
Substrate Specificity of Biofilms Proximate to Historic Shipwrecks


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Methods
2.1. Field Experiments
2.2. DNA Extraction and Metagenome Sequencing
2.3. Bioinformatics
2.4. Metagenome Taxonomic and Functional Annotation
2.5. Statistical Analysis
3. Results
3.1. Quality and Descriptive Statistics of Assembled Metagenomes
3.2. Diversity, Richness, and Evenness of Biofilm Metagenomes
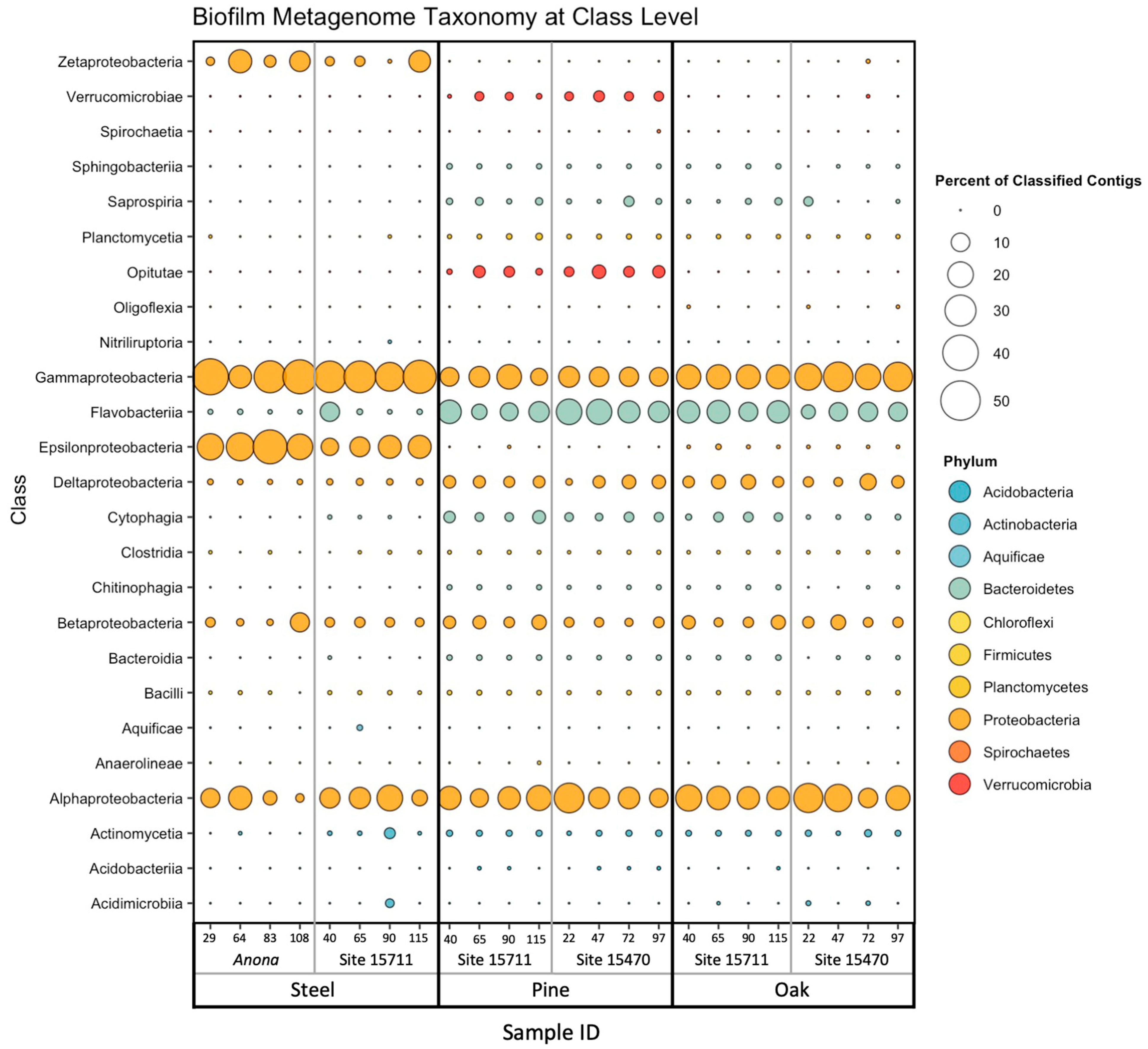
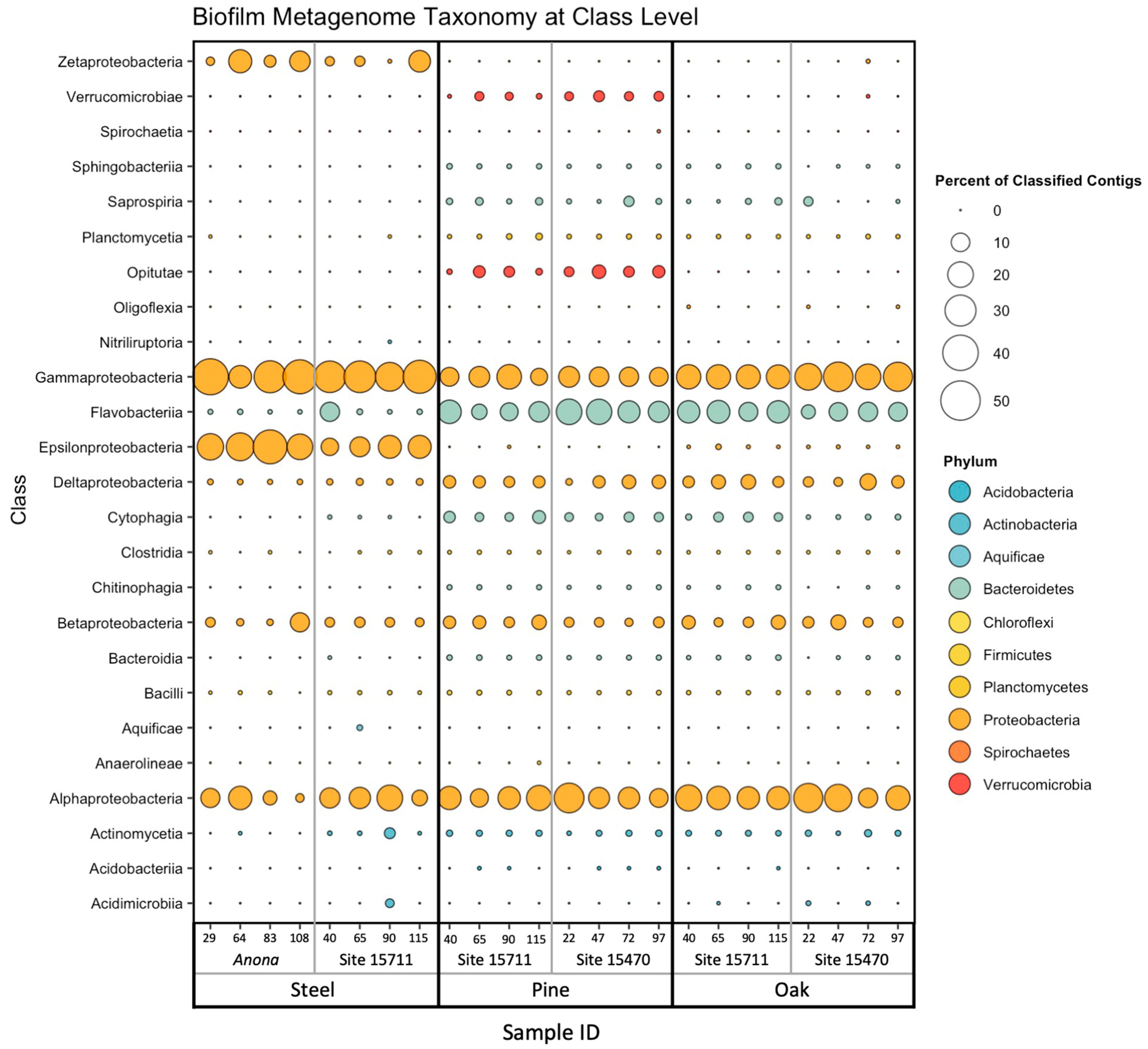
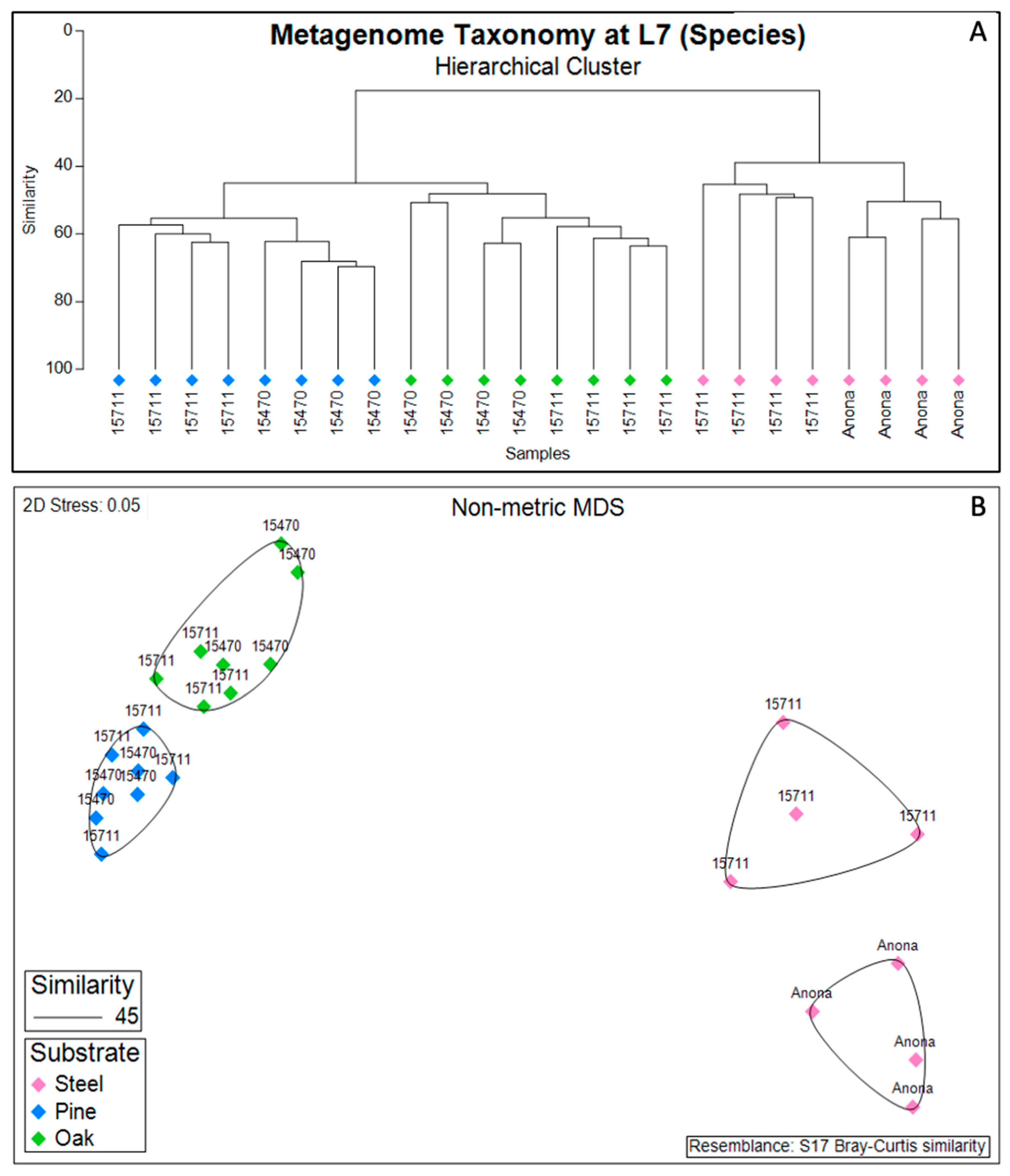
3.3. Biofilm Taxonomic Composition
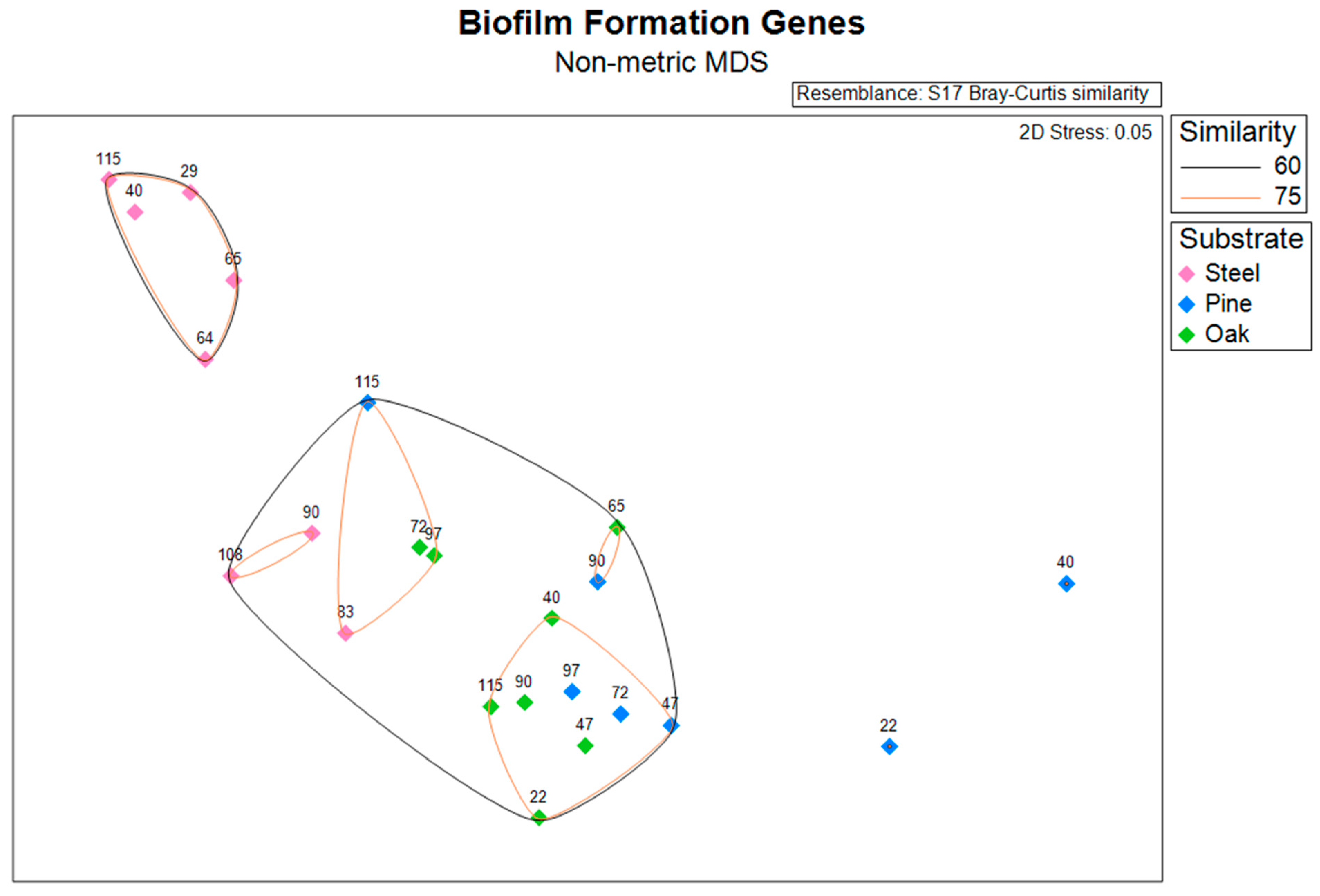
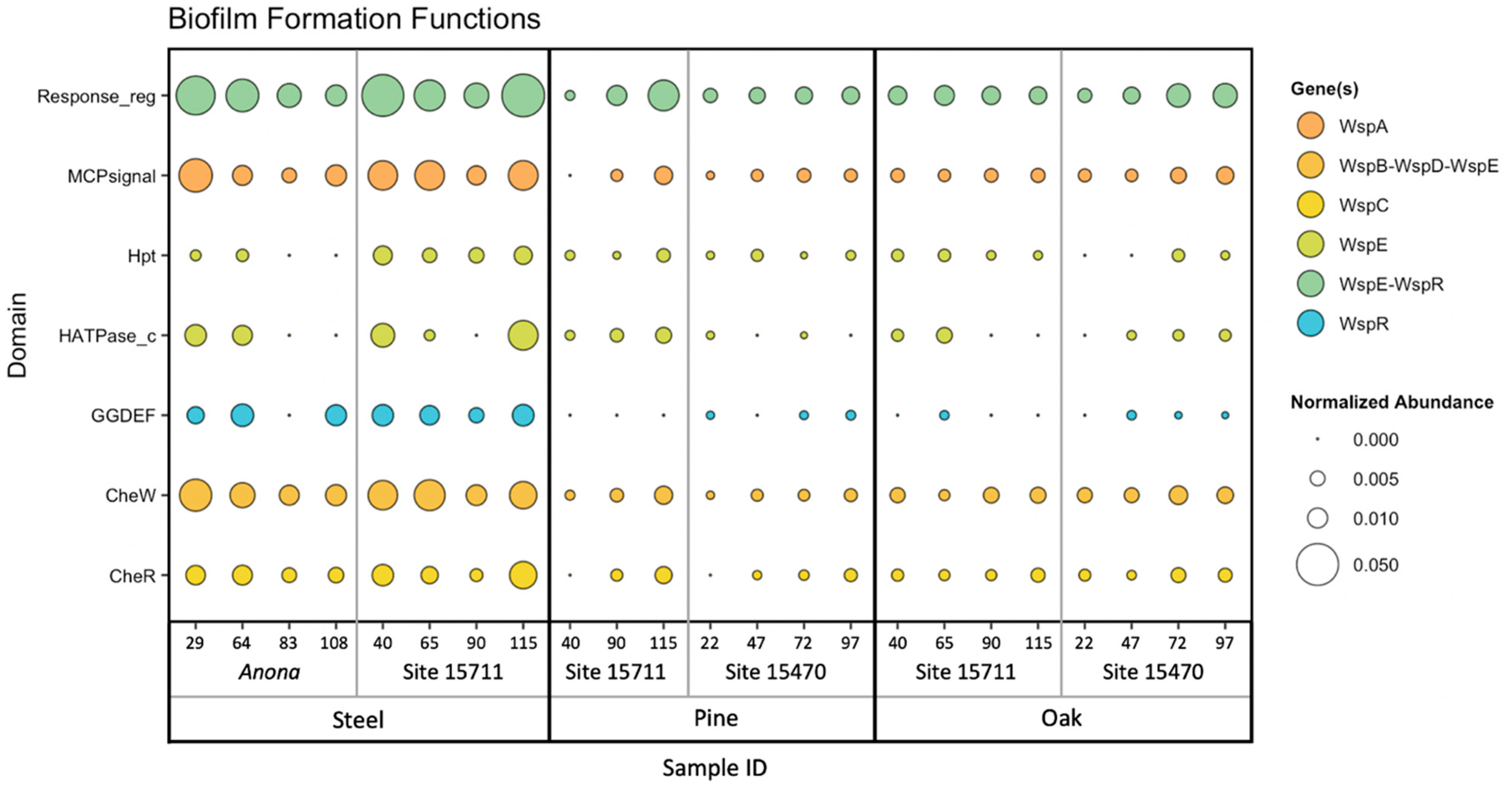
3.4. Biofilm Formation Genes
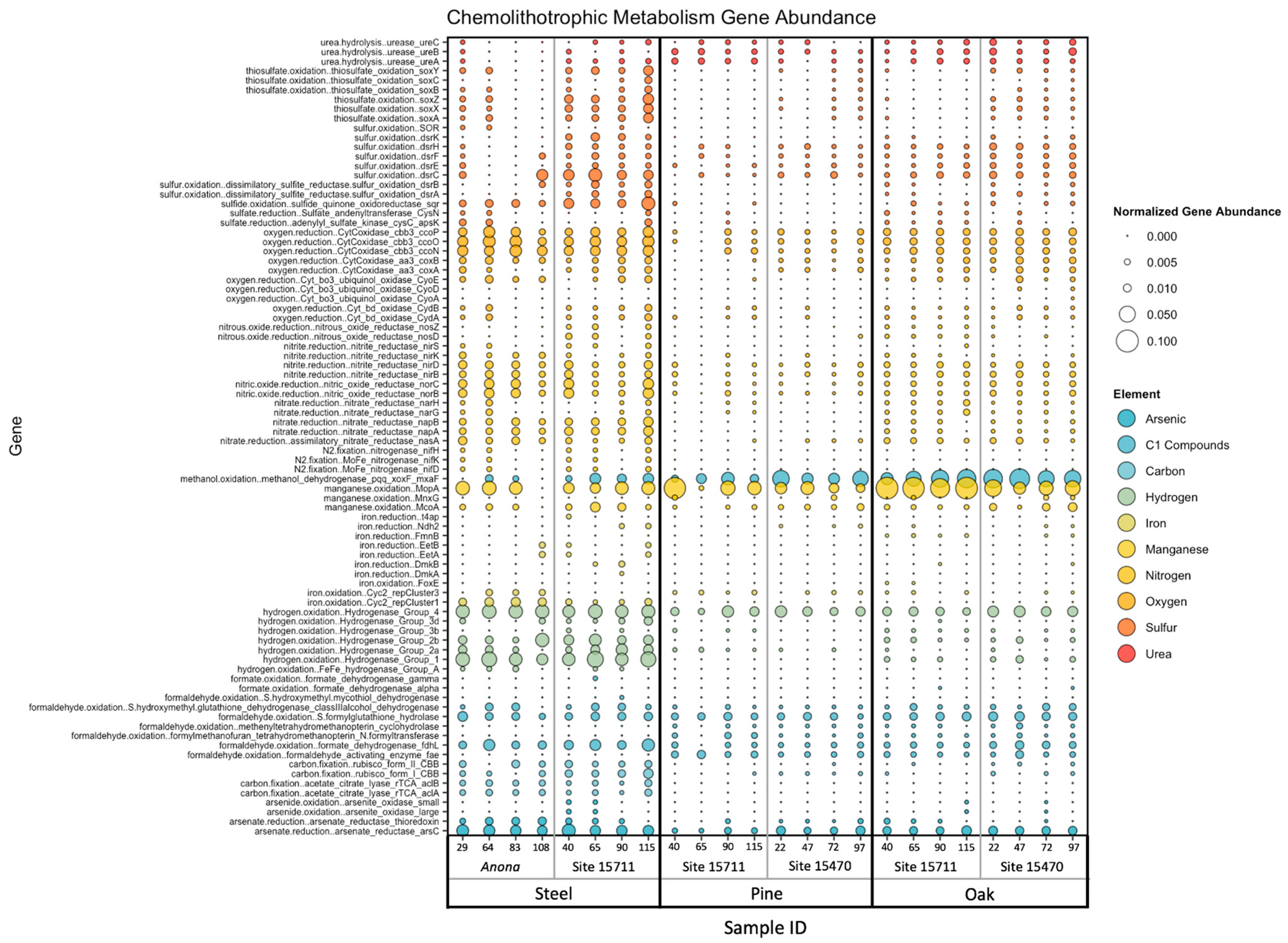
3.5. Chemolithotrophic Metabolisms
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Salta, M.; Wharton, J.A.; Blache, Y.; Stokes, K.R.; Briand, J.F. Marine biofilms on artificial surfaces: Structure and dynamics. Environ. Microbiol. 2013, 15, 2879–2893. [Google Scholar] [CrossRef] [PubMed]
- Dang, H.; Lovell, C.R. Microbial surface colonization and biofilm development in marine environments. Microbiol. Mol. Biol. Rev. 2016, 80, 91–138. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.-W.; Nam, J.-H.; Kim, Y.-H.; Lee, K.-H.; Lee, D.-H. Bacterial communities in the initial stage of marine biofilm formation on artificial surfaces. J. Microbiol. 2008, 46, 174–182. [Google Scholar] [CrossRef] [PubMed]
- Cheah, Y.T.; Chan, D.J.C. Physiology of microalgal biofilm: A review on prediction of adhesion on substrates. Bioengineered 2021, 12, 7577–7599. [Google Scholar] [CrossRef]
- Tong, C.; Derek, C. The role of substrates towards marine diatom Cylindrotheca fusiformis adhesion and biofilm development. J. Appl. Phycol. 2021, 33, 2845–2862. [Google Scholar] [CrossRef]
- Lau, S.C.; Thiyagarajan, V.; Cheung, S.C.; Qian, P.-Y. Roles of bacterial community composition in biofilms as a mediator for larval settlement of three marine invertebrates. Aquat. Microb. Ecol. 2005, 38, 41–51. [Google Scholar] [CrossRef]
- Zobell, C.E. The effect of solid surfaces upon bacterial activity. J. Bacteriol. 1943, 46, 39–56. [Google Scholar] [CrossRef]
- Harrison, J.J.; Ceri, H.; Turner, R.J. Multimetal resistance and tolerance in microbial biofilms. Nat. Rev. Microbiol. 2007, 5, 928–938. [Google Scholar] [CrossRef]
- Costerton, J.W.; Lewandowski, Z.; Caldwell, D.E.; Korber, D.R.; Lappin-Scott, H.M. Microbial biofilms. Annu. Rev. Microbiol. 1995, 49, 711–745. [Google Scholar] [CrossRef]
- Decho, A.W. Microbial biofilms in intertidal systems: An overview. Cont. Shelf Res. 2000, 20, 1257–1273. [Google Scholar] [CrossRef]
- Lee, O.O.; Wang, Y.; Tian, R.; Zhang, W.; Shek, C.S.; Bougouffa, S.; Al-Suwailem, A.; Batang, Z.B.; Xu, W.; Wang, G.C. In situ environment rather than substrate type dictates microbial community structure of biofilms in a cold seep system. Sci. Rep. 2014, 4, 3587. [Google Scholar] [CrossRef]
- Grzegorczyk, M.; Pogorzelski, S.J.; Pospiech, A.; Boniewicz-Szmyt, K. Monitoring of marine biofilm formation dynamics at submerged solid surfaces with multitechnique sensors. Front. Mar. Sci. 2018, 5, 363. [Google Scholar] [CrossRef]
- Pollet, T.; Berdjeb, L.; Garnier, C.; Durrieu, G.; Le Poupon, C.; Misson, B.; Briand, J.-F. Prokaryotic community successions and interactions in marine biofilms: The key role of Flavobacteriia. FEMS Microbiol. Ecol. 2018, 94, fiy083. [Google Scholar] [CrossRef]
- Catao, E.C.; Gallois, N.; Fay, F.; Misson, B.; Briand, J.-F. Metal resistance genes enrichment in marine biofilm communities selected by biocide-containing surfaces in temperate and tropical coastal environments. Environ. Pollut. 2021, 268, 115835. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.-Q.; Wang, R.-C.; Lu, X.-C.; Lu, J.-J.; Li, J.; Hu, H. Tolerance and biosorption of heavy metals by Cupriavidus metallidurans strain XXKD-1 isolated from a subsurface laneway in the Qixiashan Pb-Zn sulfide minery in Eastern China. Geomicrobiol. J. 2012, 29, 274–286. [Google Scholar] [CrossRef]
- Kim, H.-J.; Park, J.S.; Lee, T.-K.; Kang, D.; Kang, J.-H.; Shin, K.; Jung, S.W. Dynamics of marine bacterial biofouling communities after initial Alteromonas genovensis biofilm attachment to anti-fouling paint substrates. Mar. Pollut. Bull. 2021, 172, 112895. [Google Scholar] [CrossRef]
- Mugge, R.L.; Salerno, J.L.; Hamdan, L.J. Microbial functional responses in marine biofilms exposed to deepwater horizon spill contaminants. Front. Microbiol. 2021, 12, 636054. [Google Scholar] [CrossRef] [PubMed]
- McBeth, J.M.; Emerson, D. In situ microbial community succession on mild steel in estuarine and marine environments: Exploring the role of iron-oxidizing bacteria. Front. Microbiol. 2016, 7, 767. [Google Scholar] [CrossRef] [PubMed]
- McBeth, J.M.; Little, B.J.; Ray, R.I.; Farrar, K.M.; Emerson, D. Neutrophilic iron-oxidizing “Zetaproteobacteria” and mild steel corrosion in nearshore marine environments. Appl. Environ. Microbiol. 2011, 77, 1405–1412. [Google Scholar] [CrossRef] [PubMed]
- Little, B.J.; Lee, J.S. Microbiologically influenced corrosion: An update. Int. Mater. Rev. 2014, 59, 384–393. [Google Scholar] [CrossRef]
- Ma, Y.; Zhang, Y.; Zhang, R.; Guan, F.; Hou, B.; Duan, J. Microbiologically influenced corrosion of marine steels within the interaction between steel and biofilms: A brief view. Appl. Microbiol. Biotechnol. 2020, 104, 515–525. [Google Scholar] [PubMed]
- Procópio, L. The role of biofilms in the corrosion of steel in marine environments. World J. Microbiol. Biotechnol. 2019, 35, 73. [Google Scholar] [CrossRef] [PubMed]
- Capão, A.; Moreira-Filho, P.; Garcia, M.; Bitati, S.; Procópio, L. Marine bacterial community analysis on 316L stainless steel coupons by Illumina MiSeq sequencing. Biotechnol. Lett. 2020, 42, 1431–1448. [Google Scholar] [CrossRef] [PubMed]
- Antunes, J.T.; Sousa, A.G.; Azevedo, J.; Rego, A.; Leão, P.N.; Vasconcelos, V. Distinct temporal succession of bacterial communities in early marine biofilms in a Portuguese Atlantic Port. Front. Microbiol. 2020, 11, 1938. [Google Scholar] [CrossRef]
- Qian, P.-Y.; Cheng, A.; Wang, R.; Zhang, R. Marine biofilms: Diversity, interactions and biofouling. Nat. Rev. Microbiol. 2022, 20, 671–684. [Google Scholar]
- Bienhold, C.; Pop Ristova, P.; Wenzhöfer, F.; Dittmar, T.; Boetius, A. How deep-sea wood falls sustain chemosynthetic life. PLoS ONE 2013, 8, e53590. [Google Scholar] [CrossRef]
- Kalenitchenko, D.; Fagervold, S.K.; Pruski, A.M.; Vétion, G.; Yücel, M.; Le Bris, N.; Galand, P.E. Temporal and spatial constraints on community assembly during microbial colonization of wood in seawater. ISME J. 2015, 9, 2657–2670. [Google Scholar] [CrossRef]
- Guo, Z.; Wang, L.; Cong, W.; Jiang, Z.; Liang, Z. Comparative analysis of the ecological succession of microbial communities on two artificial reef materials. Microorganisms 2021, 9, 120. [Google Scholar] [CrossRef]
- Muthukrishnan, T.; Al Khaburi, M.; Abed, R.M. Fouling microbial communities on plastics compared with wood and steel: Are they substrate-or location-specific? Microb. Ecol. 2019, 78, 361–374. [Google Scholar]
- Moseley, R.D.; Hampel, J.J.; Mugge, R.L.; Hamdan, L.J. Historic Wooden Shipwrecks Influence Dispersal of Deep-Sea Biofilms. Front. Mar. Sci. 2022, 9, 873445. [Google Scholar] [CrossRef]
- Azam, F.; Fenchel, T.; Field, J.G.; Gray, J.S.; Meyer-Reil, L.-A.; Thingstad, F. The ecological role of water-column microbes in the sea. Mar. Ecol. Prog. Ser. 1983, 10, 257–263. [Google Scholar] [CrossRef]
- Alldredge, A.L.; Silver, M.W. Characteristics, dynamics and significance of marine snow. Prog. Oceanogr. 1988, 20, 41–82. [Google Scholar] [CrossRef]
- Azam, F. Microbial control of oceanic carbon flux: The plot thickens. Science 1998, 280, 694–696. [Google Scholar] [CrossRef]
- Azam, F.; Long, R.A. Sea snow microcosms. Nature 2001, 414, 495–498. [Google Scholar] [CrossRef]
- Klawonn, I.; Bonaglia, S.; Brüchert, V.; Ploug, H. Aerobic and anaerobic nitrogen transformation processes in N2-fixing cyanobacterial aggregates. ISME J. 2015, 9, 1456–1466. [Google Scholar] [CrossRef] [PubMed]
- Raven, M.; Keil, R.; Webb, S. Microbial sulfate reduction and organic sulfur formation in sinking marine particles. Science 2021, 371, 178–181. [Google Scholar] [CrossRef]
- Balzano, S.; Statham, P.; Pancost, R.; Lloyd, J. Role of microbial populations in the release of reduced iron to the water column from marine aggregates. Aquat. Microb. Ecol. 2009, 54, 291–303. [Google Scholar] [CrossRef]
- Duarte, C.M.; Pitt, K.A.; Lucas, C.H.; Purcell, J.E.; Uye, S.-I.; Robinson, K.; Brotz, L.; Decker, M.B.; Sutherland, K.R.; Malej, A. Is global ocean sprawl a cause of jellyfish blooms? Front. Ecol. Environ. 2013, 11, 91–97. [Google Scholar] [CrossRef]
- Firth, L.B.; Knights, A.M.; Bridger, D.; Evans, A.; Mieskowska, N.; Moore, P.J.; O’Connor, N.E.; Sheehan, E.V.; Thompson, R.C.; Hawkins, S.J. Ocean sprawl: Challenges and opportunities for biodiversity management in a changing world. Oceanogr. Mar. Biol. Annu. Rev. 2016, 54, 189–262. [Google Scholar]
- Hamdan, L.J.; Hampel, J.J.; Moseley, R.D.; Mugge, R.L.; Ray, A.; Salerno, J.L.; Damour, M. Deep-sea shipwrecks represent island-like ecosystems for marine microbiomes. ISME J. 2021, 15, 2883–2891. [Google Scholar] [CrossRef]
- Hampel, J.J.; Moseley, R.D.; Mugge, R.L.; Ray, A.; Damour, M.; Jones, D.; Hamdan, L.J. Deep-sea wooden shipwrecks influence sediment microbiome diversity. Limnol. Oceanogr. 2022, 67, 482–497. [Google Scholar] [CrossRef]
- Hampel, J.J.; Moseley, R.D.; Hamdan, L.J. Microbiomes respond predictably to built habitats on the seafloor. Mol. Ecol. 2022. [Google Scholar] [CrossRef] [PubMed]
- Mugge, R.L.; Rakocinski, C.F.; Woolsey, M.; Hamdan, L.J. Proximity to built structures on the seabed promotes biofilm development and diversity. Biofouling 2023. [Google Scholar] [CrossRef] [PubMed]
- Mugge, R.L.; Lee, J.S.; Brown, T.T.; Hamdan, L.J. Marine biofilm bacterial community response and carbon steel loss following Deepwater Horizon spill contaminant exposure. Biofouling 2019, 35, 870–882. [Google Scholar] [CrossRef]
- Salerno, J.L.; Little, B.; Lee, J.; Hamdan, L.J. Exposure to crude oil and chemical dispersant may impact marine microbial biofilm composition and steel corrosion. Front. Mar. Sci. 2018, 5, 196. [Google Scholar] [CrossRef]
- Hamdan, L.J.; Coffin, R.B.; Sikaroodi, M.; Greinert, J.; Treude, T.; Gillevet, P.M. Ocean currents shape the microbiome of Arctic marine sediments. ISME J. 2013, 7, 685–696. [Google Scholar] [CrossRef]
- Comeau, A.M.; Douglas, G.M.; Langille, M.G. Microbiome Helper: A Custom and Streamlined Workflow for Microbiome Research. mSystems 2017, 2, e00127-16. [Google Scholar] [CrossRef]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data. 2010. Available online: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 1 June 2022).
- Beghini, F.; McIver, L.J.; Blanco-Míguez, A.; Dubois, L.; Asnicar, F.; Maharjan, S.; Mailyan, A.; Manghi, P.; Scholz, M.; Thomas, A.M. Integrating taxonomic, functional, and strain-level profiling of diverse microbial communities with bioBakery 3. eLife 2021, 10, e65088. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef]
- Tange, O. Gnu parallel-the command-line power tool. USENIX Mag. 2011, 36, 42–47. [Google Scholar]
- Module 5: Assembly and Metagenome Assembled Genomes Tutorial. Available online: https://github.com/nikhilg123/Module_5_metagenome_assembly-MAGs/blob/main/Module%205_metagenome_assembly%2BMAGs.md (accessed on 1 July 2022).
- Nurk, S.; Meleshko, D.; Korobeynikov, A.; Pevzner, P.A. metaSPAdes: A new versatile metagenomic assembler. Genome Res. 2017, 27, 824–834. [Google Scholar] [CrossRef] [PubMed]
- Mikheenko, A.; Saveliev, V.; Gurevich, A. MetaQUAST: Evaluation of metagenome assemblies. Bioinformatics 2016, 32, 1088–1090. [Google Scholar] [CrossRef] [PubMed]
- Pullseq: A Utility Program for Extracting Sequences from a Fasta/Fastq File. Available online: https://github.com/bcthomas/pullseq (accessed on 1 June 2023).
- Menzel, P.; Ng, K.L.; Krogh, A. Fast and sensitive taxonomic classification for metagenomics with Kaiju. Nat. Commun. 2016, 7, 11257. [Google Scholar] [CrossRef] [PubMed]
- Pruitt, K.D.; Tatusova, T.; Maglott, D.R. NCBI Reference Sequence (RefSeq): A curated non-redundant sequence database of genomes, transcripts and proteins. Nucleic Acids Res. 2005, 33, D501–D504. [Google Scholar] [CrossRef]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef]
- Wickham, H.; François, R.; Henry, L.; Müller, K. dplyr: A grammar of data manipulation. R Package Version 0.4 2015, 3, 156. [Google Scholar]
- Wickham, H. ggplot2: Elegant Graphics for Data Analysis; Springer: Berlin/Heidelberg, Germany, 2016. [Google Scholar]
- Garber, A.; Ramirez, G.A.; Merino, N.; Pavia, M.J.; McAllister, S.M. MagicLamp: Toolkit for Annotation of ‘Omics Datasets Using Curated HMM Sets. 2020. Available online: https://github.com/Arkadiy-Garber/MagicLamp (accessed on 1 August 2022).
- Hyatt, D.; Chen, G.-L.; LoCascio, P.F.; Land, M.L.; Larimer, F.W.; Hauser, L.J. Prodigal: Prokaryotic gene recognition and translation initiation site identification. BMC Bioinform. 2010, 11, 119. [Google Scholar] [CrossRef]
- Finn, R.D.; Clements, J.; Eddy, S.R. HMMER web server: Interactive sequence similarity searching. Nucleic Acids Res. 2011, 39, W29–W37. [Google Scholar] [CrossRef]
- Bateman, A.; Coin, L.; Durbin, R.; Finn, R.D.; Hollich, V.; Griffiths-Jones, S.; Khanna, A.; Marshall, M.; Moxon, S.; Sonnhammer, E.L. The Pfam protein families database. Nucleic Acids Res. 2004, 32, D138–D141. [Google Scholar] [CrossRef]
- Haft, D.H.; Selengut, J.D.; White, O. The TIGRFAMs database of protein families. Nucleic Acids Res. 2003, 31, 371–373. [Google Scholar] [CrossRef] [PubMed]
- Anantharaman, K.; Brown, C.T.; Hug, L.A.; Sharon, I.; Castelle, C.J.; Probst, A.J.; Thomas, B.C.; Singh, A.; Wilkins, M.J.; Karaoz, U. Thousands of microbial genomes shed light on interconnected biogeochemical processes in an aquifer system. Nat. Commun. 2016, 7, 13219. [Google Scholar] [CrossRef] [PubMed]
- Garber, A.I.; Nealson, K.H.; Okamoto, A.; McAllister, S.M.; Chan, C.S.; Barco, R.A.; Merino, N. FeGenie: A comprehensive tool for the identification of iron genes and iron gene neighborhoods in genome and metagenome assemblies. Front. Microbiol. 2020, 11, 37. [Google Scholar] [CrossRef]
- Boutet, E.; Lieberherr, D.; Tognolli, M.; Schneider, M.; Bairoch, A. Uniprotkb/swiss-prot. In Plant Bioinformatics; Springer: Berlin/Heidelberg, Germany, 2007; pp. 89–112. [Google Scholar]
- Clarke, K.; Warwick, R. A further biodiversity index applicable to species lists: Variation in taxonomic distinctness. Mar. Ecol. Prog. Ser. 2001, 216, 265–278. [Google Scholar] [CrossRef]
- Winer, B.J. Statistical Principles in Experimental Design; McGraw-Hill: New York, NY, USA, 1962. [Google Scholar]
- Underwood, A.J.; Wnderwood, A. Experiments in Ecology: Their Logical Design and Interpretation Using Analysis of Variance; Cambridge University Press: Cambridge, UK, 1997. [Google Scholar]
- Alhakami, H.; Mirebrahim, H.; Lonardi, S. A comparative evaluation of genome assembly reconciliation tools. Genome Biol. 2017, 18, 93. [Google Scholar] [CrossRef] [PubMed]
- Mugge, R.L.; Brock, M.L.; Salerno, J.L.; Damour, M.; Church, R.A.; Lee, J.; Hamdan, L.J. Deep sea biofilms, historic shipwreck preservation and the Deepwater Horizon spill. Front. Mar. Sci. 2019, 6, 48. [Google Scholar] [CrossRef]
- Nakanishi, E.Y.; Palacios, J.H.; Godbout, S.; Fournel, S. Interaction between Biofilm Formation, Surface Material and Cleanability Considering Different Materials Used in Pig Facilities—An Overview. Sustainability 2021, 13, 5836. [Google Scholar] [CrossRef]
- Fagervold, S.K.; Galand, P.E.; Zbinden, M.; Gaill, F.; Lebaron, P.; Palacios, C. Sunken woods on the ocean floor provide diverse specialized habitats for microorganisms. FEMS Microbiol. Ecol. 2012, 82, 616–628. [Google Scholar] [CrossRef]
- Lawes, J.C.; Neilan, B.A.; Brown, M.V.; Clark, G.F.; Johnston, E.L. Elevated nutrients change bacterial community composition and connectivity: High throughput sequencing of young marine biofilms. Biofouling 2016, 32, 57–69. [Google Scholar] [CrossRef]
- Pang, C.M.; Liu, W.-T. Community structure analysis of reverse osmosis membrane biofilms and the significance of Rhizobiales bacteria in biofouling. Environ. Sci. Technol. 2007, 41, 4728–4734. [Google Scholar] [CrossRef]
- Elifantz, H.; Horn, G.; Ayon, M.; Cohen, Y.; Minz, D. Rhodobacteraceae are the key members of the microbial community of the initial biofilm formed in Eastern Mediterranean coastal seawater. FEMS Microbiol. Ecol. 2013, 85, 348–357. [Google Scholar] [CrossRef] [PubMed]
- Fritts, R.K.; LaSarre, B.; Stoner, A.M.; Posto, A.L.; McKinlay, J.B. A Rhizobiales-specific unipolar polysaccharide adhesin contributes to Rhodopseudomonas palustris biofilm formation across diverse photoheterotrophic conditions. Appl. Environ. Microbiol. 2017, 83, e03035-16. [Google Scholar] [CrossRef]
- Edwards, J.L.; Smith, D.L.; Connolly, J.; McDonald, J.E.; Cox, M.J.; Joint, I.; Edwards, C.; McCarthy, A.J. Identification of carbohydrate metabolism genes in the metagenome of a marine biofilm community shown to be dominated by Gammaproteobacteria and Bacteroidetes. Genes 2010, 1, 371–384. [Google Scholar] [CrossRef] [PubMed]
- Ali, S.; Jenkins, B.; Cheng, J.; Lobb, B.; Wei, X.; Egan, S.; Charles, T.C.; McConkey, B.J.; Austin, J.; Doxey, A.C. Slr4, a newly identified S-layer protein from marine Gammaproteobacteria, is a major biofilm matrix component. Mol. Microbiol. 2020, 114, 979–990. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Lu, Y.; Nawaz, M.Z.; Xu, J. Comparative genomics reveals evidence of genome reduction and high extracellular protein degradation potential in Kangiella. Front. Microbiol. 2018, 9, 1224. [Google Scholar] [CrossRef]
- Pop Ristova, P.; Bienhold, C.; Wenzhöfer, F.; Rossel, P.E.; Boetius, A. Temporal and spatial variations of bacterial and faunal communities associated with deep-sea wood falls. PLoS ONE 2017, 12, e0169906. [Google Scholar] [CrossRef]
- Kirchman, D.L. The ecology of Cytophaga–Flavobacteria in aquatic environments. FEMS Microbiol. Ecol. 2002, 39, 91–100. [Google Scholar] [CrossRef]
- Bessette, S.; Fagervold, S.K.; Romano, C.; Martin, D.; Bris, N.L.; Galand, P.E. Diversity of bacterial communities on sunken woods in the Mediterranean Sea. J. Mar. Sci. Technol. 2014, 22, 7. [Google Scholar]
- Chistoserdova, L. Methylotrophs in natural habitats: Current insights through metagenomics. Appl. Microbiol. Biotechnol. 2015, 99, 5763–5779. [Google Scholar] [CrossRef]
- Beck, D.A.; McTaggart, T.L.; Setboonsarng, U.; Vorobev, A.; Kalyuzhnaya, M.G.; Ivanova, N.; Goodwin, L.; Woyke, T.; Lidstrom, M.E.; Chistoserdova, L. The expanded diversity of Methylophilaceae from Lake Washington through cultivation and genomic sequencing of novel ecotypes. PLoS ONE 2014, 9, e102458. [Google Scholar] [CrossRef]
- Chow, M.L.; Radomski, C.C.; McDermott, J.M.; Davies, J.; Axelrood, P.E. Molecular characterization of bacterial diversity in Lodgepole pine (Pinus contorta) rhizosphere soils from British Columbia forest soils differing in disturbance and geographic source. FEMS Microbiol. Ecol. 2002, 42, 347–357. [Google Scholar] [CrossRef]
- Webster, N.S.; Negri, A.P. Site-specific variation in Antarctic marine biofilms established on artificial surfaces. Environ. Microbiol. 2006, 8, 1177–1190. [Google Scholar] [CrossRef] [PubMed]
- Rampadarath, S.; Bandhoa, K.; Puchooa, D.; Jeewon, R.; Bal, S. Early bacterial biofilm colonizers in the coastal waters of Mauritius. Electron. J. Biotechnol. 2017, 29, 13–21. [Google Scholar] [CrossRef]
- Hickman, J.W.; Tifrea, D.F.; Harwood, C.S. A chemosensory system that regulates biofilm formation through modulation of cyclic diguanylate levels. Proc. Natl. Acad. Sci. USA 2005, 102, 14422–14427. [Google Scholar] [CrossRef] [PubMed]
- Hueso-Gil, Á.; Calles, B.; de Lorenzo, V. The Wsp intermembrane complex mediates metabolic control of the swim-attach decision of Pseudomonas putida. Environ. Microbiol. 2020, 22, 3535–3547. [Google Scholar] [CrossRef]
- Kessler, C.; Mhatre, E.; Cooper, V.; Kim, W. Evolutionary divergence of the Wsp signal transduction systems in Beta-and Gammaproteobacteria. Appl. Environ. Microbiol. 2021, 87, e01306-21. [Google Scholar] [CrossRef]
- Belkaid, S.; Ladjouzi, M.A.; Hamdani, S. Effect of biofilm on naval steel corrosion in natural seawater. J. Solid State Electrochem. 2011, 15, 525–537. [Google Scholar] [CrossRef]
- Pao, G.M.; Saier, M.H. Response regulators of bacterial signal transduction systems: Selective domain shuffling during evolution. J. Mol. Evol. 1995, 40, 136–154. [Google Scholar] [CrossRef]
- Wang, R.; Zhang, W.; Ding, W.; Liang, Z.; Long, L.; Wong, W.C.; Qian, P.-Y. Profiling signal transduction in global marine biofilms. Front. Microbiol. 2021, 12, 768926. [Google Scholar] [CrossRef]
- Mohamed, M.; Fatimazahra, B.; Hassan, L.; Abdellah, H.; Fatima, H. Study of microbial adhesion on some wood species: Theoretical prediction. Microbiology 2011, 80, 43–49. [Google Scholar]
- Harrison, W.G. Nutrient regeneration and primary production in the sea. In Primary Productivity in the Sea; Springer: Berlin/Heidelberg, Germany, 1980; pp. 433–460. [Google Scholar]
- Herndl, G.J.; Reinthaler, T. Microbial control of the dark end of the biological pump. Nat. Geosci. 2013, 6, 718–724. [Google Scholar] [CrossRef] [PubMed]
- Swan, B.K.; Martinez-Garcia, M.; Preston, C.M.; Sczyrba, A.; Woyke, T.; Lamy, D.; Reinthaler, T.; Poulton, N.J.; Masland, E.D.P.; Gomez, M.L. Potential for chemolithoautotrophy among ubiquitous bacteria lineages in the dark ocean. Science 2011, 333, 1296–1300. [Google Scholar] [CrossRef] [PubMed]
- Boschker, H.T.; Vasquez-Cardenas, D.; Bolhuis, H.; Moerdijk-Poortvliet, T.W.; Moodley, L. Chemoautotrophic carbon fixation rates and active bacterial communities in intertidal marine sediments. PLoS ONE 2014, 9, e101443. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Pedersen, K.; Edlund, J.; Eriksson, L.; Åström, M.; Andersson, A.F.; Bertilsson, S.; Dopson, M. Potential for hydrogen-oxidizing chemolithoautotrophic and diazotrophic populations to initiate biofilm formation in oligotrophic, deep terrestrial subsurface waters. Microbiome 2017, 5, 37. [Google Scholar] [CrossRef] [PubMed]
- Sievert, S.M.; Scott, K.M.; Klotz, M.G.; Chain, P.S.; Hauser, L.J.; Hemp, J.; Hugler, M.; Land, M.; Lapidus, A.; Larimer, F.W. Genome of the epsilonproteobacterial chemolithoautotroph Sulfurimonas denitrificans. Appl. Environ. Microbiol. 2008, 74, 1145–1156. [Google Scholar] [CrossRef] [PubMed]
- Singer, E.; Heidelberg, J.F.; Dhillon, A.; Edwards, K.J. Metagenomic insights into the dominant Fe (II) oxidizing Zetaproteobacteria from an iron mat at Lō’ihi, Hawai´l. Front. Microbiol. 2013, 4, 52. [Google Scholar] [CrossRef] [PubMed]
- McAllister, S.M.; Polson, S.W.; Butterfield, D.A.; Glazer, B.T.; Sylvan, J.B.; Chan, C.S. Validating the Cyc2 neutrophilic iron oxidation pathway using meta-omics of Zetaproteobacteria iron mats at marine hydrothermal vents. Msystems 2020, 5, e00553-19. [Google Scholar] [CrossRef]
- Schwermer, C.U.; Lavik, G.; Abed, R.M.; Dunsmore, B.; Ferdelman, T.G.; Stoodley, P.; Gieseke, A.; de Beer, D. Impact of nitrate on the structure and function of bacterial biofilm communities in pipelines used for injection of seawater into oil fields. Appl. Environ. Microbiol. 2008, 74, 2841–2851. [Google Scholar] [CrossRef]
- Nuppunen-Puputti, M.; Kietäväinen, R.; Raulio, M.; Soro, A.; Purkamo, L.; Kukkonen, I.; Bomberg, M. Epilithic microbial community functionality in deep oligotrophic continental bedrock. Front. Microbiol. 2022, 13, 826048. [Google Scholar] [CrossRef]
- Li, H.; Zhou, E.; Ren, Y.; Zhang, D.; Xu, D.; Yang, C.; Feng, H.; Jiang, Z.; Li, X.; Gu, T. Investigation of microbiologically influenced corrosion of high nitrogen nickel-free stainless steel by Pseudomonas aeruginosa. Corros. Sci. 2016, 111, 811–821. [Google Scholar] [CrossRef]
- Ben Fekih, I.; Zhang, C.; Li, Y.P.; Zhao, Y.; Alwathnani, H.A.; Saquib, Q.; Rensing, C.; Cervantes, C. Distribution of arsenic resistance genes in prokaryotes. Front. Microbiol. 2018, 9, 2473. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Yin, N.; Du, H.; Cai, X.; Cui, Y. The fate of arsenic adsorbed on iron oxides in the presence of arsenite-oxidizing bacteria. Chemosphere 2016, 151, 108–115. [Google Scholar] [CrossRef] [PubMed]
- D’Arcy, R.; Amend, J.P.; Osburn, M.R. Microbial diversity and potential for arsenic and iron biogeochemical cycling at an arsenic rich, shallow-sea hydrothermal vent (Tutum Bay, Papua New Guinea). Chem. Geol. 2013, 348, 37–47. [Google Scholar]
- Jackson, C.R.; Dugas, S.L. Phylogenetic analysis of bacterial and archaeal arsC gene sequences suggests an ancient, common origin for arsenate reductase. BMC Evol. Biol. 2003, 3, 18. [Google Scholar] [CrossRef]
- Cho, B.; Park, M.; Shim, J.; Azam, F. Significance of bacteria in urea dynamics in coastal surface waters. Mar. Ecol. Prog. Ser. 1996, 142, 19–26. [Google Scholar] [CrossRef]
- Koper, T.E.; El-Sheikh, A.F.; Norton, J.M.; Klotz, M.G. Urease-encoding genes in ammonia-oxidizing bacteria. Appl. Environ. Microbiol. 2004, 70, 2342–2348. [Google Scholar] [CrossRef]
- Pedneault, E.; Galand, P.E.; Potvin, M.; Tremblay, J.-É.; Lovejoy, C. Archaeal amoA and ureC genes and their transcriptional activity in the Arctic Ocean. Sci. Rep. 2014, 4, 4661. [Google Scholar] [CrossRef]
- Yakimov, M.M.; Cono, V.L.; Smedile, F.; DeLuca, T.H.; Juárez, S.; Ciordia, S.; Fernández, M.; Albar, J.P.; Ferrer, M.; Golyshin, P.N. Contribution of crenarchaeal autotrophic ammonia oxidizers to the dark primary production in Tyrrhenian deep waters (Central Mediterranean Sea). ISME J. 2011, 5, 945–961. [Google Scholar] [CrossRef]
- Kielemoes, J.; Bultinck, I.; Storms, H.; Boon, N.; Verstraete, W. Occurrence of manganese-oxidizing microorganisms and manganese deposition during biofilm formation on stainless steel in a brackish surface water. FEMS Microbiol. Ecol. 2002, 39, 41–55. [Google Scholar] [CrossRef]
- Medina, M.; Rizo, A.; Dinh, D.; Chau, B.; Omidvar, M.; Juarez, A.; Ngo, J.; Johnson, H.A. MopA, the Mn oxidizing protein from Erythrobacter sp. SD-21, requires heme and NAD+ for Mn (II) oxidation. Front. Microbiol. 2018, 9, 2671. [Google Scholar] [CrossRef]
- Nercessian, O.; Noyes, E.; Kalyuzhnaya, M.G.; Lidstrom, M.E.; Chistoserdova, L. Bacterial populations active in metabolism of C1 compounds in the sediment of Lake Washington, a freshwater lake. Appl. Environ. Microbiol. 2005, 71, 6885–6899. [Google Scholar] [CrossRef] [PubMed]
- Ander, P.; Eriksson, K.-E. Methanol formation during lignin degradation by Phanerochaete chrysosporium. Appl. Microbiol. Biotechnol. 1985, 21, 96–102. [Google Scholar] [CrossRef]
- Babbitt, C.W.; Lindner, A.S. Effect of nitrogen source on methanol oxidation and genetic diversity of methylotrophic mixed cultures enriched from pulp and paper mill biofilms. Biodegradation 2011, 22, 309–320. [Google Scholar] [CrossRef] [PubMed]
- Bugnot, A.; Mayer-Pinto, M.; Airoldi, L.; Heery, E.; Johnston, E.; Critchley, L.; Strain, E.; Morris, R.; Loke, L.; Bishop, M. Current and projected global extent of marine built structures. Nat. Sustain. 2021, 4, 33–41. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mugge, R.L.; Moseley, R.D.; Hamdan, L.J. Substrate Specificity of Biofilms Proximate to Historic Shipwrecks. Microorganisms 2023, 11, 2416. https://doi.org/10.3390/microorganisms11102416
Mugge RL, Moseley RD, Hamdan LJ. Substrate Specificity of Biofilms Proximate to Historic Shipwrecks. Microorganisms. 2023; 11(10):2416. https://doi.org/10.3390/microorganisms11102416
Chicago/Turabian StyleMugge, Rachel L., Rachel D. Moseley, and Leila J. Hamdan. 2023. "Substrate Specificity of Biofilms Proximate to Historic Shipwrecks" Microorganisms 11, no. 10: 2416. https://doi.org/10.3390/microorganisms11102416
APA StyleMugge, R. L., Moseley, R. D., & Hamdan, L. J. (2023). Substrate Specificity of Biofilms Proximate to Historic Shipwrecks. Microorganisms, 11(10), 2416. https://doi.org/10.3390/microorganisms11102416