 13 | 16,268,793 | 16,268,793 | 5.32 × 10−8 | 1.223 | ITIH5 | 1 |
| 15 | 65,654,411 | 65,654,411 | 9.20 × 10−8 | 1.138 | CD44 | 1 |
| 16 | 44,888,428 | 45,019,492 | 7.68 × 10−10 | 0.613 | RERE, bta-mir-2285ck, SLC45A1 | 2 |
| 16 | 46,077,301 | 47,047,708 | 2.77 × 10−8 | 0.619 | DNAJC11, THAP3, PHF13, KLHL21, ZBTB48, TAS1R1, NOL9, PLEKHG5, TNFRSF25, ESPN, HES2, ACOT7, GPR153, ENSBTAG00000054938, ENSBTAG00000049238 | 7 |
| 16 | 72,020,440 | 72,020,440 | 4.42 × 10−7 | 1.127 | RD3, TRAF5, RCOR3 | 1 |
| 16 | 74,098,459 | 74,098,459 | 4.52 × 10−7 | 0.598 | 1 | |
| 17 | 8,929,084 | 8,929,084 | 2.27 × 10−9 | 1.338 | 1 | |
| 17 | 57,930,217 | 58,393,881 | 3.13 × 10−8 | 0.338 | FBXO21, TESC, FBXW8, RNFT2, SPRING1, U6, ENSBTAG00000053074, ENSBTAG00000037415, ENSBTAG00000051326, ENSBTAG00000053055 | 3 |
| 20 | 37,487,725 | 37,487,725 | 2.18 × 10−7 | 1.105 | 1 | |
| 20 | 38,295,510 | 38,295,510 | 5.32 × 10−8 | 1.223 | CAPSL, IL7R | 1 |
| 21 | 13,584,472 | 13,584,472 | 2.27 × 10−9 | 1.338 | 1 | |
| 21 | 58,429,839 | 58,429,839 | 1.03 × 10−7 | 0.922 | PRIMA1 | 1 |
| 22 | 12,939,505 | 12,984,148 | 3.85 × 10−7 | 0.502 | MYRIP, ENSBTAG00000049890 | 2 |
| 22 | 28,603,524 | 28,603,524 | 2.00 × 10−7 | 0.615 | 1 | |
| 22 | 33,464,942 | 33,464,942 | 5.60 × 10−8 | 0.553 | TAFA1 | 1 |
| 25 | 12,368,203 | 12,394,807 | 5.32 × 10−8 | 1.223 | 3 | |
| 26 | 8,076,021 | 8,076,021 | 1.36 × 10−7 | 0.511 | PRKG1 | 1 |
| 26 | 12,482,172 | 12,499,709 | 2.18 × 10−7 | 1.105 | HTR7, RPP30, ANKRD1 | 2 |
| 26 | 15,176,321 | 15,176,321 | 8.37 × 10−11 | 0.994 | 5S_rRNA, ASMTL, SLC25A6, ENSBTAG00000051075, ENSBTAG00000052863, ENSBTAG00000052720, ENSBTAG00000055018 | 1 |
| 28 | 38,626,680 | 38,626,680 | 2.57 × 10−7 | 0.675 | 1 |
| ID | Description | Adjusted p | Gene Code | Gene Ratio |
|---|---|---|---|---|
| bta04217 | Necroptosis | 0.008946 | PLA2G4E/PLA2G4D/PLA2G4F/ TRAF5/SLC25A6/MAPK9 | 6/42 |
| bta04730 | Long-term depression | 0.008946 | PLA2G4E/PLA2G4D/PLA2G4F/ PRKG1 | 4/42 |
| bta04611 | Platelet activation | 0.008946 | PLA2G4E/PLA2G4D/PLA2G4F/ SNAP23/PRKG1 | 5/42 |
| bta04664 | Fc epsilon RI signaling pathway | 0.008946 | PLA2G4E/PLA2G4D/PLA2G4F/ MAPK9 | 4/42 |
| bta00592 | alpha-Linolenic acid metabolism | 0.008946 | PLA2G4E/PLA2G4D/PLA2G4F | 3/42 |
| bta04912 | GnRH signaling pathway | 0.017492 | PLA2G4E/PLA2G4D/PLA2G4F/ MAPK9 | 4/42 |
| bta04014 | Ras signaling pathway | 0.017492 | PLA2G4E/PLA2G4D/PLA2G4F/ HTR7/FLT4/MAPK9 | 6/42 |
| bta00591 | Linoleic acid metabolism | 0.017492 | PLA2G4E/PLA2G4D/PLA2G4F | 3/42 |
| bta05231 | Choline metabolism in cancer | 0.017492 | PLA2G4E/PLA2G4D/PLA2G4F/ MAPK9 | 4/42 |
| bta04750 | Inflammatory mediator regulation of TRP channels | 0.023268 | PLA2G4E/PLA2G4D/PLA2G4F/ MAPK9 | 4/42 |
| bta00565 | Ether lipid metabolism | 0.025462 | PLA2G4E/PLA2G4D/PLA2G4F | 3/42 |
| bta04726 | Serotonergic synapse | 0.026458 | PLA2G4E/PLA2G4D/PLA2G4F/ HTR7 | 4/42 |
| bta04370 | VEGF signaling pathway | 0.026458 | PLA2G4E/PLA2G4D/PLA2G4F | 3/42 |
| bta04913 | Ovarian steroidogenesis | 0.031114 | PLA2G4E/PLA2G4D/PLA2G4F | 3/42 |
| bta04270 | Vascular smooth muscle contraction | 0.037248 | PLA2G4E/PLA2G4D/PLA2G4F/ PRKG1 | 4/42 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Badia-Bringué, G.; Canive, M.; Vázquez, P.; Garrido, J.M.; Fernández, A.; Juste, R.A.; Jiménez, J.A.; González-Recio, O.; Alonso-Hearn, M. Association between High Interferon-Gamma Production in Avian Tuberculin-Stimulated Blood from Mycobacterium avium subsp. paratuberculosis-Infected Cattle and Candidate Genes Implicated in Necroptosis. Microorganisms 2023, 11, 1817. https://doi.org/10.3390/microorganisms11071817
Badia-Bringué G, Canive M, Vázquez P, Garrido JM, Fernández A, Juste RA, Jiménez JA, González-Recio O, Alonso-Hearn M. Association between High Interferon-Gamma Production in Avian Tuberculin-Stimulated Blood from Mycobacterium avium subsp. paratuberculosis-Infected Cattle and Candidate Genes Implicated in Necroptosis. Microorganisms. 2023; 11(7):1817. https://doi.org/10.3390/microorganisms11071817
Chicago/Turabian StyleBadia-Bringué, Gerard, María Canive, Patricia Vázquez, Joseba M. Garrido, Almudena Fernández, Ramón A. Juste, José Antonio Jiménez, Oscar González-Recio, and Marta Alonso-Hearn. 2023. "Association between High Interferon-Gamma Production in Avian Tuberculin-Stimulated Blood from Mycobacterium avium subsp. paratuberculosis-Infected Cattle and Candidate Genes Implicated in Necroptosis" Microorganisms 11, no. 7: 1817. https://doi.org/10.3390/microorganisms11071817
APA StyleBadia-Bringué, G., Canive, M., Vázquez, P., Garrido, J. M., Fernández, A., Juste, R. A., Jiménez, J. A., González-Recio, O., & Alonso-Hearn, M. (2023). Association between High Interferon-Gamma Production in Avian Tuberculin-Stimulated Blood from Mycobacterium avium subsp. paratuberculosis-Infected Cattle and Candidate Genes Implicated in Necroptosis. Microorganisms, 11(7), 1817. https://doi.org/10.3390/microorganisms11071817