
A Robust One-Step Recombineering System for Enterohemorrhagic Escherichia coli

 , ,
, ,  , , and
, , and {kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Bacterial Strains and Plasmids Used in This Study
2.2. Construction of Plasmids and Strains
2.3. Comparison of the Recombination Efficiency of pKD46recX with pKD46recX
2.4. Test of pKD46recX in E. coli O104:H4
2.5. Measurement of SOS Response
2.6. Statistics and Data Visualization
3. Results
3.1. Construction of Plasmids
3.2. Selection of Conserved Chromosomal Loci for a Direct Comparison of Recombination Efficiency in EHEC O157:H7 EDL933 Δstx1/2 and E. coli K-12 MG1655 and Generation of Recombination Substrates
3.3. Test for the Optimal Induction Time of the λ Red Functions
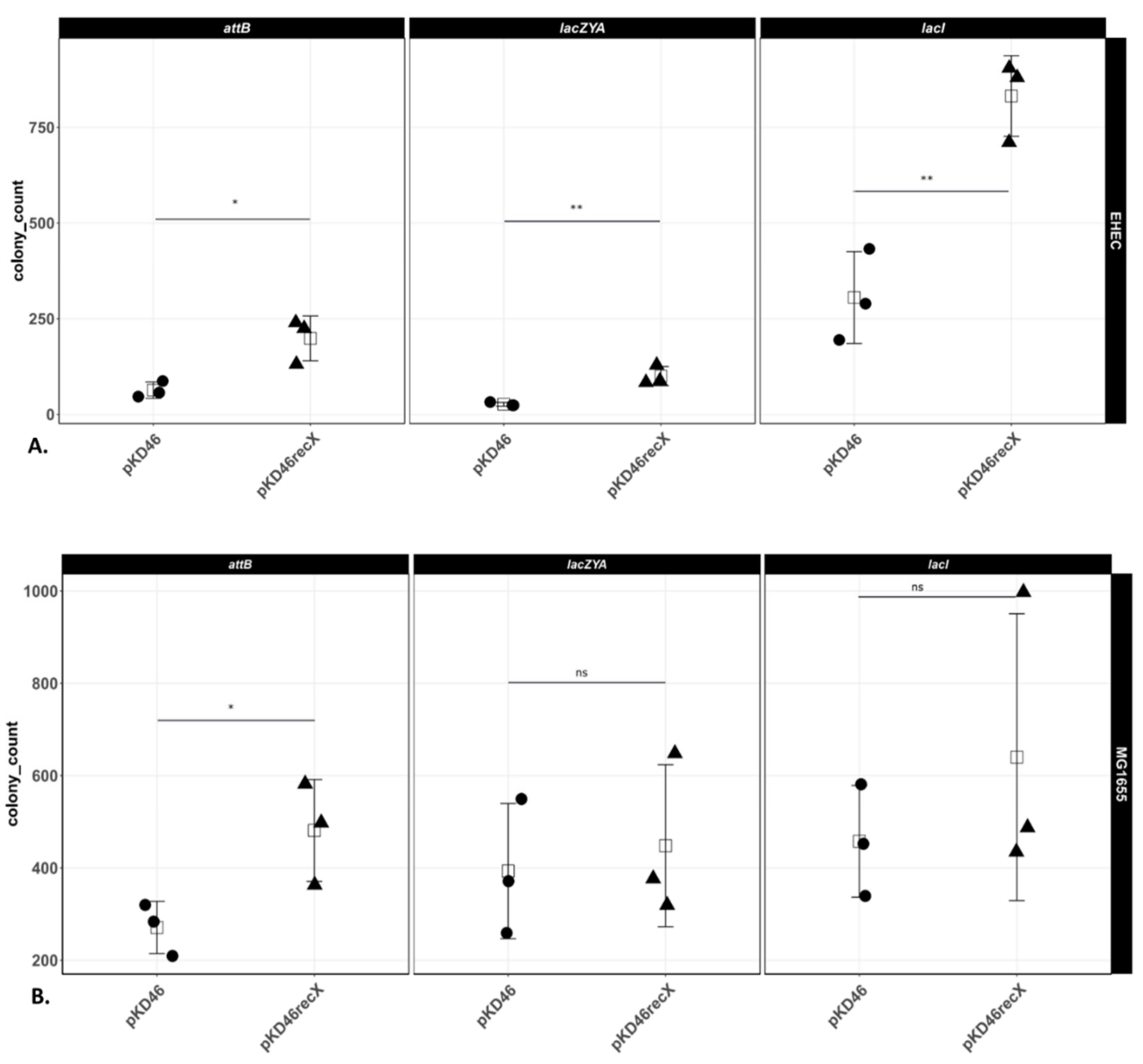
3.4. Comparison of the Numbers of Recombinants in E. coli K-12 MG1655 and EHEC O157:H7 EDL933 Δstx1/2
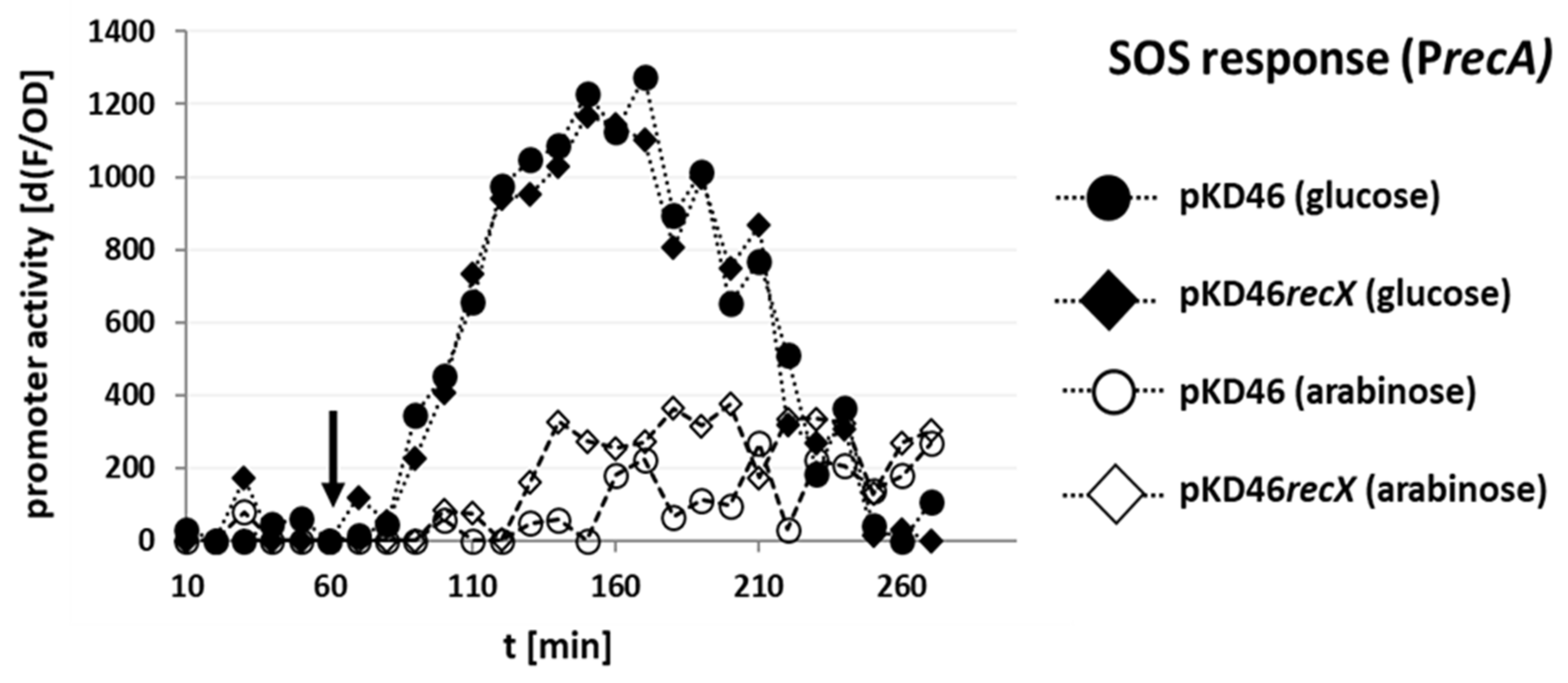
3.5. Coexpression of RecX with Gam, Beta, and Exo Does Not Further Repress the Sparfloxacin-Dependent SOS Response Induction
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Nguyen, Y.; Sperandio, V. Enterohemorrhagic E. coli (EHEC) pathogenesis. Front. Cell. Infect. Microbiol. 2012, 2, 90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kampmeier, S.; Berger, M.; Mellmann, A.; Karch, H.; Berger, P. The 2011 German enterohemorrhagic Escherichia coli O104:H4 outbreak—The danger is still out there. Curr. Top. Microbiol. Immunol. 2018, 416, 117–148. [Google Scholar] [CrossRef] [PubMed]
- Michino, H.; Araki, K.; Minami, S.; Takaya, S.; Sakai, N.; Miyazaki, M.; Ono, A.; Yanagawa, H. Massive outbreak of Escherichia coli O157:H7 infection in schoolchildren in Sakai City, Japan, associated with consumption of white radish sprouts. Am. J. Epidemiol. 1999, 150, 787–796. [Google Scholar] [CrossRef] [Green Version]
- Xiong, Y.; Wang, P.; Lan, R.; Ye, C.; Wang, H.; Ren, J.; Jing, H.; Wang, Y.; Zhou, Z.; Bai, X.; et al. A novel Escherichia coli O157:H7 clone causing a major hemolytic uremic syndrome outbreak in China. PLoS ONE 2012, 7, e36144. [Google Scholar] [CrossRef]
- Karch, H.; Denamur, E.; Dobrindt, U.; Finlay, B.B.; Hengge, R.; Johannes, L.; Ron, E.Z.; Tønjum, T.; Sansonetti, P.J.; Vicente, M. The enemy within us: Lessons from the 2011 European Escherichia coli O104:H4 outbreak. EMBO Mol. Med. 2012, 4, 841–848. [Google Scholar] [CrossRef]
- Melton-Celsa, A.R. Shiga toxin (Stx) classification, structure, and function. Microbiol. Spectr. 2014, 2, EHEC-0024-2013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, C.S.; Mooney, J.C.; Brandt, J.R.; Staples, A.O.; Jelacic, S.; Boster, D.R.; Watkins, S.L.; Tarr, P.I. Risk factors for the hemolytic uremic syndrome in children infected with Escherichia coli O157:H7: A multivariable analysis. Clin. Infect. Dis. 2012, 55, 33–41. [Google Scholar] [CrossRef] [Green Version]
- Kimmitt, P.T.; Harwood, C.R.; Barer, M.R. Toxin gene expression by Shiga toxin-producing Escherichia coli: The role of antibiotics and the bacterial SOS response. Emerg. Infect. Dis. 2000, 6, 458–465. [Google Scholar] [CrossRef] [Green Version]
- Berger, M.; Aijaz, I.; Berger, P.; Dobrindt, U.; Koudelka, G. Transcriptional and translational inhibitors block SOS response and Shiga toxin expression in enterohemorrhagic Escherichia coli. Sci. Rep. 2019, 9, 18777. [Google Scholar] [CrossRef]
- Mühlen, S.; Ramming, I.; Pils, M.C.; Koeppel, M.; Glaser, J.; Leong, J.; Flieger, A.; Stecher, B.; Dersch, P. Identification of antibiotics that diminish disease in a murine model of enterohemorrhagic Escherichia coli infection. Antimicrob. Agents Chemother. 2020, 64, e02159-19. [Google Scholar] [CrossRef]
- Murphy, K.C. Use of bacteriophage lambda recombination functions to promote gene replacement in Escherichia coli. J. Bacteriol. 1998, 180, 2063–2071. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Datsenko, K.A.; Wanner, B.L. One-Step Inactivation of Chromosomal Genes in Escherichia coli K-12 Using PCR Products. Proc. Natl. Acad. Sci. USA 2000, 97, 6640–6645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murphy, K.C.; Campellone, K.G. Lambda red-mediated recombinogenic engineering of enterohemorrhagic and enteropathogenic E. coli. BMC Mol. Biol. 2003, 4, 11. [Google Scholar] [CrossRef] [Green Version]
- Lee, D.J.; Bingle, L.E.; Heurlier, K.; Pallen, M.J.; Penn, C.W.; Busby, S.J.; Hobman, J.L. Gene doctoring: A method for recombineering in laboratory and pathogenic Escherichia coli strains. BMC Microbiol. 2009, 9, 252. [Google Scholar] [CrossRef] [Green Version]
- Koudelka, A.P.; Hufnagel, L.A.; Koudelka, G.B. Purification and characterization of the repressor of the Shiga toxin-encoding bacteriophage 933W: DNA binding, gene regulation, and autocleavage. J. Bacteriol. 2004, 186, 7659–7669. [Google Scholar] [CrossRef] [Green Version]
- Baharoglu, Z.; Mazel, D. SOS, the formidable strategy of bacteria against aggressions. FEMS Microbiol. Rev. 2014, 38, 1126–1145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muyrers, J.P.; Zhang, Y.; Buchholz, F.; Stewart, A.F. RecE/RecT and Redalpha/Redbeta initiate double-stranded break repair by specifically interacting with their respective partners. Genes Dev. 2000, 14, 1971–1982. [Google Scholar] [CrossRef]
- Wilkinson, M.; Troman, L.; Wan Nur Ismah, W.A.; Chaban, Y.; Avison, M.B.; Dillingham, M.S.; Wigley, D.B. Structural basis for the inhibition of RecBCD by gam and its synergistic antibacterial effect with quinolones. eLife 2016, 5, e22963. [Google Scholar] [CrossRef] [Green Version]
- Stohl, E.A.; Brockman, J.P.; Burkle, K.L.; Morimatsu, K.; Kowalczykowski, S.C.; Seifert, H.S. Escherichia coli RecX inhibits RecA recombinase and coprotease activities in vitro and in vivo. J. Biol. Chem. 2003, 278, 2278–2285. [Google Scholar] [CrossRef] [Green Version]
- Lusetti, S.L.; Hobbs, M.D.; Stohl, E.A.; Chitteni-Pattu, S.; Inman, R.B.; Seifert, H.S.; Cox, M.M. The RecF protein antagonizes RecX function via direct interaction. Mol. Cell 2006, 21, 41–50. [Google Scholar] [CrossRef] [Green Version]
- Yakimov, A.; Pobegalov, G.; Bakhlanova, I.; Khodorkovskii, M.; Petukhov, M.; Baitin, D. Blocking the RecA activity and SOS-response in bacteria with a short α-helical peptide. Nucleic Acids Res. 2017, 45, 9788–9796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berger, M.; Gerganova, V.; Berger, P.; Rapiteanu, R.; Lisicovas, V.; Dobrindt, U. Genes on a wire: The nucleoid-associated protein HU insulates transcription units in Escherichia coli. Sci. Rep. 2016, 6, 31512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Norrander, J.; Kempe, T.; Messing, J. Construction of improved M13 vectors using oligodeoxynucleotide-directed mutagenesis. Gene 1983, 26, 101–106. [Google Scholar] [CrossRef]
- Wang, R.F.; Kushner, S.R. Construction of versatile low-copy-number vectors for cloning, sequencing and gene expression in Escherichia coli. Gene 1991, 100, 195–199. [Google Scholar] [CrossRef]
- Chittò, M.; Berger, M.; Klotz, L.; Dobrindt, U. Sub-inhibitory concentrations of SOS-response inducing antibiotics stimulate integrase expression and excision of pathogenicity islands in uropathogenic Escherichia coli strain 536. Int. J. Med. Microbiol. 2020, 310, 151361. [Google Scholar] [CrossRef]
- Gobert, A.P.; Vareille, M.; Glasser, A.-L.; Hindré, T.; de Sablet, T.; Martin, C. Shiga toxin produced by enterohemorrhagic Escherichia coli inhibits PI3K/NF-KB signaling pathway in globotriaosylceramide-3-negative human intestinal epithelial cells. J. Immunol. 2007, 178, 8168–8174. [Google Scholar] [CrossRef] [Green Version]
- Piddock, L.J.; Zhu, M. Mechanism of action of Sparfloxacin against and mechanism of resistance in gram-negative and gram-positive bacteria. Antimicrob. Agents Chemother. 1991, 35, 2423–2427. [Google Scholar] [CrossRef] [Green Version]
- Pan, X.S.; Fisher, L.M. Targeting of DNA gyrase in Streptococcus pneumoniae by Sparfloxacin: Selective targeting of gyrase or topoisomerase IV by quinolones. Antimicrob. Agents Chemother. 1997, 41, 471–474. [Google Scholar] [CrossRef] [Green Version]
- Drlica, K.; Zhao, X. DNA gyrase, topoisomerase IV, and the 4-quinolones. Microbiol. Mol. Biol. Rev. 1997, 61, 377–392. [Google Scholar] [CrossRef]
- Guzman, L.M.; Belin, D.; Carson, M.J.; Beckwith, J. Tight regulation, modulation, and high-level expression by vectors containing the arabinose PBAD promoter. J. Bacteriol. 1995, 177, 4121–4130. [Google Scholar] [CrossRef] [Green Version]
- Tuttle, J.; Gomez, T.; Doyle, M.P.; Wells, J.G.; Zhao, T.; Tauxe, R.V.; Griffin, P.M. Lessons from a large outbreak of Escherichia coli O157:H7 infections: Insights into the infectious dose and method of widespread contamination of hamburger patties. Epidemiol. Infect. 1999, 122, 185–192. [Google Scholar] [CrossRef] [PubMed]
- Joseph, A.; Cointe, A.; Mariani Kurkdjian, P.; Rafat, C.; Hertig, A. Shiga toxin-associated hemolytic uremic syndrome: A narrative review. Toxins 2020, 12, 67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, C.S.; Jelacic, S.; Habeeb, R.L.; Watkins, S.L.; Tarr, P.I. The risk of the hemolytic-uremic syndrome after antibiotic treatment of Escherichia coli O157:H7 infections. N. Engl. J. Med. 2000, 342, 1930–1936. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murphy, K.C. λ recombination and recombineering. EcoSal Plus 2016, 7, ESP-0011-2015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vericat, J.A.; Guerrero, R.; Barbé, J. Increase in plasmid transformation efficiency in SOS-induced Escherichia coli cells. Mol. Gen. Genet. 1988, 211, 526–530. [Google Scholar] [CrossRef]
- Abbott, P.J. Stimulation of recombination between homologous sequences on carcinogen-treated plasmid DNA and chromosomal DNA by induction of the SOS response in Escherichia coli K12. Mol. Gen. Genet. 1985, 201, 129–132. [Google Scholar] [CrossRef]
- Chung, W.-J.; Cui, Y.; Chen, C.-S.; Wei, W.H.; Chang, R.-S.; Shu, W.-Y.; Hsu, I.C. Freezing shortens the lifetime of DNA molecules under tension. J. Biol. Phys. 2017, 43, 511–524. [Google Scholar] [CrossRef]
- Casjens, S. Prophages and bacterial genomics: What have we learned so far? Mol. Microbiol. 2003, 49, 277–300. [Google Scholar] [CrossRef]
- Masters, M. The frequency of P1 transduction of the genes of Escherichia coli as a function of chromosomal position: Preferential transduction of the origin of replication. Mol. Gen. Genet. 1977, 155, 197–202. [Google Scholar] [CrossRef]
- Fels, U.; Gevaert, K.; Van Damme, P. Bacterial genetic engineering by means of recombineering for reverse genetics. Front. Microbiol. 2020, 11, 548410. [Google Scholar] [CrossRef]
- Blattner, F.R.; Plunkett, G.; Bloch, C.A.; Perna, N.T.; Burland, V.; Riley, M.; Collado-Vides, J.; Glasner, J.D.; Rode, C.K.; Mayhew, G.F.; et al. The Complete Genome Sequence of Escherichia Coli K-12. Science 1997, 277, 1453–1462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grant, S.G.; Jessee, J.; Bloom, F.R.; Hanahan, D. Differential Plasmid Rescue from Transgenic Mouse DNAs into Escherichia coli Methylation-Restriction Mutants. Proc. Natl. Acad. Sci. USA 1990, 87, 4645–4649. [Google Scholar] [CrossRef] [PubMed] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Peng, L.; Dumevi, R.M.; Chitto, M.; Haarmann, N.; Berger, P.; Koudelka, G.; Schmidt, H.; Mellmann, A.; Dobrindt, U.; Berger, M. A Robust One-Step Recombineering System for Enterohemorrhagic Escherichia coli. Microorganisms 2022, 10, 1689. https://doi.org/10.3390/microorganisms10091689
Peng L, Dumevi RM, Chitto M, Haarmann N, Berger P, Koudelka G, Schmidt H, Mellmann A, Dobrindt U, Berger M. A Robust One-Step Recombineering System for Enterohemorrhagic Escherichia coli. Microorganisms. 2022; 10(9):1689. https://doi.org/10.3390/microorganisms10091689
Chicago/Turabian StylePeng, Lang, Rexford Mawunyo Dumevi, Marco Chitto, Nadja Haarmann, Petya Berger, Gerald Koudelka, Herbert Schmidt, Alexander Mellmann, Ulrich Dobrindt, and Michael Berger. 2022. "A Robust One-Step Recombineering System for Enterohemorrhagic Escherichia coli" Microorganisms 10, no. 9: 1689. https://doi.org/10.3390/microorganisms10091689
APA StylePeng, L., Dumevi, R. M., Chitto, M., Haarmann, N., Berger, P., Koudelka, G., Schmidt, H., Mellmann, A., Dobrindt, U., & Berger, M. (2022). A Robust One-Step Recombineering System for Enterohemorrhagic Escherichia coli. Microorganisms, 10(9), 1689. https://doi.org/10.3390/microorganisms10091689