
Need for a Standardized Translational Drug Development Platform: Lessons Learned from the Repurposing of Drugs for COVID-19

 , , , ,
, , , ,  ,
,  , , , , ,
, , , , ,  , , , , , and
, , , , , and
Abstract
1. Introduction
2. Experimental Verification of Repurposed Drugs against COVID-19
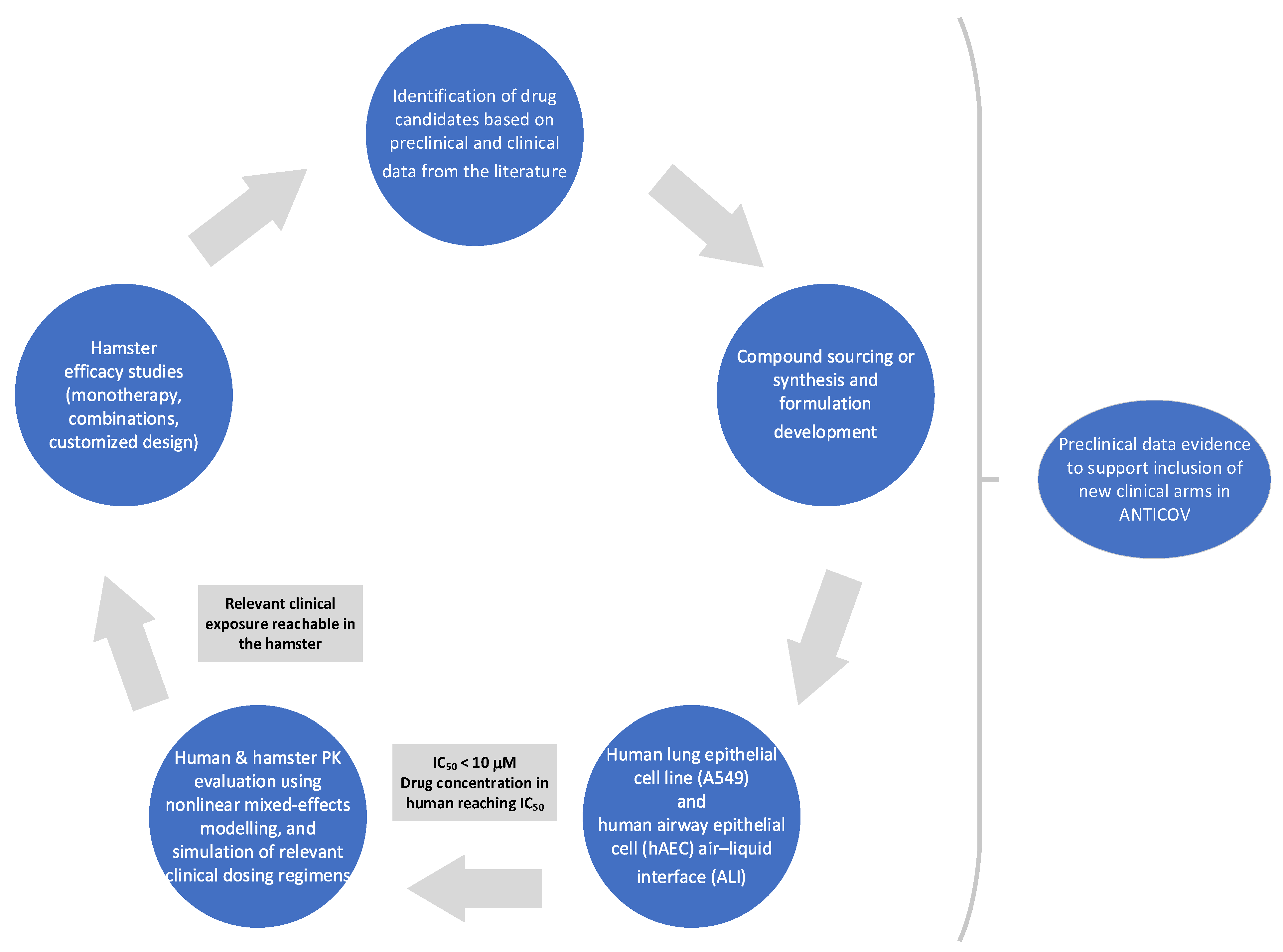
2.1. Identification and Selection of Drug Repurposing Candidates for Preclinical Studies
2.2. Generation of Preclinical Data
2.2.1. In Vitro/Ex Vivo Activity
2.2.2. In Vivo Efficacy in Hamsters
2.2.3. Protein Binding and Correction of IC50 Values for Protein Binding
2.2.4. Pharmacokinetic Studies in Hamster
2.2.5. Population Pharmacokinetic Analysis
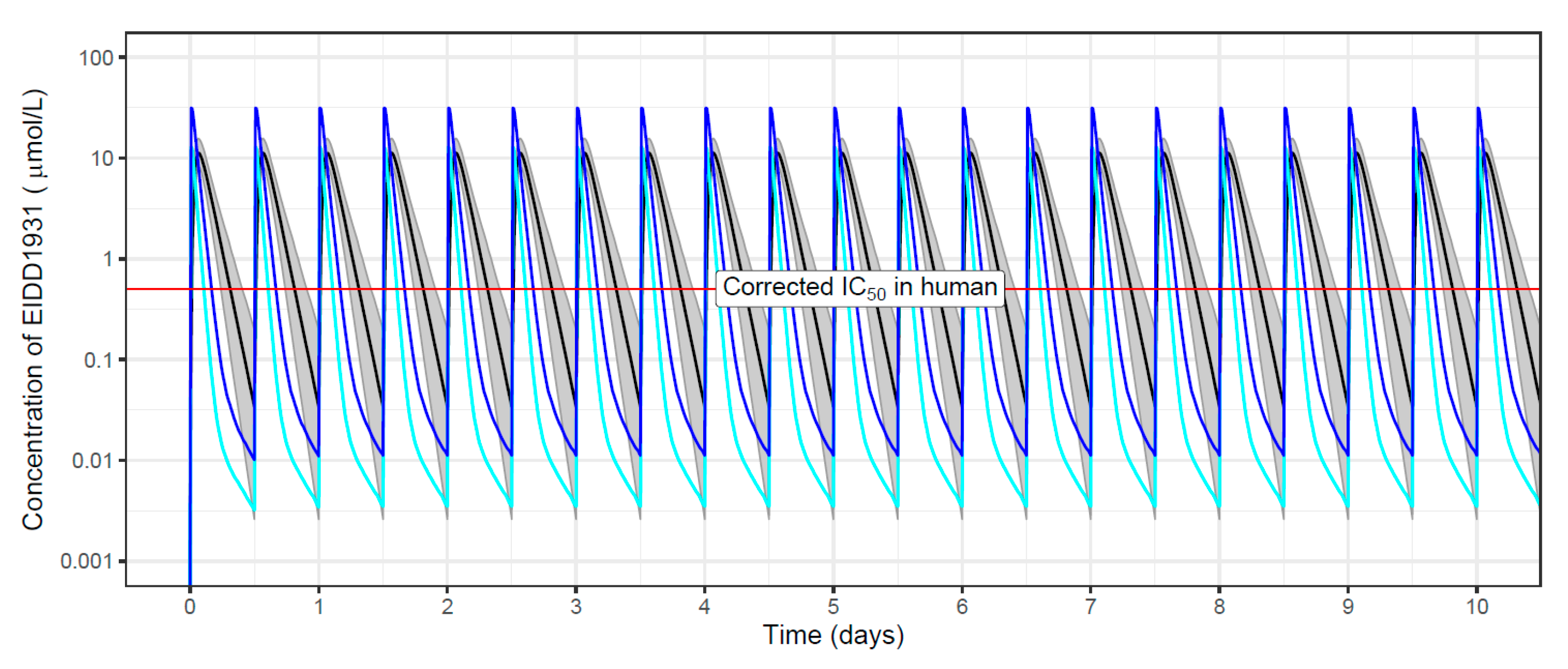
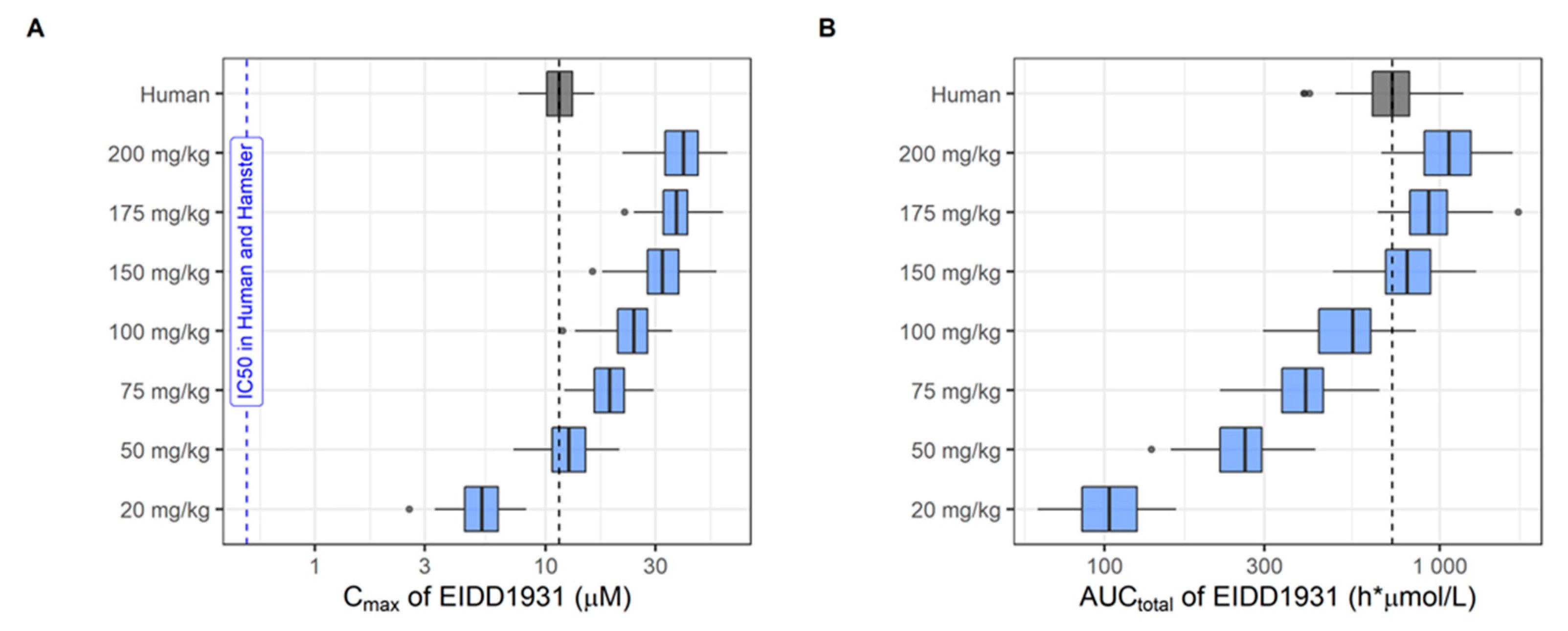
2.2.6. Population Pharmacokinetic Simulations
3. Results
3.1. Selected Repurposed Drugs for Further Evaluation
3.2. Preclinical Data Generated for DAAs
3.3. Preclinical Data Generated for IAAs
3.4. Preclinical Data Generated for Drug Combinations
4. Discussion and Lessons Learned
4.1. Selection of Repurposed Drugs
4.2. Relevance of In Vitro Assays
4.3. Relevance of In Vivo Assays
4.4. Working under Time Pressure
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Valencia, D.N. Brief Review on COVID-19: The 2020 Pandemic Caused by SARS-CoV-2. Cureus 2020, 12, e7386. [Google Scholar] [CrossRef]
- WHO Director-General’s Opening Remarks at the Media Briefing on COVID-19. 11 March 2020. Available online: https://www.who.int/director-general/speeches/detail/who-director-general-s-opening-remarks-at-the-media-briefing-on-covid-19---11-march-2020 (accessed on 14 April 2022).
- WHO Coronavirus (COVID-19) Dashboard. Available online: https://covid19.who.int/ (accessed on 15 June 2022).
- COVID-19 Dashboard by the Center for Systems Science and Engineering (CSSE) at Johns Hopkins University (JHU). Available online: https://coronavirus.jhu.edu/map.html (accessed on 14 April 2022).
- World Health Organization. Press Release. 14.9 Million Excess Deaths Associated with the COVID-19 Pandemic in 2020 and 2021. Available online: https://www.who.int/news/item/05-05-2022-14.9-million-excess-deaths-were-associated-with-the-covid-19-pandemic-in-2020-and-2021 (accessed on 5 May 2022).
- Wang, D.; Hu, B.; Hu, C.; Zhu, F.; Liu, X.; Zhang, J.; Wang, B.; Xiang, H.; Cheng, Z.; Xiong, Y.; et al. Clinical Characteristics of 138 Hospitalized Patients With 2019 Novel Coronavirus-Infected Pneumonia in Wuhan, China. JAMA 2020, 323, 1061–1069. [Google Scholar] [CrossRef]
- Schultze, J.L.; Aschenbrenner, A.C. COVID-19 and the human innate immune system. Cell 2021, 184, 1671–1692. [Google Scholar] [CrossRef]
- Hu, B.; Guo, H.; Zhou, P.; Shi, Z.L. Characteristics of SARS-CoV-2 and COVID-19. Nat. Rev. Microbiol. 2021, 19, 141–154. [Google Scholar] [CrossRef]
- COVID-19 Clinical Research Coalition. Available online: https://covid19crc.org/ (accessed on 14 April 2022).
- ANTICOV Consortium. ANTICOV Clinical Trial. Available online: https://anticov.org/ (accessed on 5 May 2022).
- Tobinick, E.L. The value of drug repositioning in the current pharmaceutical market. Drug News Perspect. 2009, 22, 119–125. [Google Scholar] [CrossRef] [PubMed]
- Sleigh, S.H.; Barton, C.L. Repurposing Strategies for Therapeutics. Pharm. Med. 2010, 24, 151–159. [Google Scholar] [CrossRef]
- Chavda, V.P.; Kapadia, C.; Soni, S.; Prajapati, R.; Chauhan, S.C.; Yallapu, M.M.; Apostolopoulos, V. A global picture: Therapeutic perspectives for COVID-19. Immunotherapy 2022, 14, 351–371. [Google Scholar] [CrossRef]
- Novac, N. Challenges and opportunities of drug repositioning. Trends Pharmacol. Sci. 2013, 34, 267–272. [Google Scholar] [CrossRef]
- Sultana, J.; Crisafulli, S.; Gabbay, F.; Lynn, E.; Shakir, S.; Trifiro, G. Challenges for Drug Repurposing in the COVID-19 Pandemic Era. Front. Pharmacol. 2020, 11, 588654. [Google Scholar] [CrossRef]
- Chen, B.; Tian, E.K.; He, B.; Tian, L.; Han, R.; Wang, S.; Xiang, Q.; Zhang, S.; El Arnaout, T.; Cheng, W. Overview of lethal human coronaviruses. Signal Transduct. Target, Ther. 2020, 5, 89. [Google Scholar] [CrossRef]
- Mercorelli, B.; Palù, G.; Loregian, A. Drug Repurposing for Viral Infectious Diseases: How Far Are We? Trends Microbiol. 2018, 26, 865–876. [Google Scholar] [CrossRef] [PubMed]
- Kim, W.Y.; Kweon, O.J.; Cha, M.J.; Baek, M.S.; Choi, S.H. Dexamethasone may improve severe COVID-19 via ameliorating endothelial injury and inflammation: A preliminary pilot study. PLoS ONE 2021, 16, e0254167. [Google Scholar] [CrossRef] [PubMed]
- Ledford, H. Coronavirus breakthrough: Dexamethasone is first drug shown to save lives. Nature 2020, 582, 469. [Google Scholar] [CrossRef]
- Mahase, E. COVID-19: Anti-inflammatory treatment baricitinib reduces deaths in patients admitted to hospital, finds trial. BMJ 2022, 376, o573. [Google Scholar] [CrossRef]
- Lawler, P.R.; Goligher, E.C.; Berger, J.S.; Neal, M.D.; McVerry, B.J.; Nicolau, J.C.; Gong, M.N.; Carrier, M.; Rosenson, R.S.; Reynolds, H.R.; et al. Therapeutic Anticoagulation with Heparin in Noncritically Ill Patients with Covid-19. N. Engl. J. Med. 2021, 385, 790–802. [Google Scholar] [CrossRef]
- Barnes, G.D.; Burnett, A.; Allen, A.; Ansell, J.; Blumenstein, M.; Clark, N.P.; Crowther, M.; Dager, W.E.; Deitelzweig, S.B.; Ellsworth, S.; et al. Thromboembolic prevention and anticoagulant therapy during the COVID-19 pandemic: Updated clinical guidance from the anticoagulation forum. J. Thromb. Thrombolysis 2022, 54, 197–210. [Google Scholar] [CrossRef]
- National Institutes of Health COVID-19 Treatment Guidelines. Antithrombotic Therapy in Patients with COVID-19. Available online: https://www.covid19treatmentguidelines.nih.gov/therapies/antithrombotic-therapy/ (accessed on 2 August 2022).
- Rannard, S.P.; McDonald, T.O.; Owen, A. Chasing COVID-19 chemotherapeutics without putting the cart before the horse. Br. J. Clin. Pharmacol. 2020. [Google Scholar] [CrossRef]
- Bayat Mokhtari, R.; Homayouni, T.S.; Baluch, N.; Morgatskaya, E.; Kumar, S.; Das, B.; Yeger, H. Combination therapy in combating cancer. Oncotarget 2017, 8, 38022–38043. [Google Scholar] [CrossRef]
- Shyr, Z.A.; Cheng, Y.S.; Lo, D.C.; Zheng, W. Drug combination therapy for emerging viral diseases. Drug Discov. Today 2021, 26, 2367–2376. [Google Scholar] [CrossRef]
- Akinbolade, S.; Coughlan, D.; Fairbairn, R.; McConkey, G.; Powell, H.; Ogunbayo, D.; Craig, D. Combination therapies for COVID-19: An overview of the clinical trials landscape. Br. J. Clin. Pharmacol. 2022, 88, 1590–1597. [Google Scholar] [CrossRef]
- Pushpakom, S.; Iorio, F.; Eyers, P.A.; Escott, K.J.; Hopper, S.; Wells, A.; Doig, A.; Guilliams, T.; Latimer, J.; McNamee, C.; et al. Drug repurposing: Progress, challenges and recommendations. Nat. Rev. Drug Discov. 2019, 18, 41–58. [Google Scholar] [CrossRef] [PubMed]
- Boffito, M.; Back, D.J.; Flexner, C.; Sjö, P.; Blaschke, T.F.; Horby, P.W.; Cattaneo, D.; Acosta, E.P.; Anderson, P.; Owen, A. Toward Consensus on Correct Interpretation of Protein Binding in Plasma and Other Biological Matrices for COVID-19 Therapeutic Development. Clin. Pharmacol. Ther. 2021, 110, 64–68. [Google Scholar] [CrossRef] [PubMed]
- Arshad, U.; Pertinez, H.; Box, H.; Tatham, L.; Rajoli, R.K.R.; Curley, P.; Neary, M.; Sharp, J.; Liptrott, N.J.; Valentijn, A.; et al. Prioritization of Anti-SARS-Cov-2 Drug Repurposing Opportunities Based on Plasma and Target Site Concentrations Derived from their Established Human Pharmacokinetics. Clin. Pharmacol. Ther. 2020, 108, 775–790. [Google Scholar] [CrossRef] [PubMed]
- Eugene, A.R. Fluoxetine pharmacokinetics and tissue distribution quantitatively supports a therapeutic role in COVID-19 at a minimum dose of 20 mg per day. F1000Research 2022, 10, 477. [Google Scholar] [CrossRef]
- Ivens, T.; Van den Eynde, C.; Van Acker, K.; Nijs, E.; Dams, G.; Bettens, E.; Ohagen, A.; Pauwels, R.; Hertogs, K. Development of a homogeneous screening assay for automated detection of antiviral agents active against severe acute respiratory syndrome-associated coronavirus. J. Virol. Methods 2005, 129, 56–63. [Google Scholar] [CrossRef]
- Do, T.N.D.; Donckers, K.; Vangeel, L.; Chatterjee, A.K.; Gallay, P.A.; Bobardt, M.D.; Bilello, J.P.; Cihlar, T.; De Jonghe, S.; Neyts, J.; et al. A robust SARS-CoV-2 replication model in primary human epithelial cells at the air liquid interface to assess antiviral agents. Antivir. Res. 2021, 192, 105122. [Google Scholar] [CrossRef]
- Drugs for Neglected Diseases Intitiative. Target Product Profile for COVID-19. Available online: https://dndi.org/diseases/covid-19/target-product-profile/ (accessed on 15 June 2022).
- Abdelnabi, R.; Foo, C.S.; Jochmans, D.; Vangeel, L.; De Jonghe, S.; Augustijns, P.; Mols, R.; Weynand, B.; Wattanakul, T.; Hoglund, R.M.; et al. The oral protease inhibitor (PF-07321332) protects Syrian hamsters against infection with SARS-CoV-2 variants of concern. Nat. Commun. 2022, 13, 719. [Google Scholar] [CrossRef]
- Boudewijns, R.; Thibaut, H.J.; Kaptein, S.J.F.; Li, R.; Vergote, V.; Seldeslachts, L.; Van Weyenbergh, J.; De Keyzer, C.; Bervoets, L.; Sharma, S.; et al. STAT2 signaling restricts viral dissemination but drives severe pneumonia in SARS-CoV-2 infected hamsters. Nat. Commun. 2020, 11, 5838. [Google Scholar] [CrossRef]
- Kaptein, S.J.F.; Jacobs, S.; Langendries, L.; Seldeslachts, L.; Ter Horst, S.; Liesenborghs, L.; Hens, B.; Vergote, V.; Heylen, E.; Barthelemy, K.; et al. Favipiravir at high doses has potent antiviral activity in SARS-CoV-2-infected hamsters, whereas hydroxychloroquine lacks activity. Proc. Natl. Acad. Sci. USA 2020, 117, 26955–26965. [Google Scholar] [CrossRef]
- Driouich, J.S.; Cochin, M.; Lingas, G.; Moureau, G.; Touret, F.; Petit, P.R.; Piorkowski, G.; Barthélémy, K.; Laprie, C.; Coutard, B.; et al. Favipiravir antiviral efficacy against SARS-CoV-2 in a hamster model. Nat. Commun. 2021, 12, 1735. [Google Scholar] [CrossRef]
- Wattanakul, T.; Chotsiri, P.; Scandale, I.; Hoglund, R.M.; Tarning, J. Pharmacometric approach to evaluate drug for potential repurposing as COVID-19 therapeutics. Expert Rev. Clin. Pharmacol. 2022; submitted. [Google Scholar]
- Lavielle, M. mlxR: Simulation of Longitudinal Data. R Package Version 4.2.0. 2021. Available online: https://cran.r-project.org/web/packages/mlxR/index.html (accessed on 15 June 2022).
- Clemency, B.M.; Varughese, R.; Gonzalez-Rojas, Y.; Morse, C.G.; Phipatanakul, W.; Koster, D.J.; Blaiss, M.S. Efficacy of Inhaled Ciclesonide for Outpatient Treatment of Adolescents and Adults With Symptomatic COVID-19: A Randomized Clinical Trial. JAMA Intern. Med. 2022, 182, 42–49. [Google Scholar] [CrossRef] [PubMed]
- Ramakrishnan, S.; Nicolau, D.V., Jr.; Langford, B.; Mahdi, M.; Jeffers, H.; Mwasuku, C.; Krassowska, K.; Fox, R.; Binnian, I.; Glover, V.; et al. Inhaled budesonide in the treatment of early COVID-19 (STOIC): A phase 2, open-label, randomised controlled trial. Lancet Respir. Med. 2021, 9, 763–772. [Google Scholar] [CrossRef]
- Yu, L.M.; Bafadhel, M.; Dorward, J.; Hayward, G.; Saville, B.R.; Gbinigie, O.; Van Hecke, O.; Ogburn, E.; Evans, P.H.; Thomas, N.P.B.; et al. Inhaled budesonide for COVID-19 in people at high risk of complications in the community in the UK (PRINCIPLE): A randomised, controlled, open-label, adaptive platform trial. Lancet 2021, 398, 843–855. [Google Scholar] [CrossRef]
- Si, L.; Bai, H.; Rodas, M.; Cao, W.; Oh, C.Y.; Jiang, A.; Moller, R.; Hoagland, D.; Oishi, K.; Horiuchi, S.; et al. A human-airway-on-a-chip for the rapid identification of candidate antiviral therapeutics and prophylactics. Nat. Biomed. Eng. 2021, 5, 815–829. [Google Scholar] [CrossRef] [PubMed]
- Ohashi, H.; Watashi, K.; Saso, W.; Shionoya, K.; Iwanami, S.; Hirokawa, T.; Shirai, T.; Kanaya, S.; Ito, Y.; Kim, K.S.; et al. Potential anti-COVID-19 agents, cepharanthine and nelfinavir, and their usage for combination treatment. iScience 2021, 24, 102367. [Google Scholar] [CrossRef]
- Foo, C.S.; Abdelnabi, R.; Kaptein, S.J.F.; Zhang, X.; Ter Horst, S.; Mols, R.; Delang, L.; Rocha-Pereira, J.; Coelmont, L.; Leyssen, P.; et al. HIV protease inhibitors Nelfinavir and Lopinavir/Ritonavir markedly improve lung pathology in SARS-CoV-2-infected Syrian hamsters despite lack of an antiviral effect. Antivir. Res. 2022, 202, 105311. [Google Scholar] [CrossRef]
- Riccardi, N.; Giacomelli, A.; Canetti, D.; Comelli, A.; Intini, E.; Gaiera, G.; Diaw, M.M.; Udwadia, Z.; Besozzi, G.; Codecasa, L.; et al. Clofazimine: An old drug for never-ending diseases. Future Microbiol. 2020, 15, 557–566. [Google Scholar] [CrossRef]
- Yuan, S.; Yin, X.; Meng, X.; Chan, J.F.; Ye, Z.W.; Riva, L.; Pache, L.; Chan, C.C.; Lai, P.M.; Chan, C.C.; et al. Clofazimine broadly inhibits coronaviruses including SARS-CoV-2. Nature 2021, 593, 418–423. [Google Scholar] [CrossRef]
- Fujifilm Press Release. Anti-Influenza Drug Avigan® Tablet Meets Primary Endpoint in Phase III Clinical Trial in Japan for COVID-19 Patients. 23 September 2020. Available online: https://www.fujifilm.com/jp/en/news/hq/5451 (accessed on 24 June 2022).
- Merck Press Release. Merck and Ridgeback to Present Data Demonstrating That Treatment With LAGEVRIO™ (Molnupiravir) Was Associated with More Rapid Elimination of Infectious SARS-CoV-2 Than Placebo. 1 April 2022. Available online: https://www.merck.com/news/merck-and-ridgeback-to-present-data-demonstrating-that-treatment-with-lagevrio-molnupiravir-was-associated-with-more-rapid-elimination-of-infectious-sars-cov-2-than-placebo/ (accessed on 24 June 2022).
- Pfizer Press Release. Pfizer Announces Additional Phase 2/3 Study Results Confirming Robust Efficacy of Novel COVID-19 Oral Antiviral Treatment Candidate in Reducing Risk of Hospitalization or Death. 14 December 2021. Available online: https://www.pfizer.com/news/press-release/press-release-detail/pfizer-announces-additional-phase-23-study-results (accessed on 24 June 2022).
- Burrell, C.J.; Howard, C.R.; Murphy, F.A. Classification of Viruses and Phylogenetic Relationships. Fenner White’s Med. Virol. 2017, 15–25. [Google Scholar] [CrossRef]
- Grobler, J.A.; Anderson, A.S.; Fernandes, P.; Diamond, M.S.; Colvis, C.M.; Menetski, J.P.; Alvarez, R.M.; Young, J.A.T.; Carter, K.L. Accelerated Preclinical Paths to Support Rapid Development of COVID-19 Therapeutics. Cell Host Microbe 2020, 28, 638–645. [Google Scholar] [CrossRef]
- Salasc, F.; Lahlali, T.; Laurent, E.; Rosa-Calatrava, M.; Pizzorno, A. Treatments for COVID-19: Lessons from 2020 and new therapeutic options. Curr. Opin. Pharmacol. 2022, 62, 43–59. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, M.; Mosbauer, K.; Hofmann-Winkler, H.; Kaul, A.; Kleine-Weber, H.; Kruger, N.; Gassen, N.C.; Muller, M.A.; Drosten, C.; Pohlmann, S. Chloroquine does not inhibit infection of human lung cells with SARS-CoV-2. Nature 2020, 585, 588–590. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, M.; Hofmann-Winkler, H.; Smith, J.C.; Krüger, N.; Arora, P.; Sørensen, L.K.; Søgaard, O.S.; Hasselstrøm, J.B.; Winkler, M.; Hempel, T.; et al. Camostat mesylate inhibits SARS-CoV-2 activation by TMPRSS2-related proteases and its metabolite GBPA exerts antiviral activity. EBioMedicine 2021, 65, 103255. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Duan, X.; Yang, L.; Nilsson-Payant, B.E.; Wang, P.; Duan, F.; Tang, X.; Yaron, T.M.; Zhang, T.; Uhl, S.; et al. Identification of SARS-CoV-2 inhibitors using lung and colonic organoids. Nature 2021, 589, 270–275. [Google Scholar] [CrossRef]
- Fintelman-Rodrigues, N.; Sacramento, C.Q.; Ribeiro Lima, C.; Souza da Silva, F.; Ferreira, A.C.; Mattos, M.; de Freitas, C.S.; Cardoso Soares, V.; da Silva Gomes Dias, S.; Temerozo, J.R.; et al. Atazanavir, Alone or in Combination with Ritonavir, Inhibits SARS-CoV-2 Replication and Proinflammatory Cytokine Production. Antimicrob. Agents Chemother. 2020, 64, e00825-20. [Google Scholar] [CrossRef]
- Yamamoto, N.; Matsuyama, S.; Hoshino, T.; Yamamoto, N. Nelfinavir inhibits replication of severe acute respiratory syndrome coronavirus 2 in vitro. bioRxiv 2020. [Google Scholar] [CrossRef]
- Mollica, V.; Rizzo, A.; Massari, F. The pivotal role of TMPRSS2 in coronavirus disease 2019 and prostate cancer. Future Oncol. 2020, 16, 2029–2033. [Google Scholar] [CrossRef]
- Kuteykin-Teplyakov, K.; Luna-Tortós, C.; Ambroziak, K.; Löscher, W. Differences in the expression of endogenous efflux transporters in MDR1-transfected versus wildtype cell lines affect P-glycoprotein mediated drug transport. Br. J. Pharmacol. 2010, 160, 1453–1463. [Google Scholar] [CrossRef]
- Tummino, T.A.; Rezelj, V.V.; Fischer, B.; Fischer, A.; O’Meara, M.J.; Monel, B.; Vallet, T.; Zhang, Z.; Alon, A.; O’Donnell, H.R.; et al. Drug-induced phospholipidosis confounds drug repurposing for SARS-CoV-2. Science 2021, 373, 541–547. [Google Scholar] [CrossRef]
- Dittmar, M.; Lee, J.S.; Whig, K.; Segrist, E.; Li, M.; Kamalia, B.; Castellana, L.; Ayyanathan, K.; Cardenas-Diaz, F.L.; Morrisey, E.E.; et al. Drug repurposing screens reveal cell-type-specific entry pathways and FDA-approved drugs active against SARS-Cov-2. Cell Rep. 2021, 35, 108959. [Google Scholar] [CrossRef]
- Lukassen, S.; Chua, R.L.; Trefzer, T.; Kahn, N.C.; Schneider, M.A.; Muley, T.; Winter, H.; Meister, M.; Veith, C.; Boots, A.W.; et al. SARS-CoV-2 receptor ACE2 and TMPRSS2 are primarily expressed in bronchial transient secretory cells. EMBO J. 2020, 39, e105114. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Krüger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280.e8. [Google Scholar] [CrossRef] [PubMed]
- Atea Pharmaceuticals Press Release. Atea Pharmaceuticals Provides Update and Topline Results for Phase 2 MOONSONG Trial Evaluating AT-527 in the Outpatient Setting. 19 October 2021. Available online: https://ir.ateapharma.com/news-releases/news-release-details/atea-pharmaceuticals-provides-update-and-topline-results-phase-2 (accessed on 24 June 2022).
- Neervannan, S. Preclinical formulations for discovery and toxicology: Physicochemical challenges. Expert Opin. Drug Metab. Toxicol. 2006, 2, 715–731. [Google Scholar] [CrossRef] [PubMed]
- Mølhave, M.; Agergaard, J.; Wejse, C. Clinical Management of COVID-19 Patients—An Update. Semin. Nucl. Med. 2022, 52, 4–10. [Google Scholar] [CrossRef] [PubMed]
- Richardson, C.; Bhagani, S.; Pollara, G. Antiviral treatment for COVID-19: The evidence supporting remdesivir. Clin. Med. 2020, 20, e215–e217. [Google Scholar] [CrossRef]
- Good, S.S.; Westover, J.; Jung, K.H.; Zhou, X.J.; Moussa, A.; La Colla, P.; Collu, G.; Canard, B.; Sommadossi, J.P. AT-527, a Double Prodrug of a Guanosine Nucleotide Analog, Is a Potent Inhibitor of SARS-CoV-2 In Vitro and a Promising Oral Antiviral for Treatment of COVID-19. Antimicrob. Agents Chemother. 2021, 65, e02479-20. [Google Scholar] [CrossRef]
- Collins, F.S. COVID-19 lessons for research. Science 2021, 371, 1081. [Google Scholar] [CrossRef]
- Mullard, A. RECOVERY 1 year on: A rare success in the COVID-19 clinical trial landscape. Nat. Rev. Drug Discov. 2021, 20, 336–337. [Google Scholar] [CrossRef]
- Axfors, C.; Schmitt, A.M.; Janiaud, P.; Van’t Hooft, J.; Abd-Elsalam, S.; Abdo, E.F.; Abella, B.S.; Akram, J.; Amaravadi, R.K.; Angus, D.C.; et al. Mortality outcomes with hydroxychloroquine and chloroquine in COVID-19 from an international collaborative meta-analysis of randomized trials. Nat. Commun. 2021, 12, 2349. [Google Scholar] [CrossRef]
- Mobarak, S.; Salasi, M.; Hormati, A.; Khodadadi, J.; Ziaee, M.; Abedi, F.; Ebrahimzadeh, A.; Azarkar, Z.; Mansour-Ghanaei, F.; Joukar, F.; et al. Evaluation of the effect of sofosbuvir and daclatasvir in hospitalized COVID-19 patients: A randomized double-blind clinical trial (DISCOVER). J. Antimicrob. Chemother. 2022, 77, 758–766. [Google Scholar] [CrossRef]
- Patel, T.K.; Patel, P.B.; Barvaliya, M.; Saurabh, M.K.; Bhalla, H.L.; Khosla, P.P. Efficacy and safety of lopinavir-ritonavir in COVID-19: A systematic review of randomized controlled trials. J. Infect. Public Health 2021, 14, 740–748. [Google Scholar] [CrossRef] [PubMed]
- Acharya, B.N. Amodiaquine as COVID-19 Mpro Inhibitor: A Theoretical Study. ChemRxiv 2020. [Google Scholar] [CrossRef]
- World Health Organization. Tracking SARS-CoV-2 Variants. Available online: https://www.who.int/activities/tracking-SARS-CoV-2-variants (accessed on 23 June 2022).
- World Health Organization. Facilitation Council Preparation. 19 March 2021. Available online: https://www.who.int/docs/default-source/coronaviruse/act-accelerator/act-a-tx-dx-fc-pre-briefing-deck_fin.pdf?sfvrsn=ce5b7dea_1&download=true (accessed on 5 May 2022).
- World Health Organization. Therapeutics and COVID-19: Living Guideline. 22 April 2022. Available online: https://www.who.int/publications/i/item/WHO-2019-nCoV-therapeutics-2022.3 (accessed on 15 June 2022).
- Jochmans, D.; Leyssen, P.; Neyts, J. A novel method for high-throughput screening to quantify antiviral activity against viruses that induce limited CPE. J. Virol. Methods 2012, 183, 176–179. [Google Scholar] [CrossRef] [PubMed]
- Touret, F.; Driouich, J.S.; Cochin, M.; Petit, P.R.; Gilles, M.; Barthélémy, K.; Moureau, G.; Mahon, F.X.; Malvy, D.; Solas, C.; et al. Preclinical evaluation of Imatinib does not support its use as an antiviral drug against SARS-CoV-2. Antivir. Res. 2021, 193, 105137. [Google Scholar] [CrossRef] [PubMed]
- Pizzorno, A.; Padey, B.; Julien, T.; Trouillet-Assant, S.; Traversier, A.; Errazuriz-Cerda, E.; Fouret, J.; Dubois, J.; Gaymard, A.; Lescure, F.X.; et al. Characterization and Treatment of SARS-CoV-2 in Nasal and Bronchial Human Airway Epithelia. Cell Rep. Med. 2020, 1, 100059. [Google Scholar] [CrossRef] [PubMed]
- Gruber, A.D.; Firsching, T.C.; Trimpert, J.; Dietert, K. Hamster models of COVID-19 pneumonia reviewed: How human can they be? Vet. Pathol. 2022, 59, 528–545. [Google Scholar] [CrossRef] [PubMed]
- Sia, S.F.; Yan, L.M.; Chin, A.W.H.; Fung, K.; Choy, K.T.; Wong, A.Y.L.; Kaewpreedee, P.; Perera, R.; Poon, L.L.M.; Nicholls, J.M.; et al. Pathogenesis and transmission of SARS-CoV-2 in golden hamsters. Nature 2020, 583, 834–838. [Google Scholar] [CrossRef] [PubMed]
- Francis, M.E.; Goncin, U.; Kroeker, A.; Swan, C.; Ralph, R.; Lu, Y.; Etzioni, A.L.; Falzarano, D.; Gerdts, V.; Machtaler, S.; et al. SARS-CoV-2 infection in the Syrian hamster model causes inflammation as well as type I interferon dysregulation in both respiratory and non-respiratory tissues including the heart and kidney. PLoS Pathog. 2021, 17, e1009705. [Google Scholar] [CrossRef] [PubMed]
- Sheahan, T.P.; Sims, A.C.; Zhou, S.; Graham, R.L.; Pruijssers, A.J.; Agostini, M.L.; Leist, S.R.; Schäfer, A.; Dinnon, K.H., 3rd; Stevens, L.J.; et al. An orally bioavailable broad-spectrum antiviral inhibits SARS-CoV-2 in human airway epithelial cell cultures and multiple coronaviruses in mice. Sci. Transl. Med. 2020, 12, eabb5883. [Google Scholar] [CrossRef] [PubMed]
- Cox, R.M.; Wolf, J.D.; Plemper, R.K. Therapeutically administered ribonucleoside analogue MK-4482/EIDD-2801 blocks SARS-CoV-2 transmission in ferrets. Nat. Microbiol. 2021, 6, 11–18. [Google Scholar] [CrossRef]
- Sheahan, T.P.; Sims, A.C.; Graham, R.L.; Menachery, V.D.; Gralinski, L.E.; Case, J.B.; Leist, S.R.; Pyrc, K.; Feng, J.Y.; Trantcheva, I.; et al. Broad-spectrum antiviral GS-5734 inhibits both epidemic and zoonotic coronaviruses. Sci. Transl. Med. 2017, 9, eaal3653. [Google Scholar] [CrossRef]
- Pruijssers, A.J.; George, A.S.; Schäfer, A.; Leist, S.R.; Gralinksi, L.E.; Dinnon, K.H., 3rd; Yount, B.L.; Agostini, M.L.; Stevens, L.J.; Chappell, J.D.; et al. Remdesivir Inhibits SARS-CoV-2 in Human Lung Cells and Chimeric SARS-CoV Expressing the SARS-CoV-2 RNA Polymerase in Mice. Cell Rep. 2020, 32, 107940. [Google Scholar] [CrossRef] [PubMed]
- Abdelnabi, R.; Boudewijns, R.; Foo, C.S.; Seldeslachts, L.; Sanchez-Felipe, L.; Zhang, X.; Delang, L.; Maes, P.; Kaptein, S.J.F.; Weynand, B.; et al. Comparing infectivity and virulence of emerging SARS-CoV-2 variants in Syrian hamsters. EBioMedicine 2021, 68, 103403. [Google Scholar] [CrossRef] [PubMed]
- Cochin, M.; Luciani, L.; Touret, F.; Driouich, J.S.; Petit, P.R.; Moureau, G.; Baronti, C.; Laprie, C.; Thirion, L.; Maes, P.; et al. The SARS-CoV-2 Alpha variant exhibits comparable fitness to the D614G strain in a Syrian hamster model. Commun. Biol. 2022, 5, 225. [Google Scholar] [CrossRef] [PubMed]
- Cochin, M.; Touret, F.; Driouich, J.S.; Moureau, G.; Petit, P.R.; Laprie, C.; Solas, C.; de Lamballerie, X.; Nougairède, A. Hydroxychloroquine and azithromycin used alone or combined are not effective against SARS-CoV-2 ex vivo and in a hamster model. Antivir. Res. 2022, 197, 105212. [Google Scholar] [CrossRef]
- Driouich, J.-S.; Cochin, M.; Touret, F.; Petit, P.-R.; Gilles, M.; Moureau, G.; Barthélémy, K.; Laprie, C.; Wattanakul, T.; Chotsiri, P.; et al. Pre-clinical evaluation of antiviral activity of nitazoxanide against Sars-CoV-2. EBioMedicine 2022, 82, 104148. [Google Scholar] [CrossRef]
- Savic, R.M.; Jonker, D.M.; Kerbusch, T.; Karlsson, M.O. Implementation of a transit compartment model for describing drug absorption in pharmacokinetic studies. J. Pharmacokinet. Pharmacodyn. 2007, 34, 711–726. [Google Scholar] [CrossRef] [PubMed]
- Dosne, A.G.; Bergstrand, M.; Karlsson, M.O. An automated sampling importance resampling procedure for estimating parameter uncertainty. J. Pharmacokinet. Pharmacodyn. 2017, 44, 509–520. [Google Scholar] [CrossRef] [PubMed]
- Ferreira Sales-Medina, D.; Rodrigues Pinto Ferreira, L.; Romera, L.M.D.; Ribeiro Gonçalves, K.; Guido, R.V.C.; Courtemanche, G.; Buckeridge, M.S.; Durigon, E.L.; Moraes, C.B.; Freitas-Junior, L.H. Discovery of clinically approved drugs capable of inhibiting SARS-CoV-2 in vitro infection using a phenotypic screening strategy and network-analysis to predict their potential to treat covid-19. bioRxiv 2020. [Google Scholar] [CrossRef]
- Jeon, S.; Ko, M.; Lee, J.; Choi, I.; Byun, S.Y.; Park, S.; Shum, D.; Kim, S. Identification of Antiviral Drug Candidates against SARS-CoV-2 from FDA-Approved Drugs. Antimicrob. Agents Chemother. 2020, 64. [Google Scholar] [CrossRef] [PubMed]
- Sacramento, C.Q.; Fintelman-Rodrigues, N.; Temerozo, J.R.; Da Silva, A.P.D.; Dias, S.; da Silva, C.D.S.; Ferreira, A.C.; Mattos, M.; Pao, C.R.R.; de Freitas, C.S.; et al. In vitro antiviral activity of the anti-HCV drugs daclatasvir and sofosbuvir against SARS-CoV-2, the aetiological agent of COVID-19. J. Antimicrob. Chemother. 2021, 76, 1874–1885. [Google Scholar] [CrossRef] [PubMed]
- Ko, M.; Jeon, S.; Ryu, W.S.; Kim, S. Comparative analysis of antiviral efficacy of FDA-approved drugs against SARS-CoV-2 in human lung cells. J. Med. Virol. 2021, 93, 1403–1408. [Google Scholar] [CrossRef] [PubMed]
- Weston, S.; Coleman, C.M.; Haupt, R.; Logue, J.; Matthews, K.; Li, Y.; Reyes, H.M.; Weiss, S.R.; Frieman, M.B. Broad Anti-coronavirus Activity of Food and Drug Administration-Approved Drugs against SARS-CoV-2 In Vitro and SARS-CoV In Vivo. J. Virol. 2020, 94, e01218-20. [Google Scholar] [CrossRef] [PubMed]
- Choy, K.T.; Wong, A.Y.; Kaewpreedee, P.; Sia, S.F.; Chen, D.; Hui, K.P.Y.; Chu, D.K.W.; Chan, M.C.W.; Cheung, P.P.; Huang, X.; et al. Remdesivir, lopinavir, emetine, and homoharringtonine inhibit SARS-CoV-2 replication in vitro. Antivir. Res. 2020, 178, 104786. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Lien, C.Z.; Selvaraj, P.; Wang, T.T. Evaluation of 19 antiviral drugs against SARS-CoV-2 Infection. BioRxiv 2020. [Google Scholar] [CrossRef]
- Shannon, A.; Selisko, B.; Le, N.T.; Huchting, J.; Touret, F.; Piorkowski, G.; Fattorini, V.; Ferron, F.; Decroly, E.; Meier, C.; et al. Rapid incorporation of Favipiravir by the fast and permissive viral RNA polymerase complex results in SARS-CoV-2 lethal mutagenesis. Nat. Commun. 2020, 11, 4682. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Cao, R.; Zhang, L.; Yang, X.; Liu, J.; Xu, M.; Shi, Z.; Hu, Z.; Zhong, W.; Xiao, G. Remdesivir and chloroquine effectively inhibit the recently emerged novel coronavirus (2019-nCoV) in vitro. Cell Res. 2020, 30, 269–271. [Google Scholar] [CrossRef] [PubMed]
- Zandi, K.; Amblard, F.; Musall, K.; Downs-Bowen, J.; Kleinbard, R.; Oo, A.; Cao, D.; Liang, B.; Russell, O.O.; McBrayer, T.; et al. Repurposing Nucleoside Analogs for Human Coronaviruses. Antimicrob. Agents Chemother. 2020, 65, e01652-20. [Google Scholar] [CrossRef]
- Caly, L.; Druce, J.D.; Catton, M.G.; Jans, D.A.; Wagstaff, K.M. The FDA-approved drug ivermectin inhibits the replication of SARS-CoV-2 in vitro. Antivir. Res. 2020, 178, 104787. [Google Scholar] [CrossRef]
- Jeffreys, L.N.; Pennington, S.H.; Duggan, J.; Caygill, C.H.; Lopeman, R.C.; Breen, A.F.; Jinks, J.B.; Ardrey, A.; Donnellan, S.; Patterson, E.I.; et al. Remdesivir-ivermectin combination displays synergistic interaction with improved in vitro activity against SARS-CoV-2. Int. J. Antimicrob. Agents 2022, 59, 106542. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, N.; Yang, R.; Yoshinaka, Y.; Amari, S.; Nakano, T.; Cinatl, J.; Rabenau, H.; Doerr, H.W.; Hunsmann, G.; Otaka, A.; et al. HIV protease inhibitor nelfinavir inhibits replication of SARS-associated coronavirus. Biochem. Biophys. Res. Commun. 2004, 318, 719–725. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Yao, H.; Shen, J.; Wu, N.; Xu, Y.; Lu, X.; Zhu, W.; Li, L.-J. Nelfinavir Is Active Against SARS-CoV-2 in Vero E6 Cells. ChemRxiv 2020. [Google Scholar]
- Rogosnitzky, M.; Danks, R. Therapeutic potential of the biscoclaurine alkaloid, cepharanthine, for a range of clinical conditions. Pharmacol. Rep. 2011, 63, 337–347. [Google Scholar] [CrossRef]
- Zimniak, M.; Kirschner, L.; Hilpert, H.; Geiger, N.; Danov, O.; Oberwinkler, H.; Steinke, M.; Sewald, K.; Seibel, J.; Bodem, J. The serotonin reuptake inhibitor Fluoxetine inhibits SARS-CoV-2 in human lung tissue. Sci. Rep. 2021, 11, 5890. [Google Scholar] [CrossRef]
- Schloer, S.; Brunotte, L.; Goretzko, J.; Mecate-Zambrano, A.; Korthals, N.; Gerke, V.; Ludwig, S.; Rescher, U. Targeting the endolysosomal host-SARS-CoV-2 interface by clinically licensed functional inhibitors of acid sphingomyelinase (FIASMA) including the antidepressant fluoxetine. Emerg. Microbes Infect. 2020, 9, 2245–2255. [Google Scholar] [CrossRef]
- Fred, S.M.; Kuivanen, S.; Ugurlu, H.; Casarotto, P.C.; Levanov, L.; Saksela, K.; Vapalahti, O.; Castren, E. Antidepressant and Antipsychotic Drugs Reduce Viral Infection by SARS-CoV-2 and Fluoxetine Shows Antiviral Activity Against the Novel Variants in vitro. Front. Pharmacol. 2021, 12, 755600. [Google Scholar] [CrossRef] [PubMed]
- Olaleye, O.A.; Kaur, M.; Onyenaka, C.C. Ambroxol Hydrochloride Inhibits the Interaction between Severe Acute Respiratory Syndrome Coronavirus 2 Spike Protein’s Receptor Binding Domain and Recombinant Human ACE2. BioRxiv 2020. [Google Scholar] [CrossRef]
- Mahoney, M.; Damalanka, V.C.; Tartell, M.A.; Chung, D.H.; Lourenco, A.L.; Pwee, D.; Mayer Bridwell, A.E.; Hoffmann, M.; Voss, J.; Karmakar, P.; et al. A novel class of TMPRSS2 inhibitors potently block SARS-CoV-2 and MERS-CoV viral entry and protect human epithelial lung cells. Proc. Natl. Acad. Sci. USA 2021, 118, e2108728118. [Google Scholar] [CrossRef] [PubMed]
- Schultz, D.C.; Johnson, R.M.; Ayyanathan, K.; Miller, J.; Whig, K.; Kamalia, B.; Dittmar, M.; Weston, S.; Hammond, H.L.; Dillen, C.; et al. Pyrimidine inhibitors synergize with nucleoside analogues to block SARS-CoV-2. Nature 2022, 604, 134–140. [Google Scholar] [CrossRef]
- Owen, D.R.; Allerton, C.M.N.; Anderson, A.S.; Aschenbrenner, L.; Avery, M.; Berritt, S.; Boras, B.; Cardin, R.D.; Carlo, A.; Coffman, K.J.; et al. An oral SARS-CoV-2 M(pro) inhibitor clinical candidate for the treatment of COVID-19. Science 2021, 374, 1586–1593. [Google Scholar] [CrossRef]
- Xiong, R.; Zhang, L.; Li, S.; Sun, Y.; Ding, M.; Wang, Y.; Zhao, Y.; Wu, Y.; Shang, W.; Jiang, X.; et al. Novel and potent inhibitors targeting DHODH are broad-spectrum antivirals against RNA viruses including newly-emerged coronavirus SARS-CoV-2. Protein Cell 2020, 11, 723–739. [Google Scholar] [CrossRef] [PubMed]
- Swaim, C.D.; Dwivedi, V.; Perng, Y.C.; Zhao, X.; Canadeo, L.A.; Harastani, H.H.; Darling, T.L.; Boon, A.C.M.; Lenschow, D.J.; Kulkarni, V.; et al. 6-Thioguanine blocks SARS-CoV-2 replication by inhibition of PLpro. iScience 2021, 24, 103213. [Google Scholar] [CrossRef] [PubMed]
- McCoy, J.; Goren, A.; Cadegiani, F.A.; Vaño-Galván, S.; Kovacevic, M.; Situm, M.; Shapiro, J.; Sinclair, R.; Tosti, A.; Stanimirovic, A.; et al. Proxalutamide Reduces the Rate of Hospitalization for COVID-19 Male Outpatients: A Randomized Double-Blinded Placebo-Controlled Trial. Front. Med. 2021, 8, 668698. [Google Scholar] [CrossRef]
- Murray, J.; Hogan, R.J.; Martin, D.E.; Blahunka, K.; Sancilio, F.D.; Balyan, R.; Lovern, M.; Still, R.; Tripp, R.A. Probenecid inhibits SARS-CoV-2 replication in vivo and in vitro. Sci. Rep. 2021, 11, 18085. [Google Scholar] [CrossRef] [PubMed]
- Box, H.; Pennington, S.H.; Kijak, E.; Tatham, L.; Caygill, C.H.; Lopeman, R.C.; Jeffreys, L.N.; Herriott, J.; Sharp, J.; Neary, M.; et al. Lack of antiviral activity of probenecid in Vero E6 cells and Syrian golden hamsters: A need for better understanding of inter-lab differences in preclinical assays. bioRxiv 2022. [Google Scholar] [CrossRef]
- Muturi, E.; Hong, W.; Li, J.; Yang, W.; He, J.; Wei, H.; Yang, H. Effects of simeprevir on the replication of SARS-CoV-2 in vitro and in transgenic hACE2 mice. Int. J. Antimicrob. Agents 2022, 59, 106499. [Google Scholar] [CrossRef]
- Abdelnabi, R.; Foo, C.S.; Kaptein, S.J.F.; Zhang, X.; Do, T.N.D.; Langendries, L.; Vangeel, L.; Breuer, J.; Pang, J.; Williams, R.; et al. The combined treatment of Molnupiravir and Favipiravir results in a potentiation of antiviral efficacy in a SARS-CoV-2 hamster infection model. EBioMedicine 2021, 72, 103595. [Google Scholar] [CrossRef]
- Jan, J.T.; Cheng, T.R.; Juang, Y.P.; Ma, H.H.; Wu, Y.T.; Yang, W.B.; Cheng, C.W.; Chen, X.; Chou, T.H.; Shie, J.J.; et al. Identification of existing pharmaceuticals and herbal medicines as inhibitors of SARS-CoV-2 infection. Proc. Natl. Acad. Sci. USA 2021, 118, e2021579118. [Google Scholar] [CrossRef]
- Arevalo, A.P.; Pagotto, R.; Porfido, J.L.; Daghero, H.; Segovia, M.; Yamasaki, K.; Varela, B.; Hill, M.; Verdes, J.M.; Duhalde Vega, M.; et al. Ivermectin reduces in vivo coronavirus infection in a mouse experimental model. Sci. Rep. 2021, 11, 7132. [Google Scholar] [CrossRef]
- Pussard, E.; Verdier, F. Antimalarial 4-aminoquinolines: Mode of action and pharmacokinetics. Fundam. Clin. Pharmacol. 1994, 8, 1–17. [Google Scholar] [CrossRef]
- Atazanavir PK Fact Sheet. Available online: https://liverpool-hiv-hep.s3.amazonaws.com/fact_sheets/pdfs/000/000/087/original/HIV_FactSheet_ATV_2016_Mar.pdf (accessed on 1 July 2022).
- Gandhi, Y.; Eley, T.; Fura, A.; Li, W.; Bertz, R.J.; Garimella, T. Daclatasvir: A Review of Preclinical and Clinical Pharmacokinetics. Clin. Pharmacokinet. 2018, 57, 911–928. [Google Scholar] [CrossRef] [PubMed]
- Hayden, F.G.; Shindo, N. Influenza virus polymerase inhibitors in clinical development. Curr. Opin. Infect. Dis. 2019, 32, 176–186. [Google Scholar] [CrossRef]
- González Canga, A.; Sahagún Prieto, A.M.; Diez Liébana, M.J.; Fernández Martínez, N.; Sierra Vega, M.; García Vieitez, J.J. The pharmacokinetics and interactions of ivermectin in humans—A mini-review. AAPS J. 2008, 10, 42–46. [Google Scholar] [CrossRef] [PubMed]
- Romark Pharmaceuticals, Alinia®, Product Monograph. 2005. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2005/021818lbl.pdf (accessed on 1 July 2022).
- Cada, D.J.; Cong, J.; Baker, D.E. Sofosbuvir. Hosp. Pharm. 2014, 49, 466–478. [Google Scholar] [CrossRef]
- Lee, H.J.; Joung, S.K.; Kim, Y.G.; Yoo, J.Y.; Han, S.B. Bioequivalence assessment of ambroxol tablet after a single oral dose administration to healthy male volunteers. Pharmacol. Res. 2004, 49, 93–98. [Google Scholar] [CrossRef] [PubMed]
- Tarning, J.; Chotsiri, P.; Jullien, V.; Rijken, M.J.; Bergstrand, M.; Cammas, M.; McGready, R.; Singhasivanon, P.; Day, N.P.; White, N.J.; et al. Population pharmacokinetic and pharmacodynamic modeling of amodiaquine and desethylamodiaquine in women with Plasmodium vivax malaria during and after pregnancy. Antimicrob. Agents Chemother. 2012, 56, 5764–5773. [Google Scholar] [CrossRef] [PubMed]
- Punyawudho, B.; Thammajaruk, N.; Ruxrungtham, K.; Avihingsanon, A. Population pharmacokinetics and dose optimisation of ritonavir-boosted atazanavir in Thai HIV-infected patients. Int. J. Antimicrob. Agents 2017, 49, 327–332. [Google Scholar] [CrossRef]
- Hao, G.; Liang, H.; Li, Y.; Li, H.; Gao, H.; Liu, G.; Liu, Z. Simple, sensitive and rapid HPLC-MS/MS method for the determination of cepharanthine in human plasma. J. Chromatogr. B 2010, 878, 2923–2927. [Google Scholar] [CrossRef]
- Yasuda, K.; Moro, M.; Akasu, M.; Ohnishi, A. Pharmacokinetic disposition of cepharanthin following single and multiple intravenous doses in healthy subjects. Jpn. J. Clin. Pharmacol. Ther. 1989, 20, 741–749. [Google Scholar] [CrossRef]
- Yasuda, K.; Moro, M.; Ohnishi, A.; Akasu, M.; Shishido, A.; Tsunoo, M. Pharmacokinetic study of cepharanthin following single oral doses in healthy subjects. Jpn. J. Clin. Pharmacol. Ther. 1989, 20, 735–740. [Google Scholar] [CrossRef][Green Version]
- Faraj, A.; Svensson, R.J.; Diacon, A.H.; Simonsson, U.S.H. Drug Effect of Clofazimine on Persisters Explains an Unexpected Increase in Bacterial Load in Patients. Antimicrob. Agents Chemother. 2020, 64, e01905-19. [Google Scholar] [CrossRef] [PubMed]
- Chan, P.; Li, H.; Zhu, L.; Bifano, M.; Eley, T.; Osawa, M.; Ueno, T.; Hughes, E.; Bertz, R.; Garimella, T.; et al. Population Pharmacokinetic Analysis of Daclatasvir in Subjects with Chronic Hepatitis C Virus Infection. Clin. Pharmacokinet. 2017, 56, 1173–1183. [Google Scholar] [CrossRef]
- Wang, Y.; Zhong, W.; Salam, A.; Tarning, J.; Zhan, Q.; Huang, J.A.; Weng, H.; Bai, C.; Ren, Y.; Yamada, K.; et al. Phase 2a, open-label, dose-escalating, multi-center pharmacokinetic study of favipiravir (T-705) in combination with oseltamivir in patients with severe influenza. EBioMedicine 2020, 62, 103125. [Google Scholar] [CrossRef] [PubMed]
- Sagahón-Azúa, J.; Medellín-Garibay, S.E.; Chávez-Castillo, C.E.; González-Salinas, C.G.; Milán-Segovia, R.D.C.; Romano-Moreno, S. Factors associated with fluoxetine and norfluoxetine plasma concentrations and clinical response in Mexican patients with mental disorders. Pharmacol. Res. Perspect. 2021, 9, e00864. [Google Scholar] [CrossRef] [PubMed]
- Orlando, R.; De Martin, S.; Andrighetto, L.; Floreani, M.; Palatini, P. Fluvoxamine pharmacokinetics in healthy elderly subjects and elderly patients with chronic heart failure. Br. J. Clin. Pharmacol. 2010, 69, 279–286. [Google Scholar] [CrossRef]
- Kobylinski, K.C.; Ubalee, R.; Ponlawat, A.; Nitatsukprasert, C.; Phasomkulsolsil, S.; Wattanakul, T.; Tarning, J.; Na-Bangchang, K.; McCardle, P.W.; Davidson, S.A.; et al. Ivermectin susceptibility and sporontocidal effect in Greater Mekong Subregion Anopheles. Malar. J. 2017, 16, 280. [Google Scholar] [CrossRef]
- Painter, W.P.; Holman, W.; Bush, J.A.; Almazedi, F.; Malik, H.; Eraut, N.; Morin, M.J.; Szewczyk, L.J.; Painter, G.R. Human Safety, Tolerability, and Pharmacokinetics of Molnupiravir, a Novel Broad-Spectrum Oral Antiviral Agent with Activity Against SARS-CoV-2. Antimicrob. Agents Chemother. 2021, 65, e02428-20. [Google Scholar] [CrossRef]
- Hirt, D.; Treluyer, J.M.; Jullien, V.; Firtion, G.; Chappuy, H.; Rey, E.; Pons, G.; Mandelbrot, L.; Urien, S. Pregnancy-related effects on nelfinavir-M8 pharmacokinetics: A population study with 133 women. Antimicrob. Agents Chemother. 2006, 50, 2079–2086. [Google Scholar] [CrossRef]
- Rajoli, R.K.R.; Pertinez, H.; Arshad, U.; Box, H.; Tatham, L.; Curley, P.; Neary, M.; Sharp, J.; Liptrott, N.J.; Valentijn, A.; et al. Dose prediction for repurposing nitazoxanide in SARS-CoV-2 treatment or chemoprophylaxis. Br. J. Clin. Pharmacol. 2021, 87, 2078–2088. [Google Scholar] [CrossRef]
- Balderas-Acata, J.; Bueno, E.; Pérez-Becerril, F.; Espinosa-Martínez, C.; Burkefraga, V.; González-de la Parra, M. Bioavailability of Two Oral-Suspension Formulations of a Single Dose of Nitazoxanide 500 mg: An Open-Label, Randomized-Sequence, Two-Period Crossover, Comparison in Healthy Fasted Mexican Adult Volunteers. J. Bioequiv. Bioavailab. 2011, 3. [Google Scholar] [CrossRef]
- Jin, F.; Kirby, B.; Gao, Y.; Kearney, B.; Mathias, A. Population Pharmacokinetic Modeling of Sofosbuvir, an NS5B Polymerase Inhibitor, and Its Metabolites in Patients with Hepatitis C Virus Infection, Poster presentation at PAGE-Meeting. 2015. Hersonissos, Crete, Greece. Available online: https://www.page-meeting.org/pdf_assets/3129-PAGE%20poster.pdf (accessed on 1 July 2022).
- Rogosnitzky, M.; Okediji, P.; Koman, I. Cepharanthine: A review of the antiviral potential of a Japanese-approved alopecia drug in COVID-19. Pharmacol. Rep. 2020, 72, 1509–1516. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Repurposed Drug/Experimental Compound | Mechanism of Action; Target | Activity In Vitro; Potency EC50 or IC50 < 10 µM | Activity Ex Vivo Human Airway Epithelia (HAE) | Exposure in Human at Clinically Relevant Dose and Matching Doses in Hamster a | Activity and Exposure in Hamster Infection Model of SARS-CoV-2 | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Vero Cells | Calu-3 Cells | A549 Cells | Cmax Human Above IC50 at Clinically Relevant Dose | Cmax Hamster Above IC50 for Dose Matching Cmax in Human | Cmax Hamster Above IC50 for Dose Matching AUC in Human | Activity in SARS-CoV-2 Hamster Model | Dose(s) per Occasion, Frequency, Duration | Cmax Hamster Above IC50 b | Cmin Hamster Above IC50 for >24 h b | |||
| Direct Acting Antivirals (DAA) | ||||||||||||
| Atazanavir (ritonavir-boosted) | HIV protease inhibitors | No | No | No | No | No | No (24 mg/kg) | No (72 mg/kg) | No | 48 mg/kg (16 mg/kg ritonavir), BID, 3–4 days | No (48/16 ritonavir mg/kg, BID) | No (48/16 mg/kg ritonavir, BID) |
| Bemnifosbuvir (AT-527/AT-511) (experimental cpd) c | RdRp (guanosine nucleotide analogue) | No | No | No | No | Yes | ND | No d | 150–250 mg/kg, BID, 3 days | No (250 mg/kg, BID) | No (250 mg/kg, BID) | |
| Daclatasvir | HCV NS5A inhibitor (polymerase inhibitor) | Yes Pgp; T | No | No T | No | No | No (10 mg/kg) | No (15 mg/kg) | No | 25 mg/kg, BID, 3 days | No (25 mg/kg, BID) | No (25 mg/kg, BID) |
| Favipiravir | RNA-dependent RNA polymerase (RdRp), | No | No | No | No | No | Yes (25 mg/kg) | Yes (25 mg/kg) | Yes | 300 mg/kg, BID, 4 days; 462.5 mg/kg, BID, 3 days | Yes (300 mg/kg, BID) | No (300 mg/kg, BID) |
| Molnupiravir and metabolite EIDD-1931, experimental cpd/Merck) | RdRp (hydroxy-cytidine nucleotide analogue) | Yes Pgp | Yes | Yes | Yes (10 µM) | Yes | Yes (50 mg/kg) | Yes (150 mg/kg) | Yes | 75–200 mg/kg, BID, 3–4 days | Yes (150 mg/kg BID) | No (150 mg/kg BID) |
| Nelfinavir | HIV protease inhibitors | Yes T | Yes (borderline, 13 µM) | Yes T | Yes | No | No (70 mg/kg) | No (100 mg/kg) | No e | 100 mg/kg, QD, cepharanthine boosted (50 mg/kg, BID) | No (100 mg/kg) | No (100 mg/kg) |
| Nirmatrelvir (experimental cpd/Pfizer) [35] | Mpro inhibitor | Yes | Yes | Yes | Yes (1 µM) | ND | Yes | 125–250 mg/kg, BID, 4 days | Yes (125 mg/kg, BID) | Yes (125 mg/kg, BID) | ||
| Sofosbuvir | HCV NS5B inhibitor (polymerase inhibitor) | No | No | No | No | No | No (>200 mg/kg) | No (>200 mg/kg) | No | 100 mg/kg, QD, 3 days | No (100 mg/kg) | No (100 mg/kg) |
| Other Mechanisms of Action—Indirect Acting Antivirals | ||||||||||||
| Ambroxol | Mucolytic/prevent virus to bind to ACE-2 receptor | No | No | No | No | No | No (30 mg/kg) | No (50 mg/kg) | No | 50 mg/kg, BID, 3 days | No (50 mg/kg BID) | No (50 mg/kg BID) |
| Amodiaquine f | Anti-malarial | Yes (~10 µM) | No | No | No | No | No (<5 mg/kg) | No (<5 mg/kg) | No (parent) | 50–100 mg/kg, QD, 4–5 days | No (100 mg/kg) | No (100 mg/kg) |
| Cepharanthine | Block virus entry/ ACE2 binding | Yes T | No | No | No | No | No (1 mg/kg) | No (1 mg/kg) | No | 100 mg/kg, QD, 4 days | No (100 mg/kg) | No (100 mg/kg) |
| Camostat mesylate | TMPRSS2 inhibitor | No | Yes | No | Yes (10 µM) | ND | No | 200 mg/kg, BID, 4 days | ND | |||
| Clofazimine | TB inhibitor/ accumulate in lungs | Yes T | Yes | Yes T | No | No | No (1 mg/kg) | No (1 mg/kg) | No | 25 mg/kg, QD, 4 days | Yes (25 mg/kg) | No (25 mg/kg) |
| Colchicine | Anti-inflammatory | Yes T | Yes T | Yes T | No | ND | NT | |||||
| Fluoxetine | SSRI (selective serotonin reuptake inhibitor | Yes T | No | No | No | No | No (10 mg/kg) | No (10 mg/kg) | No | 10–100 mg/kg, QD, 4 days | No (100 mg/kg) | No (100 mg/kg) |
| Fluvoxamine maleate | SSRI (selective serotonin reuptake inhibitor | No | No | No | No | No | No (12 mg/kg) | No (20 mg/kg) | No | 100 mg/kg, QD, 3 days; 200 mg, BID, 4 days | No g (100 mg/kg) | No g (100 mg/kg) |
| Ivermectin (oral) | Anti-parasitic drug, Anti-inflammatory | Yes T | Yes | Yes T | Yes T | No | No (0.1 mg/kg) | No (0.1 mg/kg) | NT | |||
| Ivermectin (s.c.) | No | No (0.1 mg/kg) | No (0.1 mg/kg) | No | 0.4 mg/kg, QD, 1 day or 4 days | No (0.4 mg/kg) | No (0.4 mg/kg) | |||||
| Mefloquine | Anti-malarial | Yes T | No | No | No | ND | NT | |||||
| Nitazoxanide (and metabolite tizoxanide) | Antiprotozoal agent (Giardia, Cryptosporidium infections) | Yes | Yes Pgp | No | Yes | No | No (25 mg/kg) | No (150 mg/kg) | No | 250 mg/kg, BID, 3–4 days | No (250 mg/kg BID) | No (250 mg/kg BID) |
| Pentoxyfilline | Vasodilatator; anti-inflammatory | No | No | No | No | ND | NT | |||||
| Probenecid | Anti-gout? | No | No | No | No | ND | NT | |||||
| Proxalutamide | Androgen receptor antagonist, Anti-inflammatory | No T | No | No T | No | ND | NT | |||||
| Mode of Action | Dose (mg/kg/day) | Efficacy In Vivo SARS-CoV-2 Hamster Model | Comments | |
|---|---|---|---|---|
| Atazanavir /ritonavir (ATZ/r) /Nitazoxanide | DAA/IAA combination | 96/32/500 mg/kg/day | No | |
| FAV/ATZ/r | DAAs combination | 600/96/32 mg/kg/day | Yes | No additive or synergistic activity as compared with FAV alone |
| FAV/Nitazoxanide | DAA/IAA combination | 600/500 mg/kg/day | Yes | No additive or synergistic activity as compared with FAV alone |
| Sofosbuvir/daclatasvir | DAAs combination | 100/100 mg/kg/day | No | |
| Nelfinavir/Cepharantine | DAA/IAA combination | 100/100 mg/kg/day | No | |
| Ivermectin/Amodiaquine | DAA (considered)/IAA combination | 0.4/50 mg/kg/day | No | |
| Molnupiravir/Clofazimine | DAA/IAA combination | 150/25 mg/kg/day | Yes | No additive or synergistic activity as compared with Molnupiravir alone |
| Molnupiravir/Nirmatrelvir | DAA/DAA combination | 150/250 mg/kg/day | Yes | No additive or synergistic activity as compared with nirmatrelvir alone Additive effect as compared with Molnupiravir alone |
| Cmax in Hamster Above Corrected IC50 | Activity in Hamster Infection Model of SARS-CoV-2 | Total | |
|---|---|---|---|
| Yes | No | ||
| Yes | TRUE POSITIVE (TP) n = 3 (favipiravir, molnupiravir, nirmatrelvir) | FALSE POSITIVE (FP) n = 1 (clofazimine) | 4 |
| No | FALSE NEGATIVE (FN) n = 0 | TRUE NEGATIVE (TN) n = 12 (atazanavir, bemnifosbuvir, daclatasvir, nelfinavir *, sofosbuvir, ambroxol, amodiaquine, cepharanthine, fluoxetine, fluvoxamine, ivermectin, nitazoxanide | 12 |
| Total | 3 | 13 | 16 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Assmus, F.; Driouich, J.-S.; Abdelnabi, R.; Vangeel, L.; Touret, F.; Adehin, A.; Chotsiri, P.; Cochin, M.; Foo, C.S.; Jochmans, D.; et al. Need for a Standardized Translational Drug Development Platform: Lessons Learned from the Repurposing of Drugs for COVID-19. Microorganisms 2022, 10, 1639. https://doi.org/10.3390/microorganisms10081639
Assmus F, Driouich J-S, Abdelnabi R, Vangeel L, Touret F, Adehin A, Chotsiri P, Cochin M, Foo CS, Jochmans D, et al. Need for a Standardized Translational Drug Development Platform: Lessons Learned from the Repurposing of Drugs for COVID-19. Microorganisms. 2022; 10(8):1639. https://doi.org/10.3390/microorganisms10081639
Chicago/Turabian StyleAssmus, Frauke, Jean-Sélim Driouich, Rana Abdelnabi, Laura Vangeel, Franck Touret, Ayorinde Adehin, Palang Chotsiri, Maxime Cochin, Caroline S. Foo, Dirk Jochmans, and et al. 2022. "Need for a Standardized Translational Drug Development Platform: Lessons Learned from the Repurposing of Drugs for COVID-19" Microorganisms 10, no. 8: 1639. https://doi.org/10.3390/microorganisms10081639
APA StyleAssmus, F., Driouich, J.-S., Abdelnabi, R., Vangeel, L., Touret, F., Adehin, A., Chotsiri, P., Cochin, M., Foo, C. S., Jochmans, D., Kim, S., Luciani, L., Moureau, G., Park, S., Pétit, P.-R., Shum, D., Wattanakul, T., Weynand, B., Fraisse, L., ... Chatelain, E. (2022). Need for a Standardized Translational Drug Development Platform: Lessons Learned from the Repurposing of Drugs for COVID-19. Microorganisms, 10(8), 1639. https://doi.org/10.3390/microorganisms10081639