

Revealing an Iranian Isolate of Tomato Brown Rugose Fruit Virus: Complete Genome Analysis and Mechanical Transmission





Abstract
:1. Introduction
2. Materials and Methods
2.1. Sampling
2.2. Sequencing
2.3. Phylogenetic Analysis

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer Name | Sequence (5′ to 3′) | Ta (°C) | Size (bp) | Reference |
|---|---|---|---|---|
| ToBRFV-1F | TTTTACAACATATACCAACAACAACAAACAAC | 54 | 1193 | [26] |
| ToBRFV-1R | GAACTATGACCATATCTCTCATTTTTGG | |||
| ToBRFV-2F | AGCAATTTTACAGCGCAATG | 54 | 1338 | [26] |
| ToBRFV-2R | CAACGTGGTACTTCCTAGCATGTG | |||
| ToBRFV-3F | CAAGATCCTAAAGGATACAGCTGCTATAG | 56 | 1352 | [26] |
| ToBRFV-3R | TGACATTAAGAGAAATGTCAGTCAACC | |||
| ToBRFV-4F | AGGTTCTAATCTTTTTGTTGCAGC | 54 | 1349 | [26] |
| ToBRFV-4R | CCAACTGCGTGTAATACGCACA | |||
| ToBRFV-5F | ATTGTTTATTATGACCCTTTGAAGTTG | 54 | 1339 | [26] |
| ToBRFV-5R | CTATAATCCTATTTCTAGTATCGAAAGCTC | |||
| TBRFV-F-5722 | CACAATCGCAACTCCATCGC | 54 | 623 | [12,26] |
| ToBRFV-R (6344) | GTGCCTACGGATGTGTATGA |
2.4. Population Genetic Parameters
2.5. Molecular Dating Analysis of ToBRFV
2.6. Mechanical Transmission
3. Results
3.1. Greenhouse Observation and Complete Genome Sequencing of ToBRFV
3.2. Phylogenetic Relationships of ToBRFV Isolates
3.3. Population Genetic Structure
3.4. Molecular Dating Analysis of ToBRFV
3.5. Mechanical Transmission
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ali, M.Y.; Sina, A.A.I.; Khandker, S.S.; Neesa, L.; Tanvir, E.; Kabir, A.; Khalil, M.I.; Gan, S.H. Nutritional composition and bioactive compounds in tomatoes and their impact on human health and disease: A review. Foods 2020, 10, 45. [Google Scholar] [CrossRef] [PubMed]
- Salem, N.; Mansour, A.; Ciuffo, M.; Falk, B.W.; Turina, M. A new tobamovirus infecting tomato crops in Jordan. Arch. Virol. 2016, 161, 503–506. [Google Scholar] [CrossRef] [PubMed]
- Luria, N.; Smith, E.; Reingold, V.; Bekelman, I.; Lapidot, M.; Levin, I.; Elad, N.; Tam, Y.; Sela, N.; Abu-Ras, A.; et al. A new Israeli Tobamovirus isolate infects tomato plants harboring Tm-22 resistance genes. PLoS ONE 2017, 12, e0170429. [Google Scholar] [CrossRef]
- González-Concha, L.F.; Ramírez-Gil, J.G.; García-Estrada, R.S.; Rebollar-Alviter, A.; Tovar-Pedraza, J.M. Spatiotemporal analyses of tomato brown rugose fruit virus in commercial tomato greenhouses. Agronomy 2021, 11, 1268. [Google Scholar] [CrossRef]
- Avni, B.; Gelbart, D.; Sufrin-Ringwald, T.; Zinger, A.; Chen, L.; Machbash, Z.; Bekelman, I.; Segoli, M.; Dombrovsky, A.; Kamenetsky, R.; et al. Tomato genetic resistance to tobamoviruses is compromised. Acta Hortic. 2021, 1316, 89–98. [Google Scholar] [CrossRef]
- Rizzo, D.; Da Lio, D.; Panattoni, A.; Salemi, C.; Cappellini, G.; Bartolini, L.; Parrella, G. Rapid and sensitive detection of tomato brown rugose fruit virus in tomato and pepper seeds by reverse transcription loop-mediated isothermal amplification assays (real time and visual) and comparison with RT-PCR end-point and RT-qPCR methods. Front. Microbiol. 2021, 12, 640932. [Google Scholar] [CrossRef] [PubMed]
- Abrahamian, P.; Cai, W.; Nunziata, S.O.; Ling, K.; Jaiswal, N.; Mavrodieva, V.A.; Rivera, Y.; Nakhla, M.K. Comparative analysis of tomato brown rugose fruit virus isolates shows limited genetic diversity. Viruses 2022, 14, 2816. [Google Scholar] [CrossRef]
- Cambrón-Crisantos, J.M.; Rodríguez-Mendoza, J.; Valencia-Luna, J.B.; Rangel, S.A.; de Jesús García-Ávila, C.; López-Buenfil, J.A.; Ochoa-Martínez, D.L. First report of tomato brown rugose fruit virus (ToBRFV) in Michoacan, Mexico. Mex. J. Phytopathol. 2018, 37, 185–192. [Google Scholar] [CrossRef]
- Menzel, W.; Knierim, D.; Winter, S.; Hamacher, J.; Heupel, M. First report of tomato brown rugose fruit virus infecting tomato in Germany. New Dis. Rep. 2019, 39, 1. [Google Scholar] [CrossRef]
- Panno, S.; Caruso, A.G.; Davino, S. First report of tomato brown rugose fruit virus on tomato crops in Italy. Plant Dis. 2019, 103, 1443. [Google Scholar] [CrossRef]
- Fidan, H.; Sarikaya, P.; Calis, O. First report of Tomato brown rugose fruit virus on tomato in Turkey. New Dis. Rep. 2019, 39, 18. [Google Scholar] [CrossRef]
- Ling, K.-S.; Tian, T.; Gurung, S.; Salati, R.; Gilliard, A. First report of tomato brown rugose fruit virus infecting greenhouse tomato in the United States. Plant Dis. 2019, 103, 1439. [Google Scholar] [CrossRef]
- van de Vossenberg, B.T.L.H.; Visser, M.; Bruinsma, M.; Koenraadt, H.M.S.; Westenberg, M.; Botermans, M. Real-time tracking of tomato brown rugose fruit virus (ToBRFV) outbreaks in the Netherlands using Nextstrain. PLoS ONE 2020, 15, e0234671. [Google Scholar] [CrossRef]
- Çelik, A.; Coşkan, S.; Morca, A.F.; Santosa, A.I.; Koolivand, D. Insight into population structure and evolutionary analysis of the emerging tomato brown rugose fruit virus. Plants 2022, 11, 3279. [Google Scholar] [CrossRef]
- Ghorbani, A.; Rostami, M.; Seifi, S.; Izadpanah, K. First report of tomato brown rugose fruit virus in greenhouse tomato in Iran. New Dis. Rep. 2021, 44, e12040. [Google Scholar] [CrossRef]
- Esmaeilzadeh, F.; Koolivand, D. Occurrence of tomato brown rugose fruit virus in tomato in Iran. J. Plant Pathol. 2022, 104, 457. [Google Scholar] [CrossRef]
- Esmaeilzadeh, F.; Koolivand, D. First report of tomato brown rugose fruit virus infecting bell pepper in Iran. J. Plant Pathol. 2022, 104, 893. [Google Scholar] [CrossRef]
- Topkaya, Ş.; Çelik, A.; Santosa, A.I.; Jones, R.A.C. Molecular analysis of the global population of potato virus S redefines its phylogeny, and has crop biosecurity implications. Viruses 2023, 15, 1104. [Google Scholar] [CrossRef]
- Gambino, G.; Perrone, I.; Gribaudo, I. A rapid and effective method for RNA extraction from different tissues of grapevine and other woody plants. Phytochem. Anal. 2008, 19, 520–525. [Google Scholar] [CrossRef]
- Hall, T.A. BioEdit: A user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp. Ser. 1999, 41, 95–98. [Google Scholar]
- Tamura, K.; Stecher, G.; Kumar, S. MEGA11: Molecular Evolutionary Genetics Analysis Version 11. Mol. Biol. Evol. 2021, 38, 3022–3027. [Google Scholar] [CrossRef] [PubMed]
- Darriba, D.; Taboada, G.L.; Doallo, R.; Posada, D. jModelTest 2: More models, new heuristics and parallel computing. Nature Methods 2012, 9, 772. [Google Scholar] [CrossRef] [PubMed]
- Muhire, B.M.; Varsani, A.; Martin, D.P. SDT: A virus classification tool based on pairwise sequence alignment and identity calculation. PLoS ONE 2014, 9, e108277. [Google Scholar] [CrossRef]
- Martin, D.P.; Murrell, B.; Golden, M.; Khoosal, A.; Muhire, B. RDP4: Detection and analysis of recombination patterns in virus genomes. Virus Evol. 2015, 1, vev003. [Google Scholar] [CrossRef]
- Huson, D. SplitsTree: Analyzing and visualizing evolutionary data. Bioinformatics 1998, 14, 68–73. [Google Scholar] [CrossRef] [PubMed]
- Yan, Z.-Y.; Zhao, M.-S.; Ma, H.-Y.; Liu, L.-Z.; Yang, G.-L.; Geng, C.; Tian, Y.; Li, X.-D. Biological and molecular characterization of tomato brown rugose fruit virus and development of quadruplex RT-PCR detection. J. Integr. Agric. 2021, 20, 1871–1879. [Google Scholar] [CrossRef]
- Yang, Z.; Nielsen, R.; Goldman, N.; Pedersen, A.-M. Codon-substitution models for heterogeneous selection pressure at amino acid sites. Genetics 2000, 155, 431–449. [Google Scholar] [CrossRef]
- Tajima, F. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 1989, 123, 585–595. [Google Scholar] [CrossRef]
- Fu, Y.-X.; Li, W.-H. Statistical tests of neutrality of mutations. Genetics 1993, 133, 693–709. [Google Scholar] [CrossRef]
- Rozas, J.; Ferrer-Mata, A.; Sánchez-DelBarrio, J.C.; Guirao-Rico, S.; Librado, P.; Ramos-Onsins, S.E.; Sánchez-Gracia, A. DnaSP 6: DNA sequence polymorphism analysis of large data sets. Mol. Biol. Evol. 2017, 34, 3299–3302. [Google Scholar] [CrossRef]
- Çelik, A.; Morca, A.F.; Coşkan, S.; Santosa, A.I. Global population structure of apple mosaic virus (ApMV, Genus Ilarvirus). Viruses 2023, 15, 1221. [Google Scholar] [CrossRef] [PubMed]
- Mello, B. Estimating TimeTrees with MEGA and the TimeTree resource. Mol. Biol. Evol. 2018, 35, 2334–2342. [Google Scholar] [CrossRef] [PubMed]
- Branthôme, F.-X. ToBRFV: Latest Update. 2020. Available online: http://www.tomatonews.com/en/tobrfv-latest-update_2_997.html (accessed on 30 May 2023).
- HortiDaily. Netherlands: Peruvian Tomato Seed with ToBRFV Intercepted. 2020. Available online: https://www.hortidaily.com/article/9189118/netherlands-peruvian-tomato-seed-withtobrfv-intercepted (accessed on 11 May 2023).
- García-Arenal, F.; Fraile, A.; Malpica, J.M. Variability and genetic structure of plant virus populations. Annu. Rev. Phytopathol. 2001, 39, 157–186. [Google Scholar] [CrossRef] [PubMed]
- Nouri, S.; Arevalo, R.; Falk, B.W.; Groves, R.L. Genetic structure and molecular variability of cucumber mosaic virus isolates in the United States. PLoS ONE 2014, 9, e96582. [Google Scholar] [CrossRef] [PubMed]

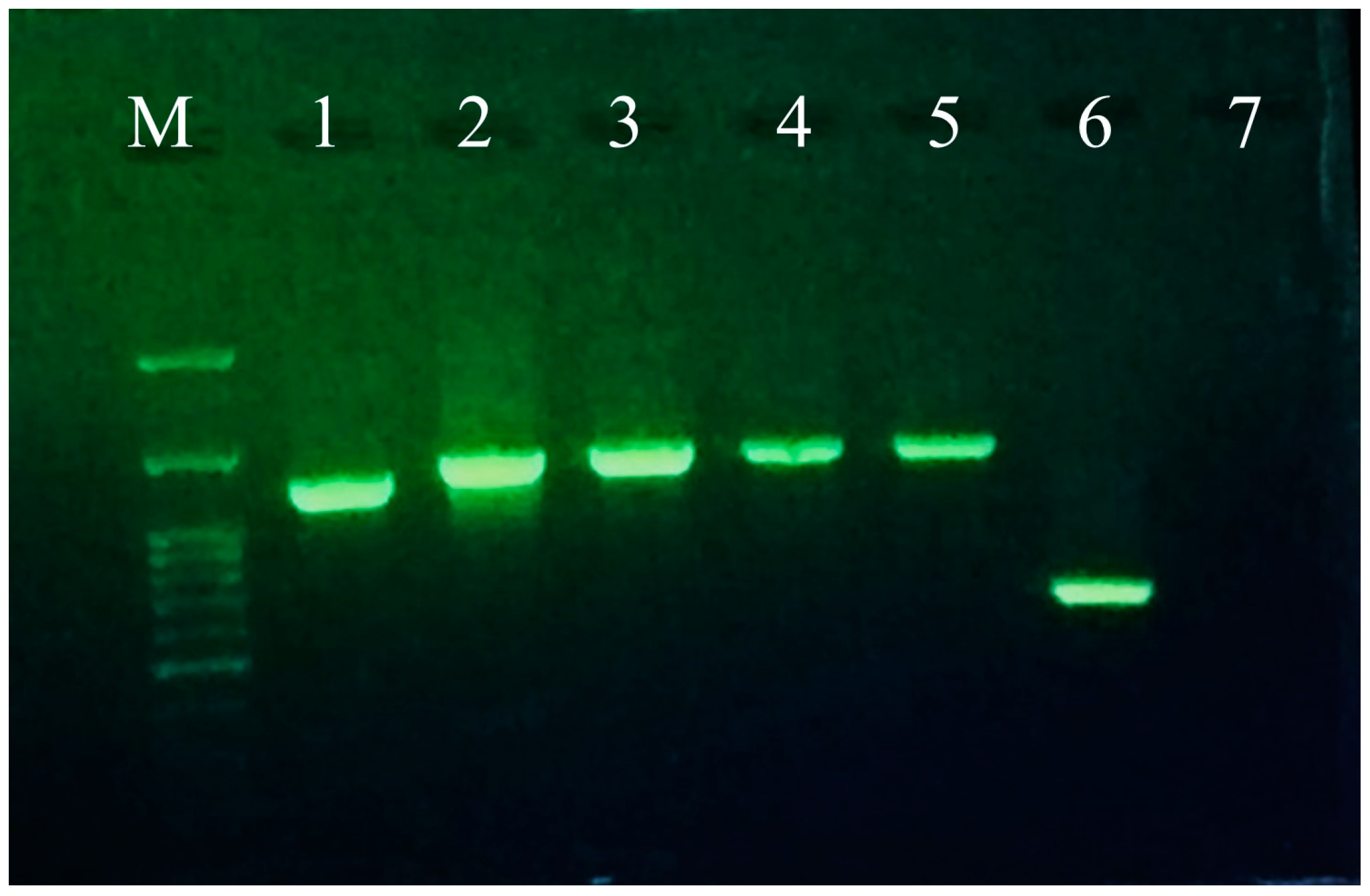

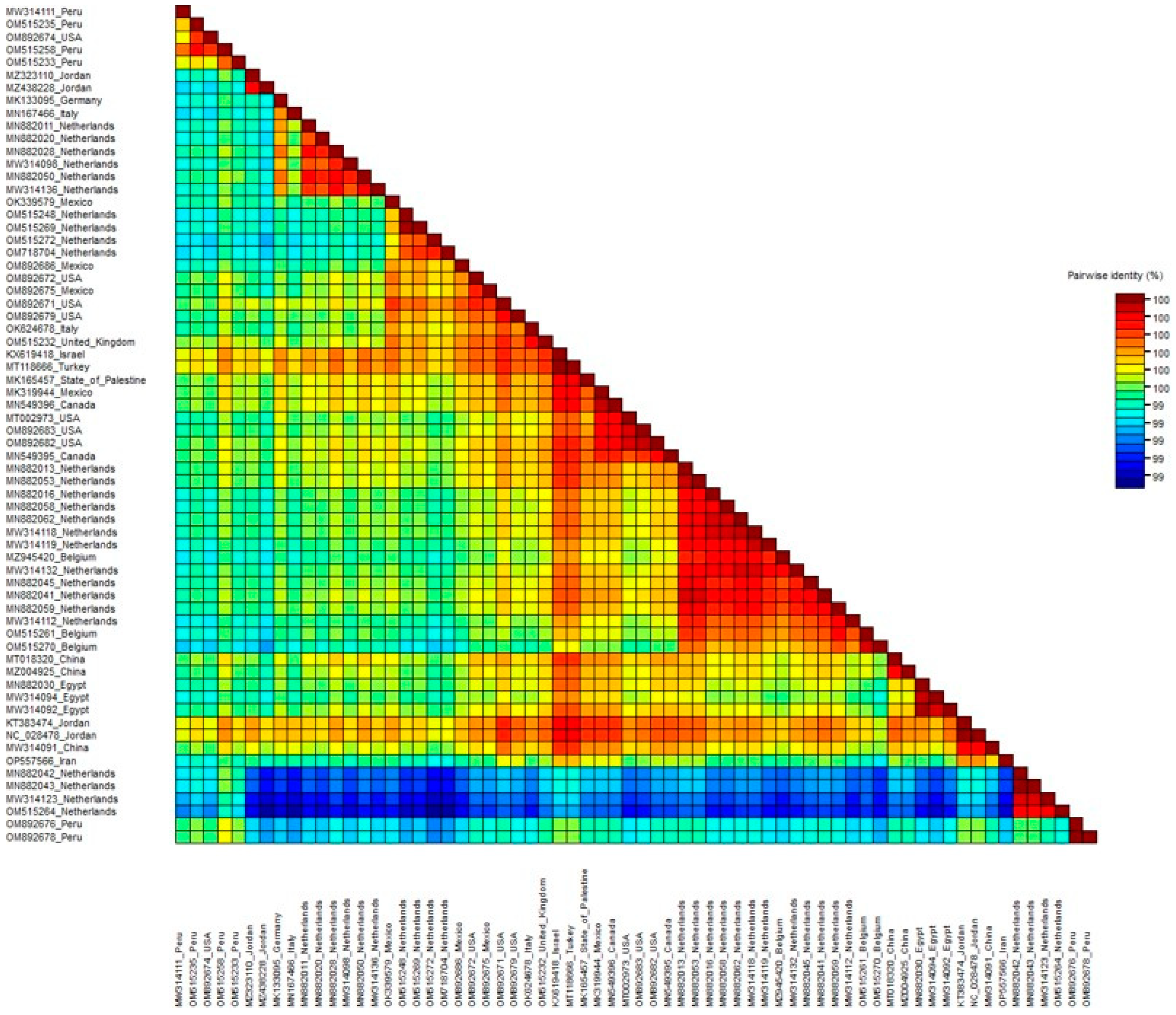

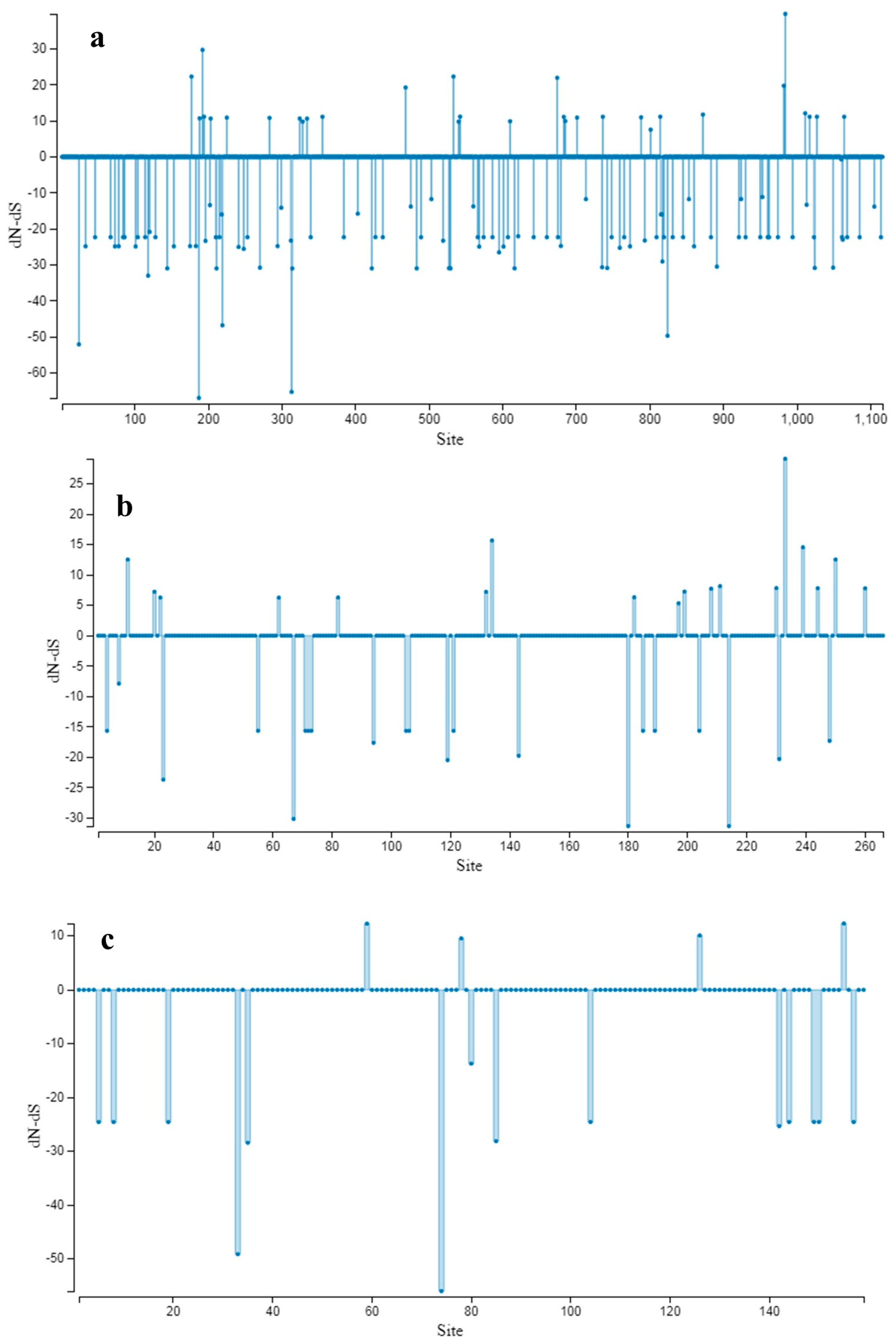


| Population | N | H | Hd | S | η | k | π | SS | NS | dS | dN | ω |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Phylogroup | ||||||||||||
| Clade I | 49 | 48 | 0.998 | 171 | 172 | 16.660 | 0.00272 | 1365.22 | 4745.78 | 0.00760 | 0.00132 | 0.1736 |
| Clade II | 6 | 5 | 0.933 | 33 | 33 | 13.400 | 0.00219 | 1366.94 | 4750.06 | 0.00697 | 0.00081 | 0.1162 |
| Clade III | 11 | 9 | 0.964 | 61 | 62 | 20.236 | 0.00331 | 1366.94 | 4750.06 | 0.00954 | 0.00152 | 0.1593 |
| Continent | ||||||||||||
| Europe | 33 | 32 | 0.998 | 136 | 138 | 21.303 | 0.00348 | 1366.76 | 4750.24 | 0.00972 | 0.00169 | 0.1738 |
| South_America | 6 | 5 | 0.933 | 37 | 38 | 16.000 | 0.00262 | 1366.44 | 4750.56 | 0.00742 | 0.00123 | 0.1657 |
| Asia | 11 | 9 | 0.964 | 51 | 51 | 12.727 | 0.00208 | 1365.12 | 4745.88 | 0.00583 | 0.00100 | 0.1715 |
| Africa | 4 | 3 | 0.833 | 3 | 3 | 1.500 | 0.00025 | 1366.92 | 4750.08 | 0.00073 | 0.00011 | 0.1506 |
| North America | 13 | 13 | 1.000 | 62 | 63 | 13.846 | 0.00226 | 1366.56 | 4750.44 | 0.00711 | 0.00087 | 0.1223 |
| Population | π | Tajima’s D | Fu and Li’s D* | Fu and Li’s F* |
|---|---|---|---|---|
| Phylogroups | ||||
| Clade I | 0.00272 | −2.03856 * | −3.49583 * | −3.52417 * |
| Clade II | 0.00219 | −0.46253 ns | −0.36366 ns | −0.42092 ns |
| Clade III | 0.00331 | −0.20899 ns | −0.33262 ns | −0.34157 ns |
| Continents | ||||
| Europe | 0.00348 | −1.41629 ns | −1.85488 ns | −2.02230 ns |
| South America | 0.00262 | −0.24596 ns | −0.08919 ns | −0.13431 ns |
| Asia | 0.00208 | −1.27138 ns | −1.00970 ns | −1.22239 ns |
| Africa | 0.00025 | −0.75445 ns | −0.75445 ns | −0.67466 ns |
| North America | 0.00226 | −1.43378 ns | −2.03804 ns | −2.14714 ns |
| Population | KS* | KST* | KS*, KST* p-Value | Z* | p-Value | Snn | p-Value | FST |
|---|---|---|---|---|---|---|---|---|
| Phylogroups | ||||||||
| Clade I (n = 50)/Clade II (n = 6) | 2.77039 | 0.01671 | 0.0000 *** | 6.25047 | 0.0010 ** | 1.000 | 0.0000 *** | 0.197 |
| Clade I (n = 50)/Clade III (n = 11) | 2.80635 | 0.05540 | 0.0000 *** | 6.21615 | 0.0000 *** | 1.000 | 0.0000 *** | 0.360 |
| Clade II (n = 6)/Clade III (n = 11) | 2.74113 | 0.09697 | 0.0000 *** | 3.50621 | 0.0000 *** | 1.000 | 0.0000 *** | 0.387 |
| Continents | ||||||||
| Europe (n = 33)/Asia (n = 11) | 2.831 | 0.032 | 0.0000 *** | 5.678 | 0.0000 *** | 1.000 | 0.0000 *** | 0.140 |
| Europe (n = 33)/Africa (n = 4) | 2.812 | 0.049 | 0.0000 *** | 5.266 | 0.0000 *** | 1.000 | 0.0000 *** | 0.488 |
| Europe (n = 33)/S. America (n = 6) | 2.908 | 0.039 | 0.0000 *** | 5.388 | 0.0000 *** | 1.000 | 0.0000 *** | 0.283 |
| Europe (n = 33)/N. America (n = 13) | 2.847 | 0.035 | 0.0000 *** | 5.744 | 0.0000 *** | 0.978 | 0.0000 *** | 0.144 |
| S. America (n = 6)/Asia (n = 11) | 2.515 | 0.098 | 0.0000 *** | 3.511 | 0.0000 *** | 1.000 | 0.0010 ** | 0.338 |
| S. America (n = 6)/Africa (n = 4) | 2.049 | 0.253 | 0.0010 ** | 2.163 | 0.0020 ** | 1.000 | 0.0100 * | 0.657 |
| S. America (n = 6)/N. America (n = 13) | 2.605 | 0.086 | 0.0000 *** | 3.713 | 0.0000 *** | 0.947 | 0.0000 *** | 0.344 |
| Asia (n = 11)/Africa (n = 4) | 2.156 | 0.141 | 0.0000 *** | 3.236 | 0.0000 *** | 1.000 | 0.0020 ** | 0.564 |
| Asia (n = 11)/N. America (n = 13) | 2.525 | 0.042 | 0.0000 *** | 4.415 | 0.0000 *** | 0.958 | 0.0000 *** | 0.138 |
| Africa (n = 4)/N. America (n = 13) | 2.315 | 0.121 | 0.0000 *** | 3.428 | 0.0010 ** | 1.000 | 0.0010 ** | 0.555 |
| Species | Mean Patristic Distance | Ratios of ToBRFV and Other Patristic Distance |
|---|---|---|
| TMV | 0.16 | 0.8 |
| RheMV | 0.16 | 0.8 |
| ToBRFV | 0.20 | 1 |
| ToMV | 0.22 | 1.1 |
| ToMMV | 0.22 | 1.1 |
| BPMV | 0.28 | 1.4 |
| PMMoV | 0.42 | 2.1 |
| TMGMV | 0.52 | 2.6 |
| YTMMV | 0.54 | 2.7 |
| ObPV | 0.54 | 2.7 |
| PaMMV | 0.54 | 2.7 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Esmaeilzadeh, F.; Santosa, A.I.; Çelik, A.; Koolivand, D. Revealing an Iranian Isolate of Tomato Brown Rugose Fruit Virus: Complete Genome Analysis and Mechanical Transmission. Microorganisms 2023, 11, 2434. https://doi.org/10.3390/microorganisms11102434
Esmaeilzadeh F, Santosa AI, Çelik A, Koolivand D. Revealing an Iranian Isolate of Tomato Brown Rugose Fruit Virus: Complete Genome Analysis and Mechanical Transmission. Microorganisms. 2023; 11(10):2434. https://doi.org/10.3390/microorganisms11102434
Chicago/Turabian StyleEsmaeilzadeh, Fereshteh, Adyatma Irawan Santosa, Ali Çelik, and Davoud Koolivand. 2023. "Revealing an Iranian Isolate of Tomato Brown Rugose Fruit Virus: Complete Genome Analysis and Mechanical Transmission" Microorganisms 11, no. 10: 2434. https://doi.org/10.3390/microorganisms11102434
APA StyleEsmaeilzadeh, F., Santosa, A. I., Çelik, A., & Koolivand, D. (2023). Revealing an Iranian Isolate of Tomato Brown Rugose Fruit Virus: Complete Genome Analysis and Mechanical Transmission. Microorganisms, 11(10), 2434. https://doi.org/10.3390/microorganisms11102434