
Comparative Study of Intestinal Microbiome in Patients with Ulcerative Colitis and Healthy Controls in Korea


Abstract
:1. Introduction
2. Materials and Methods
2.1. Ethics Statement
2.2. Patients and Healthy Controls
2.3. DNA Extraction
2.4. 16S rRNA Gene Sequencing
2.5. Bioinformatic Analysis and Statistical Analysis
3. Results
3.1. Clinical Data of Patients with Ulcerative Colitis and Healthy Controls
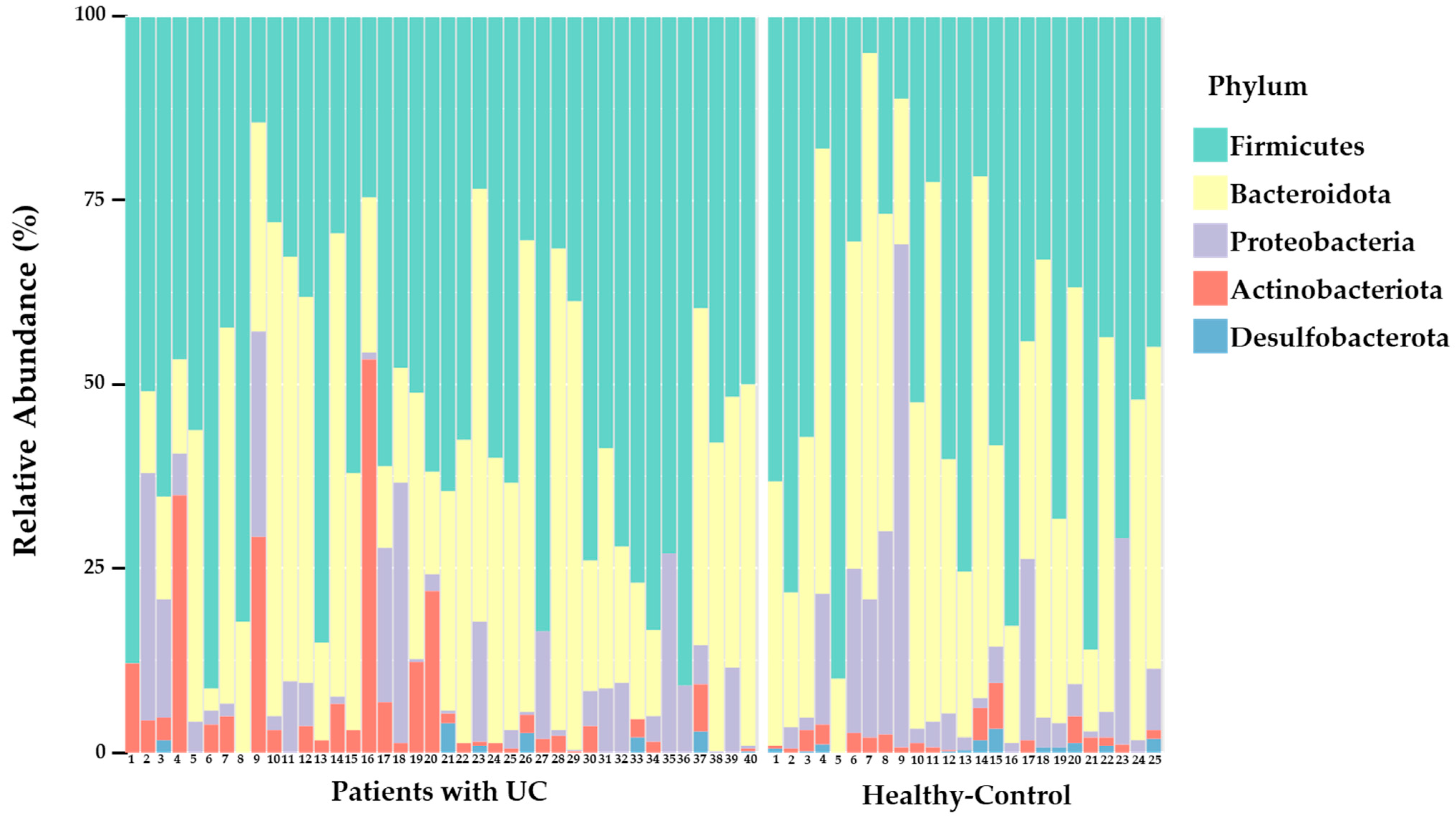
3.2. Overall Composition of the Intestinal Microbiota in UC Patients and Healthy Controls
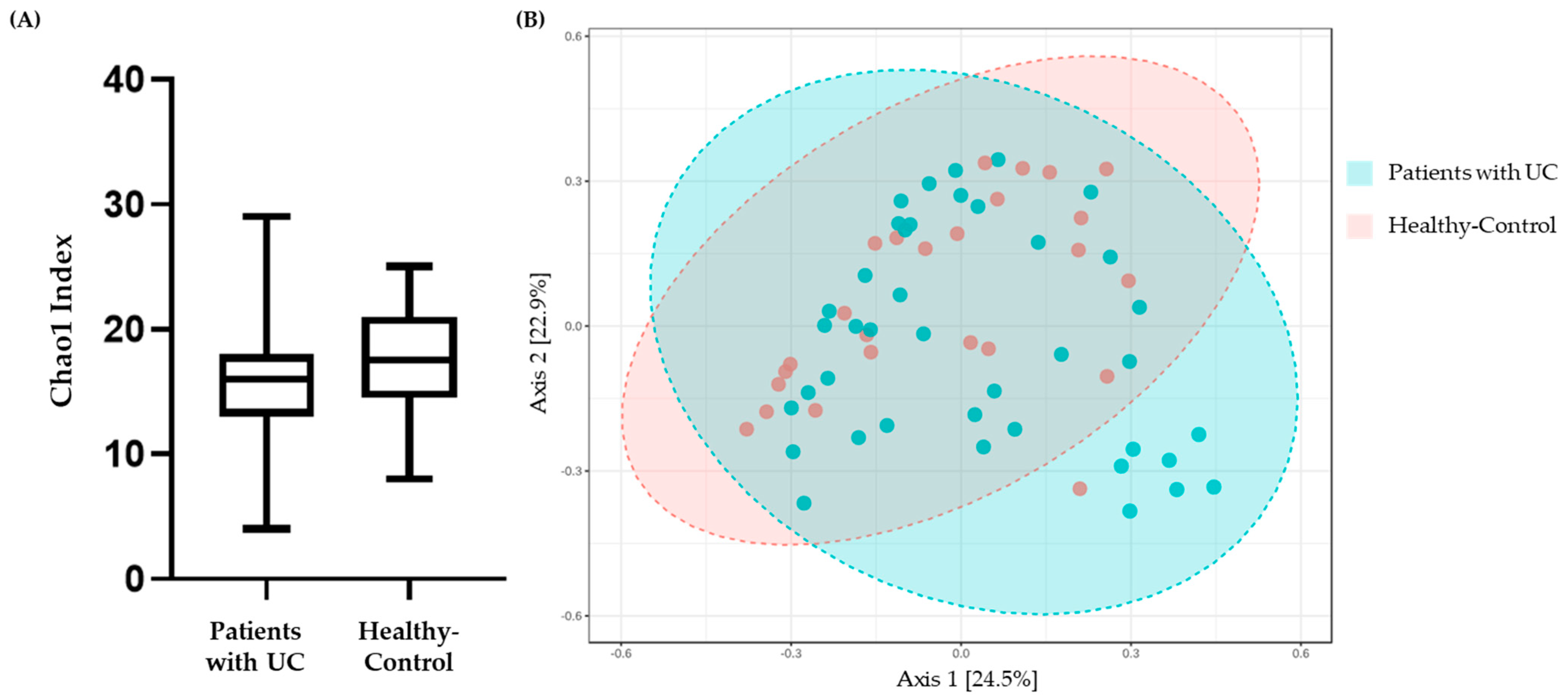
3.3. Alpha Diversity and Beta Diversity
4. Discussion
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Ungaro, R.; Mehandru, S.; Allen, P.B.; Peyrin-Biroulet, L.; Colombel, J.F. Ulcerative colitis. Lancet 2017, 389, 1756–1770. [Google Scholar] [CrossRef]
- Mozdiak, E.; O’Malley, J.; Arasaradnam, R. Inflammatory bowel disease. BMJ 2015, 351, h4416. [Google Scholar] [CrossRef]
- Gryaznova, M.V.; Solodskikh, S.A.; Panevina, A.V.; Syromyatnikov, M.Y.; Dvoretskaya, Y.D.; Sviridova, T.N.; Popov, E.S.; Popov, V.N. Study of microbiome changes in patients with ulcerative colitis in the Central European part of Russia. Heliyon 2021, 7, e06432. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, G.G. The global burden of IBD: From 2015 to 2025. Nat. Rev. Gastroenterol. Hepatol. 2015, 12, 720–727. [Google Scholar] [CrossRef]
- Ng, S.C.; Shi, H.Y.; Hamidi, N.; Underwood, F.E.; Tang, W.; Benchimol, E.I.; Panaccione, R.; Ghosh, S.; Wu, J.C.Y.; Chan, F.K.L.; et al. Worldwide incidence and prevalence of inflammatory bowel disease in the 21st century: A systematic review of population-based studies. Lancet 2017, 390, 2769–2778. [Google Scholar] [CrossRef] [PubMed]
- Rhen, T.; Cidlowski, J.A. Antiinflammatory action of glucocorticoids-new mechanisms for old drugs. N. Engl. J. Med. 2005, 353, 1711–1723. [Google Scholar] [CrossRef] [PubMed]
- Habens, F.; Srinivasan, N.; Oakley, F.; Mann, D.A.; Ganesan, A.; Packham, G. Novel sulfasalazine analogues with enhanced NF-kB inhibitory and apoptosis promoting activity. Apoptosis 2005, 10, 481–491. [Google Scholar] [CrossRef] [PubMed]
- Nugent, S.G.; Kumar, D.; Rampton, D.S.; Evans, D.F. Intestinal luminal pH in inflammatory bowel disease: Possible determinants and implications for therapy with aminosalicylates and other drugs. Gut 2001, 48, 571–577. [Google Scholar] [CrossRef]
- Van Dieren, J.M.; Kuipers, E.J.; Samsom, J.N.; Nieuwenhuis, E.E.; van der Woude, C.J. Revisiting the immunomodulators tacrolimus, methotrexate, and mycophenolate mofetil: Their mechanisms of action and role in the treatment of IBD. Inflamm. Bowel Dis. 2006, 12, 311–327. [Google Scholar] [CrossRef]
- Sartor, R.B. Mechanisms of disease: Pathogenesis of Crohn’s disease and ulcerative colitis. Nat. Rev. Gastroenterol. Hepatol. 2006, 3, 390–407. [Google Scholar] [CrossRef]
- McGovern, D.P.B.; Kugathasan, S.; Cho, J.H. Genetics of inflammatory bowel diseases. Gastroenterology 2015, 149, 1163–1176.e2. [Google Scholar] [CrossRef] [PubMed]
- Ananthakrishnan, A.N. Environmental risk factors for inflammatory bowel disease. Gastroenterol. Hepatol. 2013, 9, 367–374. [Google Scholar] [CrossRef]
- Sheehan, D.; Moran, C.; Shanahan, F. The microbiota in inflammatory bowel disease. J. Gastroenterol. 2015, 50, 495–507. [Google Scholar] [CrossRef] [PubMed]
- Khor, B.; Gardet, A.; Xavier, R.J. Genetics and pathogenesis of inflammatory bowel disease. Nature 2011, 474, 307–317. [Google Scholar] [CrossRef]
- Human Microbiome Project Consortium. Structure, function and diversity of the healthy human microbiome. Nature 2012, 486, 207–214. [Google Scholar] [CrossRef]
- Zakerska-Banaszak, O.; Tomczak, H.; Gabryel, M.; Baturo, A.; Wolko, L.; Michalak, M.; Malinska, N.; Mankowska-Wierzbicka, D.; Eder, P.; Dobrowolska, A.; et al. Dysbiosis of gut microbiota in Polish patients with ulcerative colitis: A pilot study. Sci. Rep. 2021, 11, 2166. [Google Scholar] [CrossRef]
- Arumugam, M.; Raes, J.; Pelletier, E.; Le Paslier, D.; Yamada, T.; Mende, D.R.; Fernandes, G.R.; Tap, J.; Bruls, T.; Batto, J.M.; et al. Enterotypes of the human gut microbiome. Nature 2011, 473, 174–180. [Google Scholar] [CrossRef]
- Xavier, R.J.; Podolsky, D.K. Unravelling the pathogenesis of inflammatory bowel disease. Nature 2007, 448, 427–434. [Google Scholar] [CrossRef] [PubMed]
- Hörmannsperger, G.; Schaubeck, M.; Haller, D. Intestinal microbiota in animal models of inflammatory diseases. ILAR J. 2015, 56, 179–191. [Google Scholar] [CrossRef]
- He, X.X.; Li, Y.H.; Yan, P.G.; Meng, X.C.; Chen, C.Y.; Li, K.M.; Li, J.N. Relationship between clinical features and intestinal microbiota in Chinese patients with ulcerative colitis. World J. Gastroenterol. 2021, 27, 4722–4737. [Google Scholar] [CrossRef]
- Sepehri, S.; Kotlowski, R.; Bernstein, C.N.; Krause, D.O. Microbial diversity of inflamed and noninflamed gut biopsy tissues in inflammatory bowel disease. Inflamm. Bowel Dis. 2007, 13, 675–683. [Google Scholar] [CrossRef] [PubMed]
- Imhann, F.; Vich Vila, A.; Bonder, M.J.; Fu, J.; Gevers, D.; Visschedijk, M.C.; Spekhorst, L.M.; Alberts, R.; Franke, L.; van Dullemen, H.M.; et al. Interplay of host genetics and gut microbiota underlying the onset and clinical presentation of inflammatory bowel disease. Gut 2018, 67, 108–119. [Google Scholar] [CrossRef] [PubMed]
- Casén, C.; Vebø, H.C.; Sekelja, M.; Hegge, F.T.; Karlsson, M.K.; Ciemniejewska, E.; Dzankovic, S.; Frøyland, C.; Nestestog, R.; Engstrand, L.; et al. Deviations in human gut microbiota: A novel diagnostic test for determining dysbiosis in patients with IBS or IBD. Aliment. Pharmacol. Ther. 2015, 42, 71–83. [Google Scholar] [CrossRef] [PubMed]
- Walker, A.W.; Sanderson, J.D.; Churcher, C.; Parkes, G.C.; Hudspith, B.N.; Rayment, N.; Brostoff, J.; Parkhill, J.; Dougan, G.; Petrovska, L. High-throughput clone library analysis of the mucosa-associated microbiota reveals dysbiosis and differences between inflamed and non-inflamed regions of the intestine in inflammatory bowel disease. BMC Microbiol. 2011, 11, 7. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Liu, B.; Zhang, Y.; Wei, H.; Lei, Y.; Zhao, L. Structural shifts of mucosa-associated lactobacilli and Clostridium leptum subgroup in patients with ulcerative colitis. J. Clin. Microbiol. 2007, 45, 496–500. [Google Scholar] [CrossRef]
- Zhang, S.L.; Wang, S.N.; Miao, C.Y. Influence of microbiota on intestinal immune system in Ulcerative colitis and its intervention. Front. Immunol. 2017, 8, 1674. [Google Scholar] [CrossRef]
- Ng, Q.X.; Yau, C.E.; Yaow, C.Y.L.; Chong, R.I.H.; Chong, N.Z.-Y.; Teoh, S.E.; Lim, Y.L.; Soh, A.Y.S.; Ng, W.K.; Thumboo, J. What Has Longitudinal ‘Omics’ Studies Taught Us about Irritable Bowel Syndrome? A Systematic Review. Metabolites 2023, 13, 484. [Google Scholar]
- Willing, B.P.; Dicksved, J.; Halfvarson, J.; Andersson, A.F.; Lucio, M.; Zheng, Z.; Jarnerot, G.; Tysk, C.; Jansson, J.K.; Engstrand, L. A pyrosequencing study in twins shows that gastrointestinal microbial profiles vary with inflammatory bowel disease phenotypes. Gastroenterology 2010, 139, 1844–1854. [Google Scholar] [PubMed]
- Khan, I.; Ullah, N.; Zha, L.; Bai, Y.; Khan, A.; Zhao, T.; Che, T.; Zhang, C. Alteration of gut microbiota in inflammatory bowel disease (IBD): Cause or consequence? IBD treatment targeting the gut microbiome. Pathogens 2019, 8, 126. [Google Scholar] [CrossRef] [PubMed]
- Eckburg, P.B.; Bik, E.M.; Bernstein, C.N.; Purdom, E.; Dethlefsen, L.; Sargent, M.; Gill, S.R.; Nelson, K.E.; Relman, D.A. Diversity of the human intestinal microbial flora. Science 2005, 308, 1635–1638. [Google Scholar] [CrossRef]
- Pittayanon, R.; Lau, J.T.; Leontiadis, G.I.; Tse, F.; Yuan, Y.; Surette, M.; Moayyedi, P. Differences in gut microbiota in patients with vs without inflammatory bowel diseases: A systematic review. Gastroenterology 2020, 158, 930–946.e1. [Google Scholar] [CrossRef]
- Frank, D.N.; St Amand, A.L.; Feldman, R.A.; Boedeker, E.C.; Harpaz, N.; Pace, N.R. Molecular-phylogenetic characterization of microbial community imbalances in human inflammatory bowel diseases. Proc. Natl. Acad. Sci. USA 2007, 104, 13780–13785. [Google Scholar] [CrossRef]
- Nagao-Kitamoto, H.; Kamada, N. Host-microbial Cross-talk in inflammatory bowel Disease. Immune Netw. 2017, 17, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Forbes, J.D.; Chen, C.Y.; Knox, N.C.; Marrie, R.A.; El-Gabalawy, H.; de Kievit, T.; Alfa, M.; Bernstein, C.N.; Van Domselaar, G. A comparative study of the gut microbiota in immune-mediated inflammatory diseases—Does a common dysbiosis exist? Microbiome 2018, 6, 221. [Google Scholar] [CrossRef]
- Nemoto, H.; Kataoka, K.; Ishikawa, H.; Ikata, K.; Arimochi, H.; Iwasaki, T.; Ohnishi, Y.; Kuwahara, T.; Yasutomo, K. Reduced diversity and imbalance of fecal microbiota in patients with ulcerative colitis. Dig. Dis. Sci. 2012, 57, 2955–2964. [Google Scholar] [CrossRef] [PubMed]
- Nomura, K.; Ishikawa, D.; Okahara, K.; Ito, S.; Haga, K.; Takahashi, M.; Arakawa, A.; Shibuya, T.; Osada, T.; Kuwahara-Arai, K.; et al. Bacteroidetes species are correlated with disease activity in ulcerative colitis. J. Clin. Med. 2021, 10, 1749. [Google Scholar] [CrossRef]
- Smith, P.M.; Howitt, M.R.; Panikov, N.; Michaud, M.; Gallini, C.A.; Bohlooly, Y.M.; Glickman, J.N.; Garrett, W.S. The microbial metabolites, short-chain fatty acids, regulate colonic Treg cell homeostasis. Science 2013, 1979, 569–573. [Google Scholar] [CrossRef] [PubMed]
- Alam, M.T.; Amos, G.C.A.; Murphy, A.R.J.; Murch, S.; Wellington, E.M.H.; Arasaradnam, R.P. Microbial imbalance in inflammatory bowel disease patients at different taxonomic levels. Gut Pathog. 2020, 12, 1. [Google Scholar] [CrossRef]
- Rehman, A.; Rausch, P.; Wang, J.; Skieceviciene, J.; Kiudelis, G.; Bhagalia, K.; Amarapurkar, D.; Kupcinskas, L.; Schreiber, S.; Rosenstiel, P.; et al. Geographical patterns of the standing and active human gut microbiome in health and IBD. Gut 2016, 65, 238–248. [Google Scholar] [CrossRef]
- De Meij, T.G.J.; De Groot, E.F.J.; Peeters, C.F.W.; De Boer, N.K.H.; Kneepkens, C.M.F.; Eck, A.; Benninga, M.A.; Savelkoul, P.H.M.; Van Bodegraven, A.A.; Budding, A.E. Variability of core microbiota in newly diagnosed treatment-naïve paediatric inflammatory bowel disease patients. PLoS ONE 2018, 13, e0197649. [Google Scholar] [CrossRef]
- Do, M.H.; Lee, E.; Oh, M.-J.; Kim, Y.; Park, H.-Y. High-glucose or -fructose diet cause changes of the gut microbiota and metabolic disorders in mice without body weight change. Nutrients 2018, 10, 761. [Google Scholar] [CrossRef]
- Méndez-Salazar, E.O.; Ortiz-López, M.G.; Granados-Silvestre, M.D.L.Á.; Palacios-González, B.; Menjivar, M. Altered gut microbiota and compositional changes in firmicutes and proteobacteria in Mexican undernourished and obese children. Front. Microbiol. 2018, 9, 2494. [Google Scholar]
- Aldars-García, L.; Chaparro, M.; Gisbert, J.P. Systematic review: The gut microbiome and its potential clinical application in inflammatory bowel disease. Microorganisms 2021, 9, 977. [Google Scholar] [CrossRef]
- Lavelle, A.; Lennon, G.; O’Sullivan, O.; Docherty, N.; Balfe, A.; Maguire, A.; Mulcahy, H.E.; Doherty, G.; O’Donoghue, D.; Hyland, J.; et al. Spatial variation of the colonic microbiota in patients with ulcerative colitis and control volunteers. Gut 2015, 64, 1553–1561. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Chen, L.; Zhou, R.; Wang, X.; Song, L.; Huang, S.; Wang, G.; Xia, B. Increased proportions of Bifidobacterium and the Lactobacillus group and loss of butyrate-producing bacteria in inflammatory bowel disease. J. Clin. Microbiol. 2014, 52, 398–406. [Google Scholar] [CrossRef] [PubMed]
- Gargari, G.; Taverniti, V.; Balzaretti, S.; Ferrario, C.; Gardana, C.; Simonetti, P.; Guglielmetti, S. Consumption of a Bifidobacterium bifidum strain for 4 weeks modulates dominant intestinal bacterial taxa and fecal butyrate in healthy adults. Appl. Environ. Microbiol. 2016, 82, 5850–5859. [Google Scholar] [CrossRef] [PubMed]
- Naschla, G.; Marcela, A.H.; Martin, G. Butyrate and the Fine-Tuning of Colonic Homeostatis: Implication for Inflammatory Bowel Diseases. Int. J. Mol. Sci. 2020, 22, 3061. [Google Scholar] [CrossRef]
- Morotomi, M.; Nagai, F.; Sakon, H.; Tanaka, R. Dialister succinatiphilus sp. nov. and Barnesiella intestinihominis sp. nov., isolated from human faeces. Int. J. Syst. Evol. Microbiol. 2008, 58, 2716–2720. [Google Scholar] [CrossRef]
- Michail, S.; Durbin, M.; Turner, D.; Griffiths, A.M.; Mack, D.R.; Hyams, J.; Leleiko, N.; Kenche, H.; Stolfi, A.; Wine, E. Alterations in the gut microbiome of children with severe ulcerative colitis. Inflamm. Bowel Dis. 2012, 18, 1799–1808. [Google Scholar] [CrossRef]
- Clooney, A.G.; Eckenberger, J.; Laserna-Mendieta, E.; Sexton, K.A.; Bernstein, M.T.; Vagianos, K.; Sargent, M.; Ryan, F.J.; Moran, C.; Sheehan, D.; et al. Ranking microbiome variance in inflammatory bowel disease: A large longitudinal intercontinental study. Gut 2021, 70, 499–510. [Google Scholar] [CrossRef]
- Nishida, A.; Inoue, R.; Inatomi, O.; Bamba, S.; Naito, Y.; Andoh, A. Gut microbiota in the pathogenesis of inflammatory bowel disease. Clin. J. Gastroenterol. 2018, 11, 1–10. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variable | Patients with Ulcerative Colitis (n = 40) | Healthy Controls (n = 25) | p-Value | Statistical Significance |
|---|---|---|---|---|
| Age (years), mean ± S.D. | 48.5 ± 17.3 | 43.8 ± 10.7 | 0.237 | - |
| Female/male (%) | 19/21 (47.5/52.5) | 17/8 (68.0/32.0) | 0.129 | - |
| Weight (kg), mean ± S.D. | 66.10 ± 14.41 | 66.10 ± 11.28 | 0.445 | - |
| Height (cm), mean ± S.D. | 164.01 ± 7.19 | 165.31 ± 9.65 | 0.593 | - |
| Body mass index, mean ± S.D. | 24.38 ± 3.81 | 23.30 ± 3.86 | 0.305 | - |
| White blood cell, mean ± S.D. | 6.34 ± 2.01 | 6.76 ± 1.71 | 0.398 | - |
| Red blood cell, mean ± S.D. | 4.48 ± 0.51 | 4.54 ± 0.49 | 0.642 | - |
| Hemoglobin, mean ± S.D. | 13.76 ± 2.00 | 13.84 ± 1.76 | 0.868 | - |
| Hematocrit, mean ± S.D. | 39.15 ± 9.50 | 43.16 ± 5.57 | 0.061 | - |
| Platelet, mean ± S.D. | 251.79 ± 56.39 | 294.08 ± 56.84 | 0.005 | ** |
| Segmented neutrophil | 58.67 ± 9.51 | -(6) | - | - |
| Lymphocyte | 31.49 ± 8.37 | - | - | - |
| Monocyte | 8.10 ± 5.16 | - | - | - |
| Eosinophil | 2.17 ± 1.62 | - | - | - |
| Basophil | 0.68 ± 0.45 | - | - | - |
| Calcium, mean ± S.D. | 16.49 ± 44.53 | 9.52 ± 0.37 | 0.438 | - |
| Phosphorus, mean ± S.D. | 3.67 ± 1.29 | 4.34 ± 2.12 | 0.165 | - |
| Glucose, mean ± S.D. | 101.88 ± 39.66 | 94.04 ± 11.82 | 0.264 | - |
| Creatinine, mean ± S.D. | 3.30 ± 15.33 | 0.72 ± 0.19 | 0.404 | - |
| eGFR (1), mean ± S.D. | 101.46 ± 13.23 | 104.96 ± 26.74 | 0.549 | - |
| BUN (2), mean ± S.D. | 15.76 ± 12.54 | 11.87 ± 3.00 | 0.134 | - |
| Uric acid, mean ± S.D. | 5.11 ± 1.64 | 4.83 ± 1.24 | 0.476 | - |
| Cholesterol, mean ± S.D. | 187.43 ± 37.77 | 210.16 ± 49.21 | 0.044 | * |
| Protein, mean ± S.D. | 10.59 ± 22.72 | 7.45 ± 0.47 | 0.493 | - |
| Albumin, mean ± S.D. | 4.48 ± 0.61 | 4.81 ± 0.31 | 0.017 | * |
| AST (3), mean ± S.D. | 21.77 ± 9.81 | 22.48 ± 5.72 | 0.746 | - |
| ALT (4), mean ± S.D. | 21.22 ± 11.68 | 19.60 ± 10.05 | 0.574 | - |
| ALP (5), mean ± S.D. | 69.78 ± 18.93 | 63.80 ± 15.68 | 0.197 | - |
| Total bilirubin, mean ± S.D. | 0.56 ± 0.22 | 1.01 ± 1.94 | 0.256 | - |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Do, K.-H.; Ko, S.-H.; Kim, K.B.; Seo, K.; Lee, W.-K. Comparative Study of Intestinal Microbiome in Patients with Ulcerative Colitis and Healthy Controls in Korea. Microorganisms 2023, 11, 2750. https://doi.org/10.3390/microorganisms11112750
Do K-H, Ko S-H, Kim KB, Seo K, Lee W-K. Comparative Study of Intestinal Microbiome in Patients with Ulcerative Colitis and Healthy Controls in Korea. Microorganisms. 2023; 11(11):2750. https://doi.org/10.3390/microorganisms11112750
Chicago/Turabian StyleDo, Kyung-Hyo, Seung-Hyun Ko, Ki Bae Kim, Kwangwon Seo, and Wan-Kyu Lee. 2023. "Comparative Study of Intestinal Microbiome in Patients with Ulcerative Colitis and Healthy Controls in Korea" Microorganisms 11, no. 11: 2750. https://doi.org/10.3390/microorganisms11112750
APA StyleDo, K.-H., Ko, S.-H., Kim, K. B., Seo, K., & Lee, W.-K. (2023). Comparative Study of Intestinal Microbiome in Patients with Ulcerative Colitis and Healthy Controls in Korea. Microorganisms, 11(11), 2750. https://doi.org/10.3390/microorganisms11112750