
The Microbial Nitrogen Cycling, Bacterial Community Composition, and Functional Potential in a Natural Grassland Are Stable from Breaking Dormancy to Being Dormant Again

 , ,
, ,  and
and 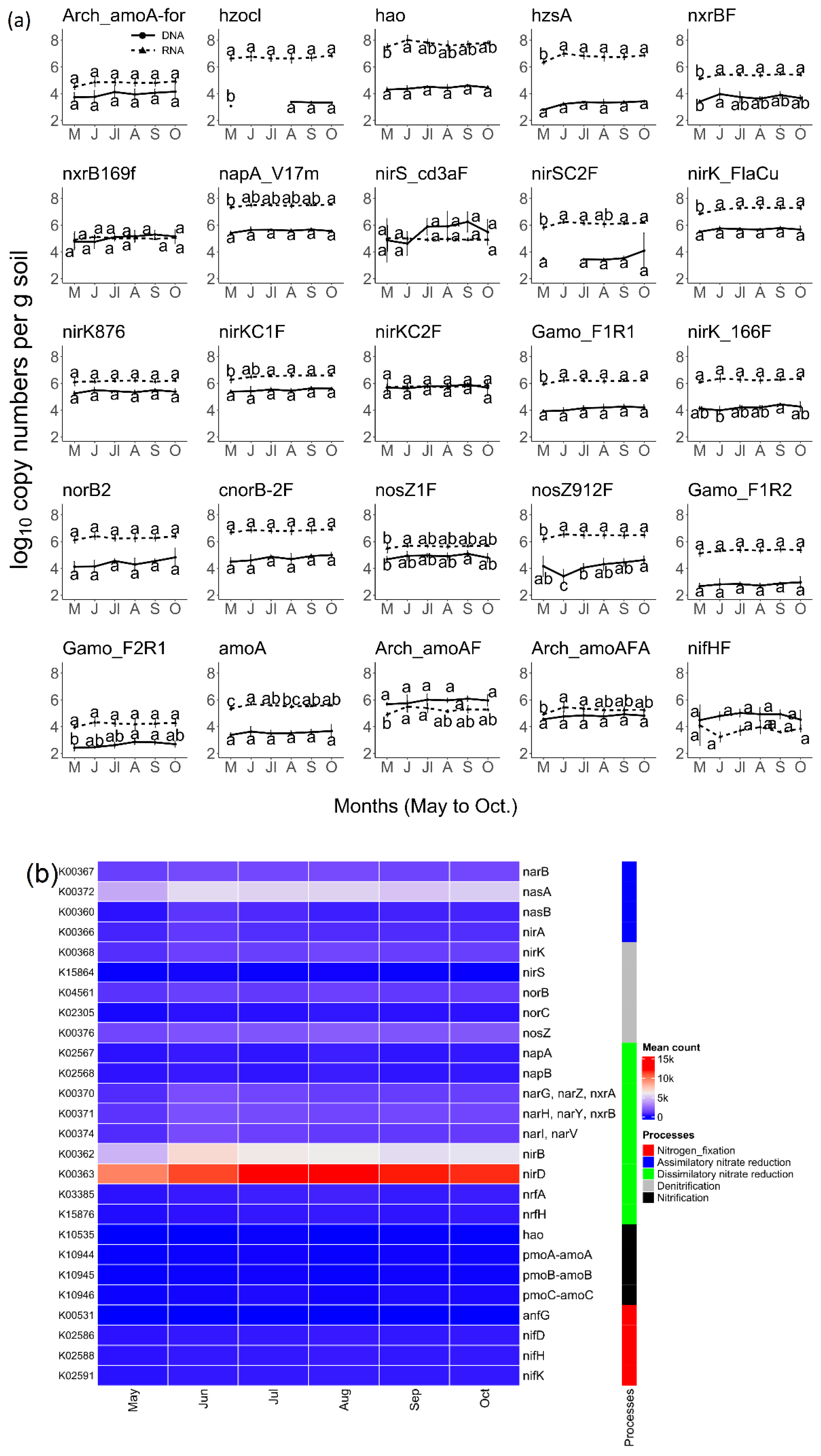{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Sites
2.2. Extraction of Nucleic Acids
2.3. cDNA Preparation
2.4. Standards Preparation for the NiCE Chip
2.5. Specific Target Amplification for the NiCE Chip
2.6. NiCE Chip Amplicon Quantification
2.7. 16S rRNA Gene Sequence-Based Community Profiling
2.8. Data Analysis
3. Results
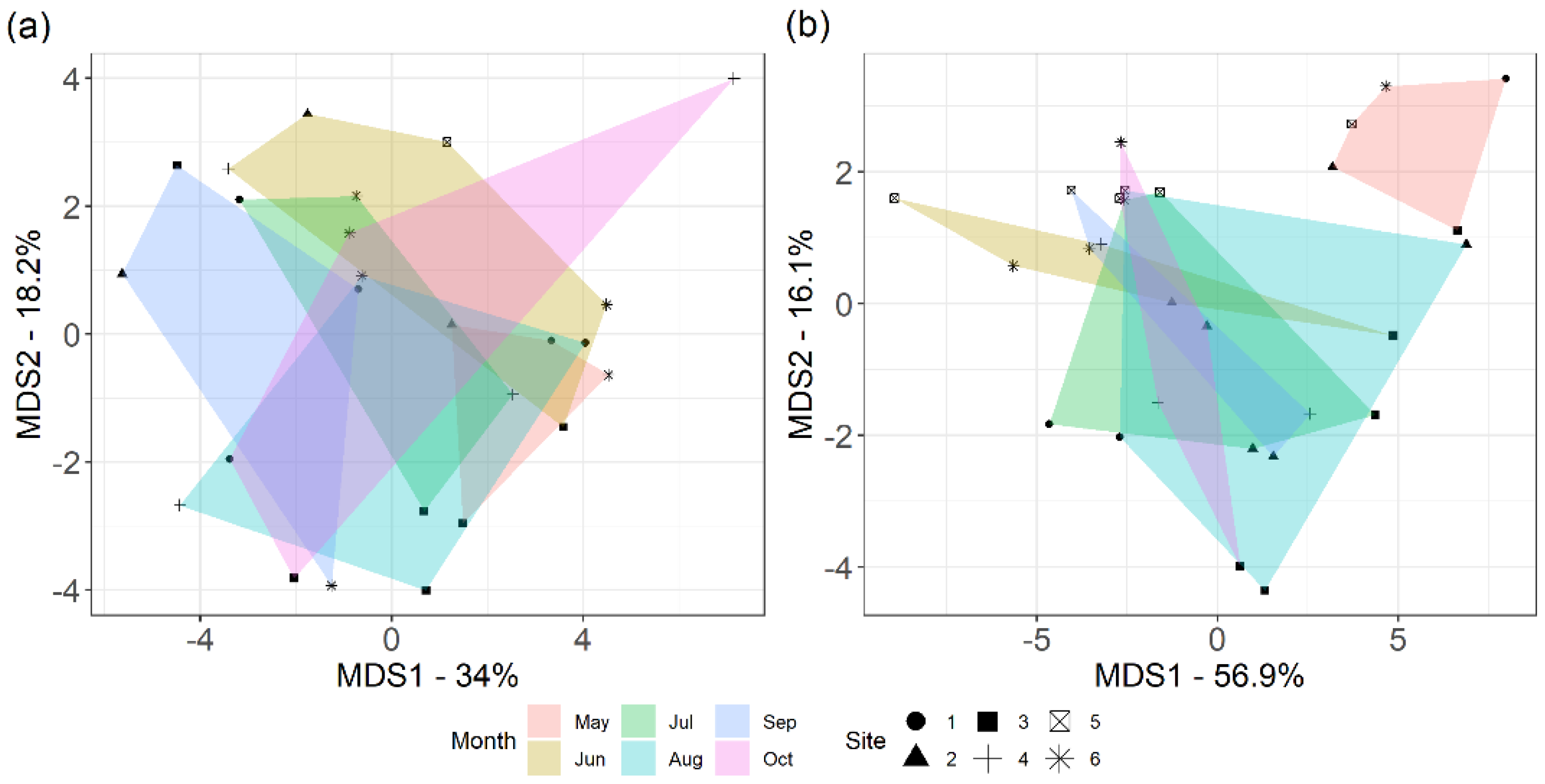
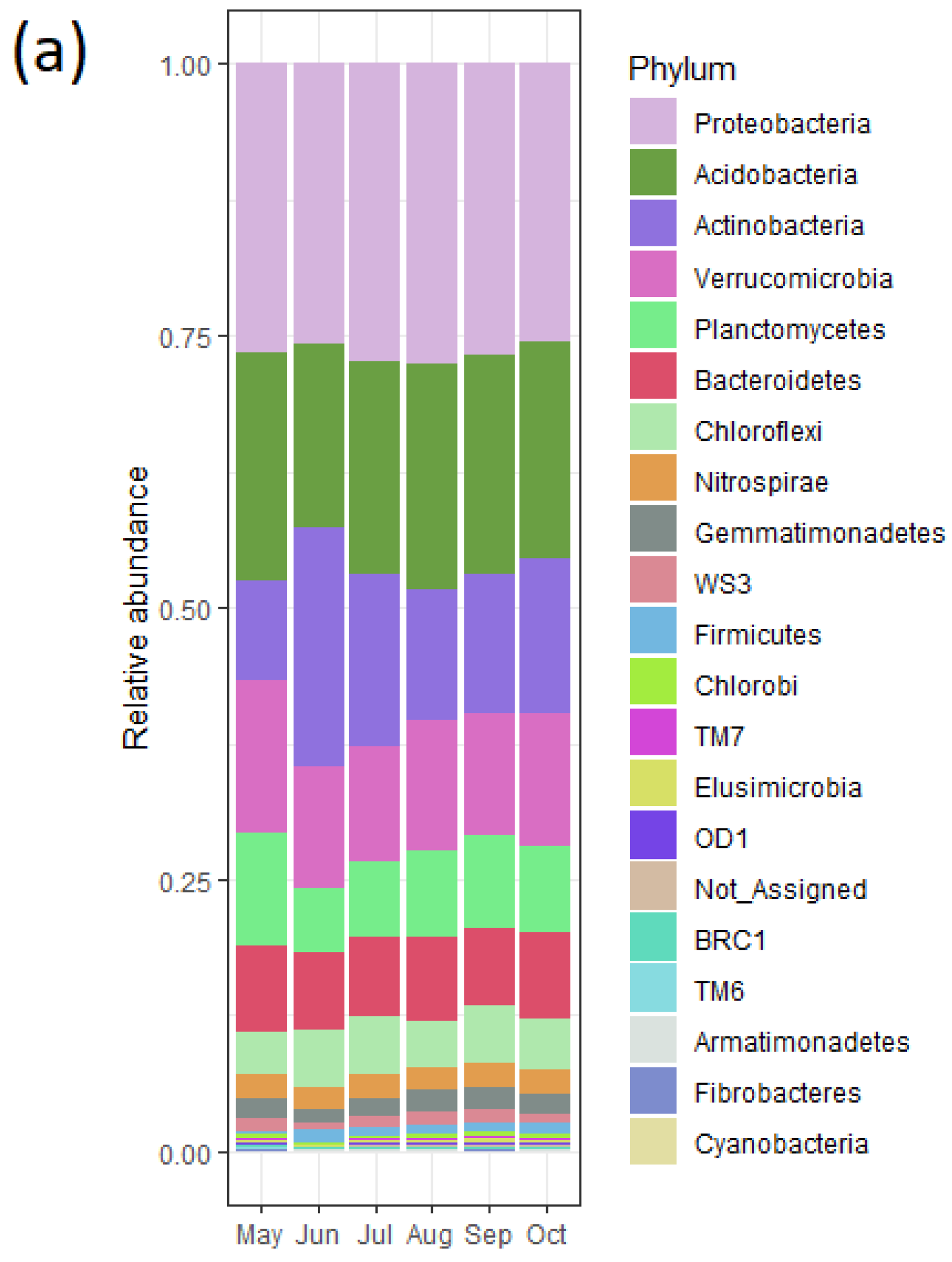
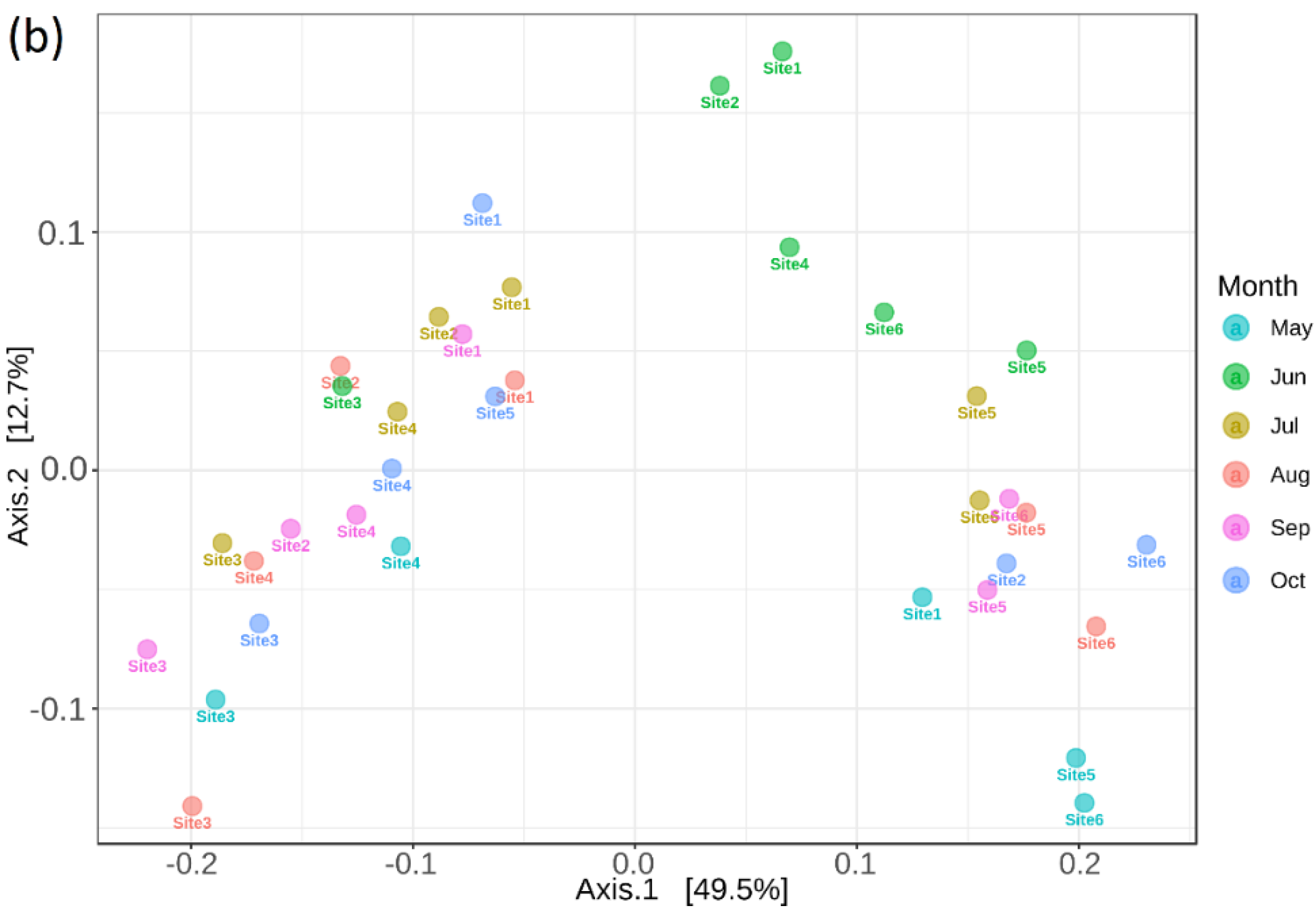
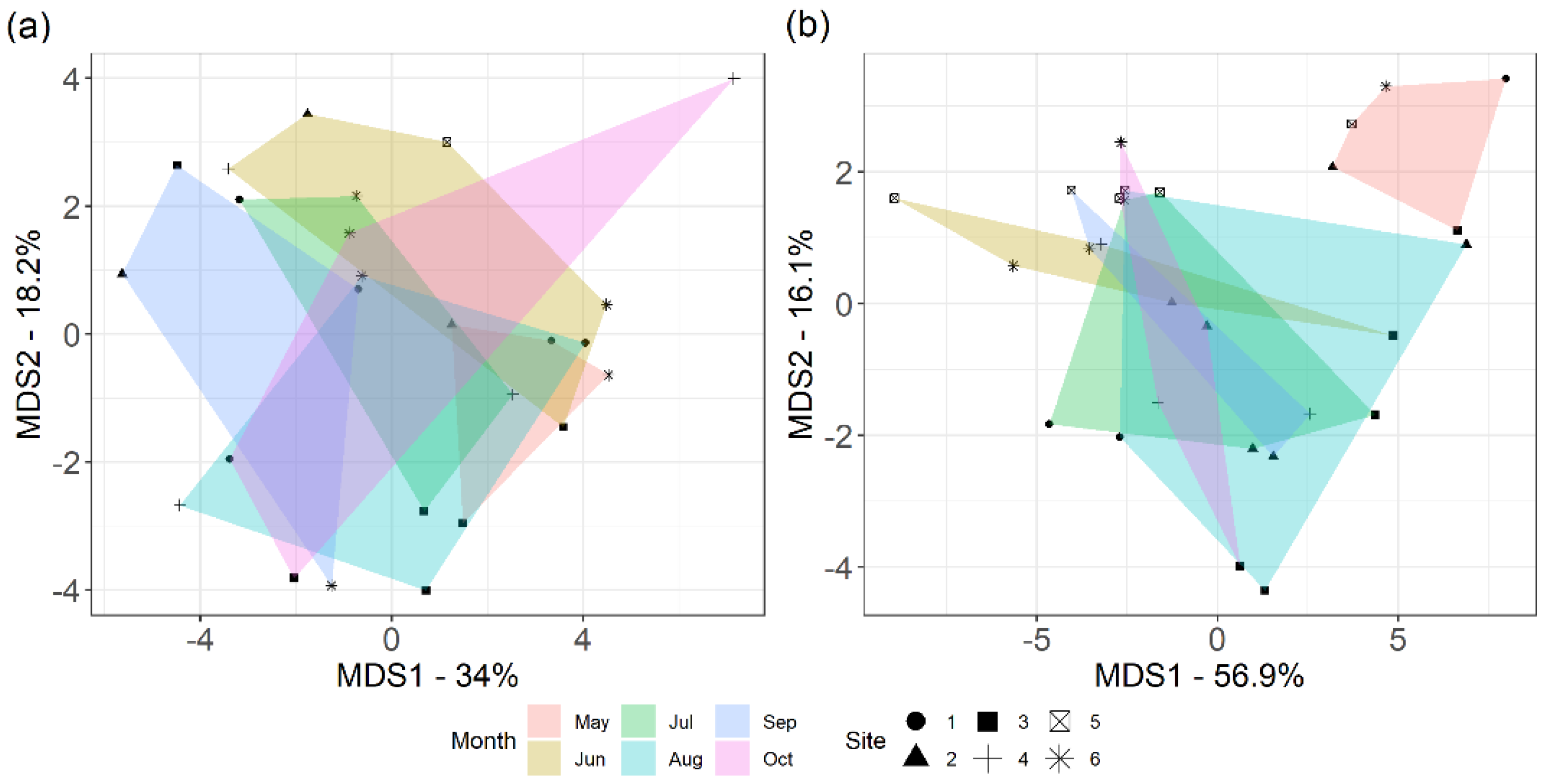
3.1. Heterogeneity of Prairie
3.2. Prairie N Cycle Functional Genes and Transcripts across the Growing Season
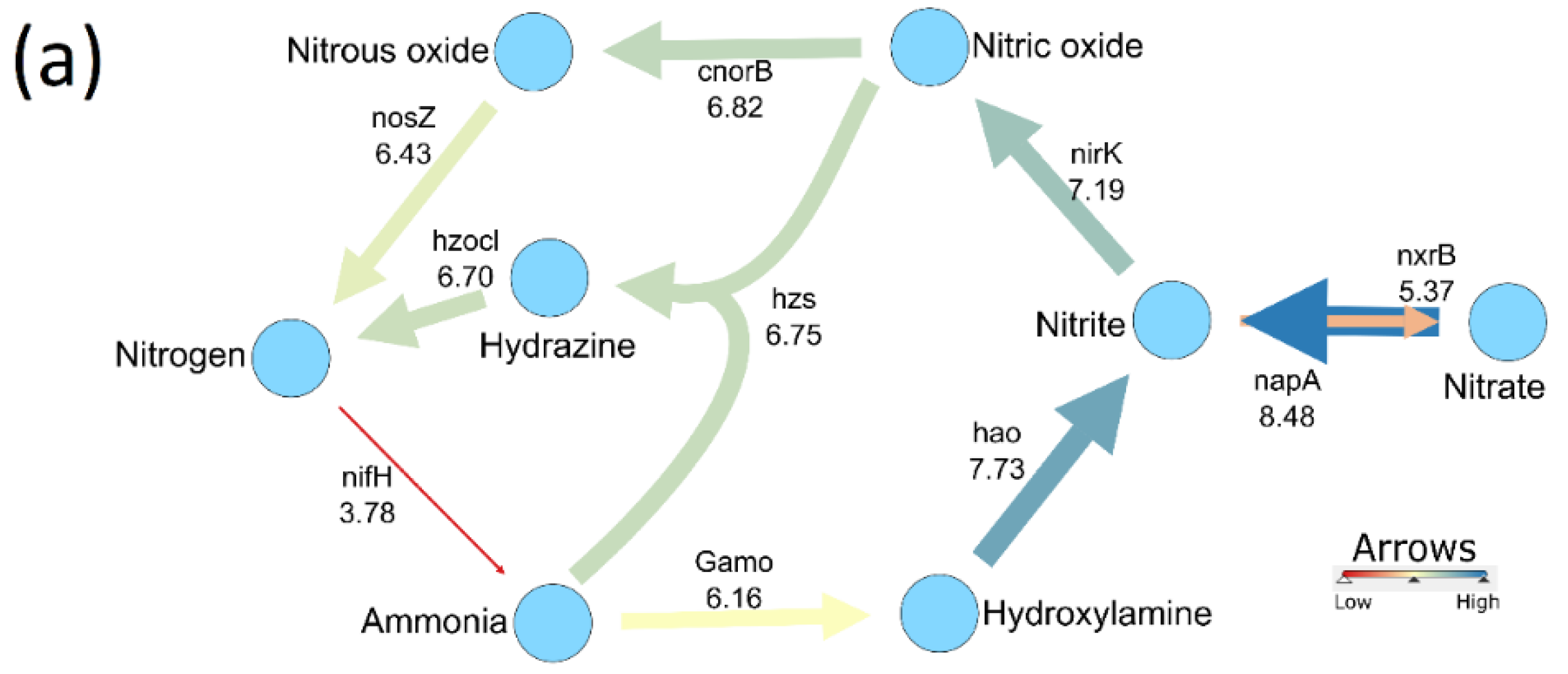
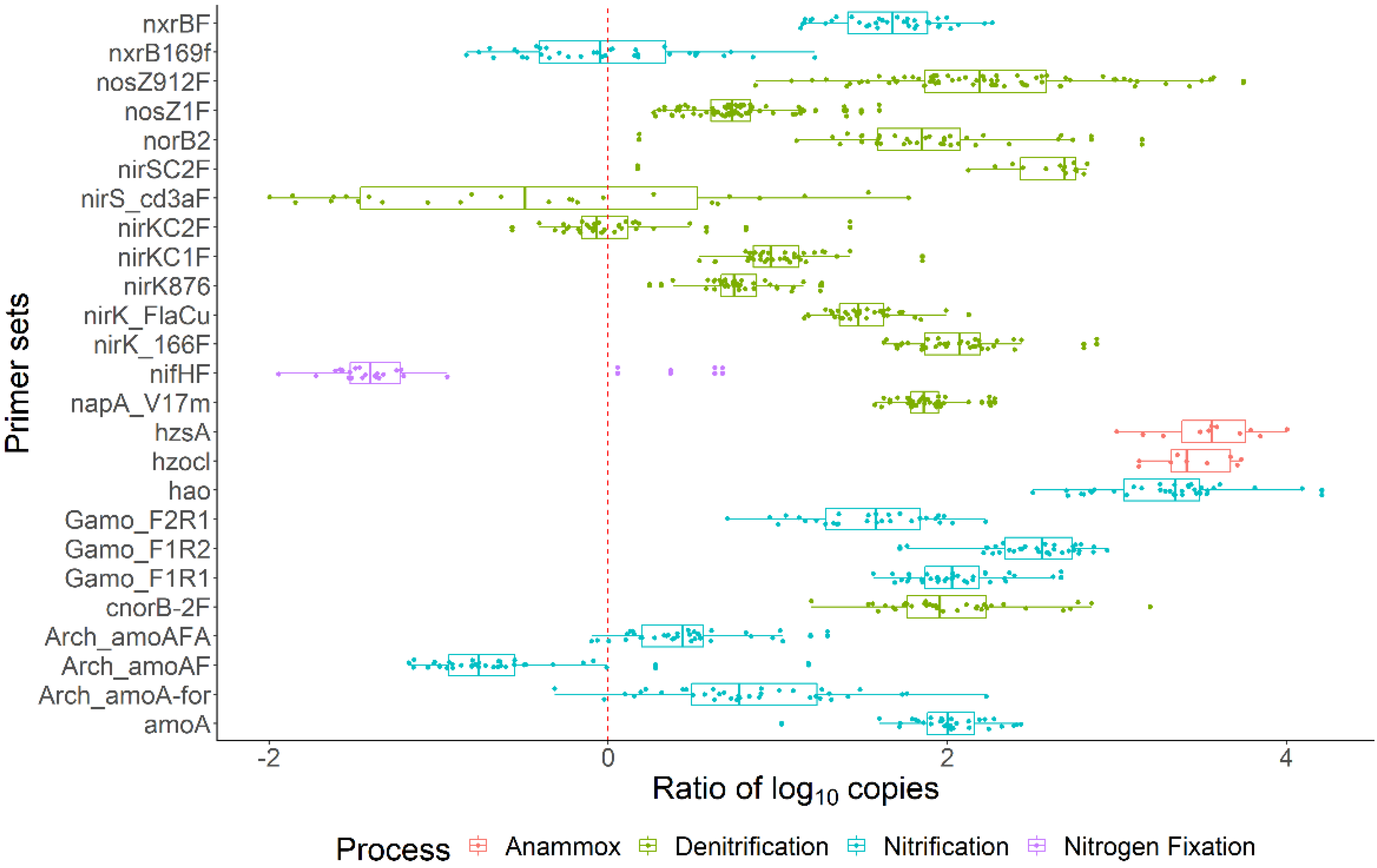
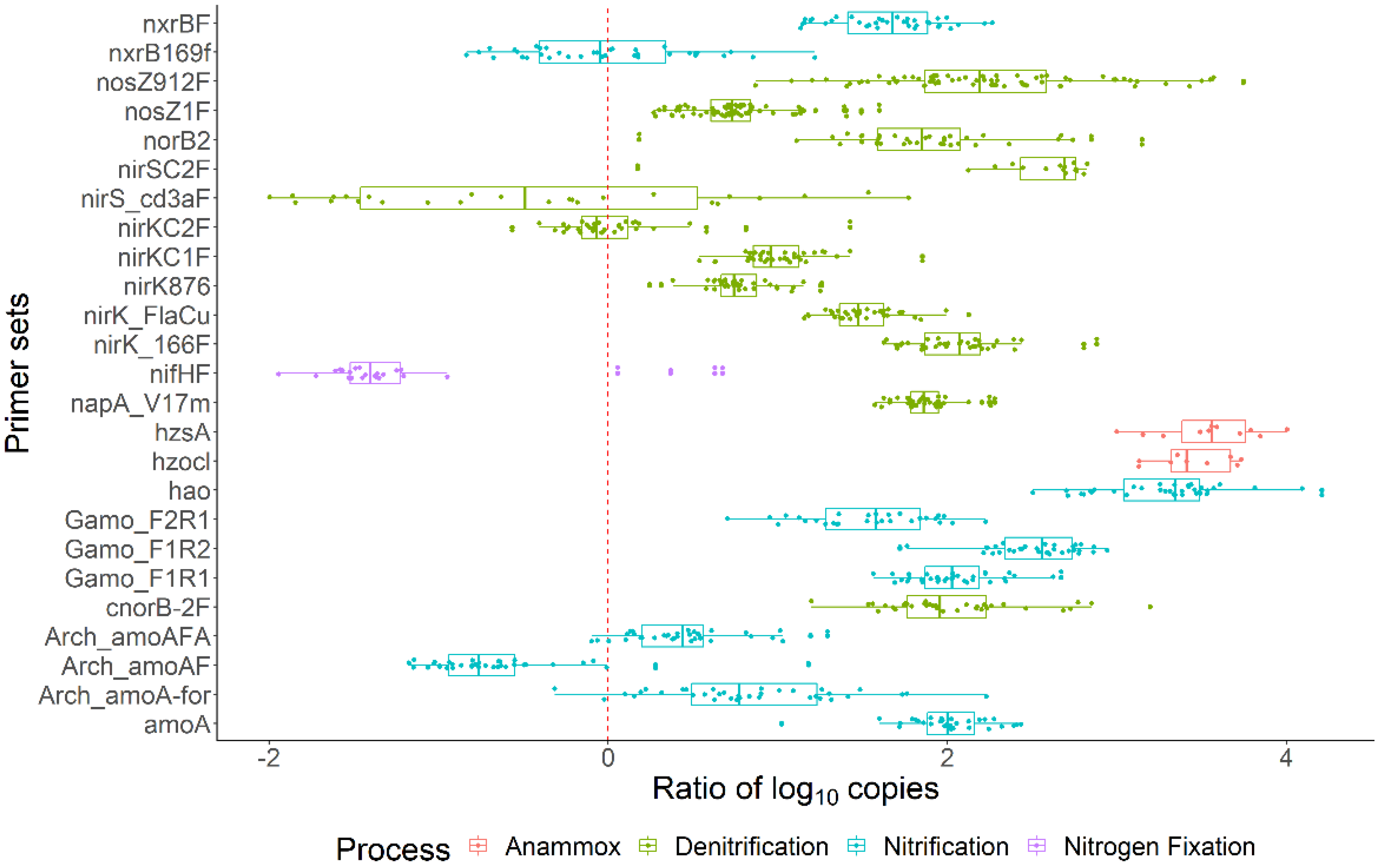
3.3. Bacterial Activity in the Nitrogen Cycle
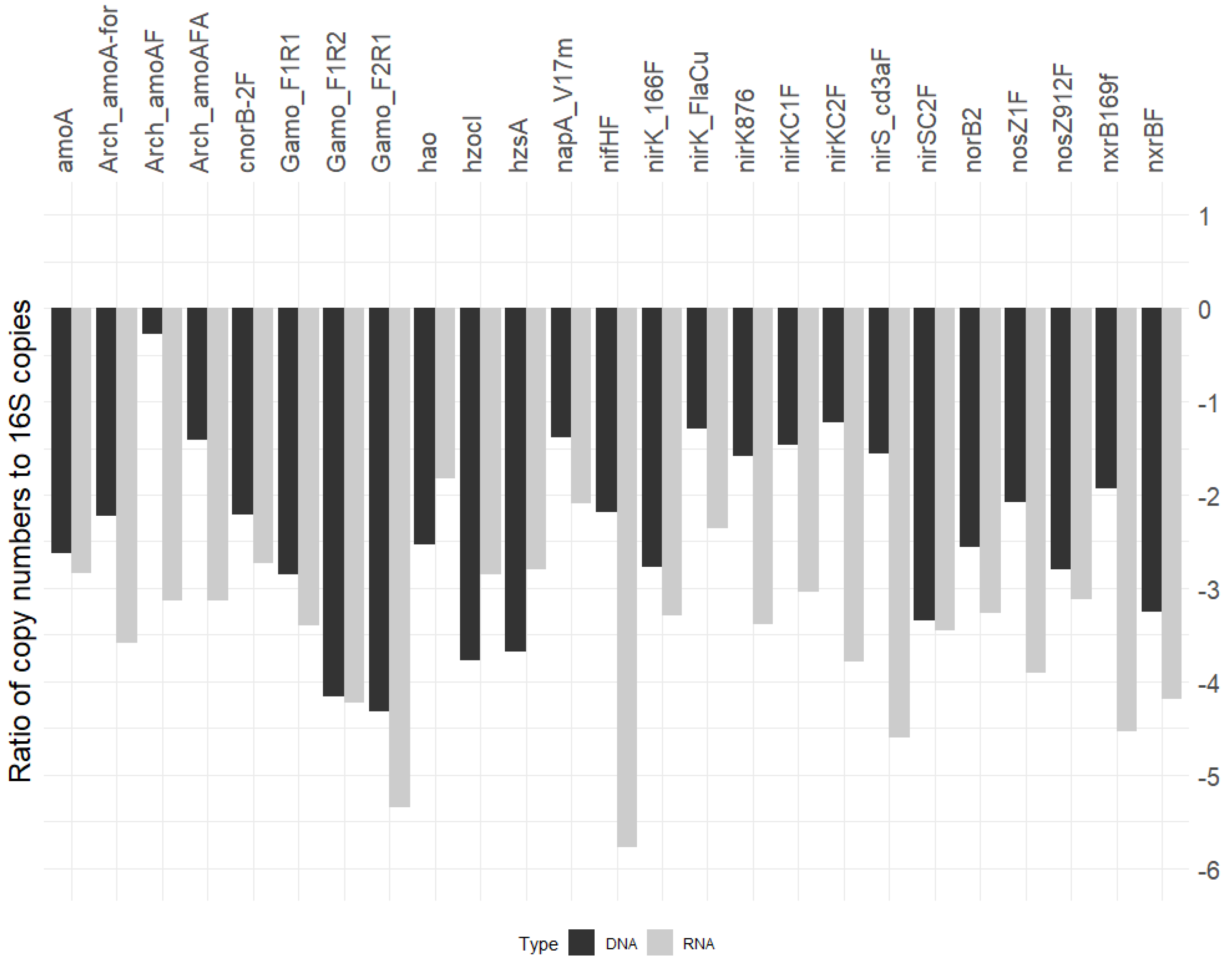
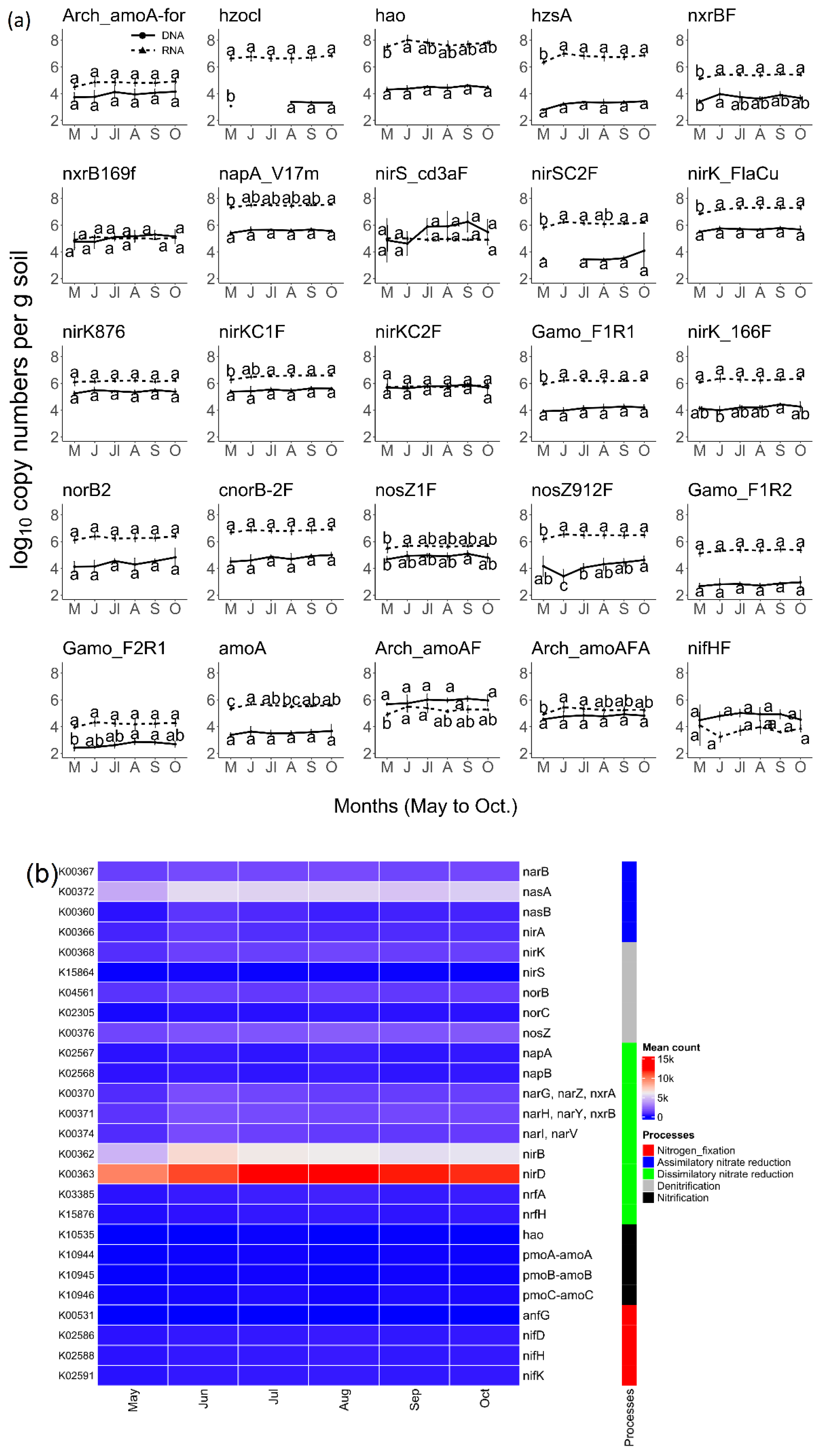
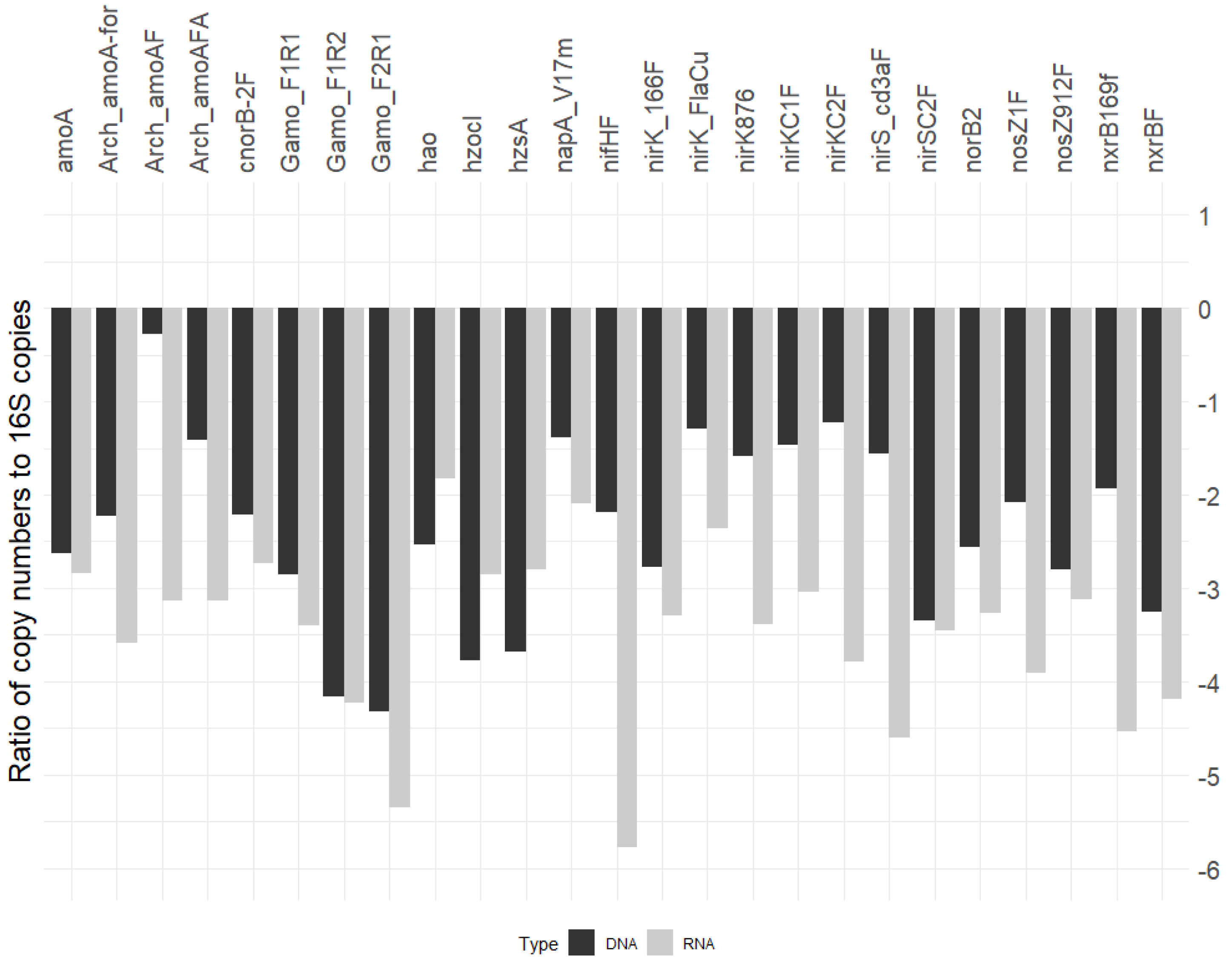
3.4. N-Cycle Gene Copy and Transcript Numbers Are Both Similar across Seasons
3.5. N-Cycle Genes Are Strongly Transcribed except nifH
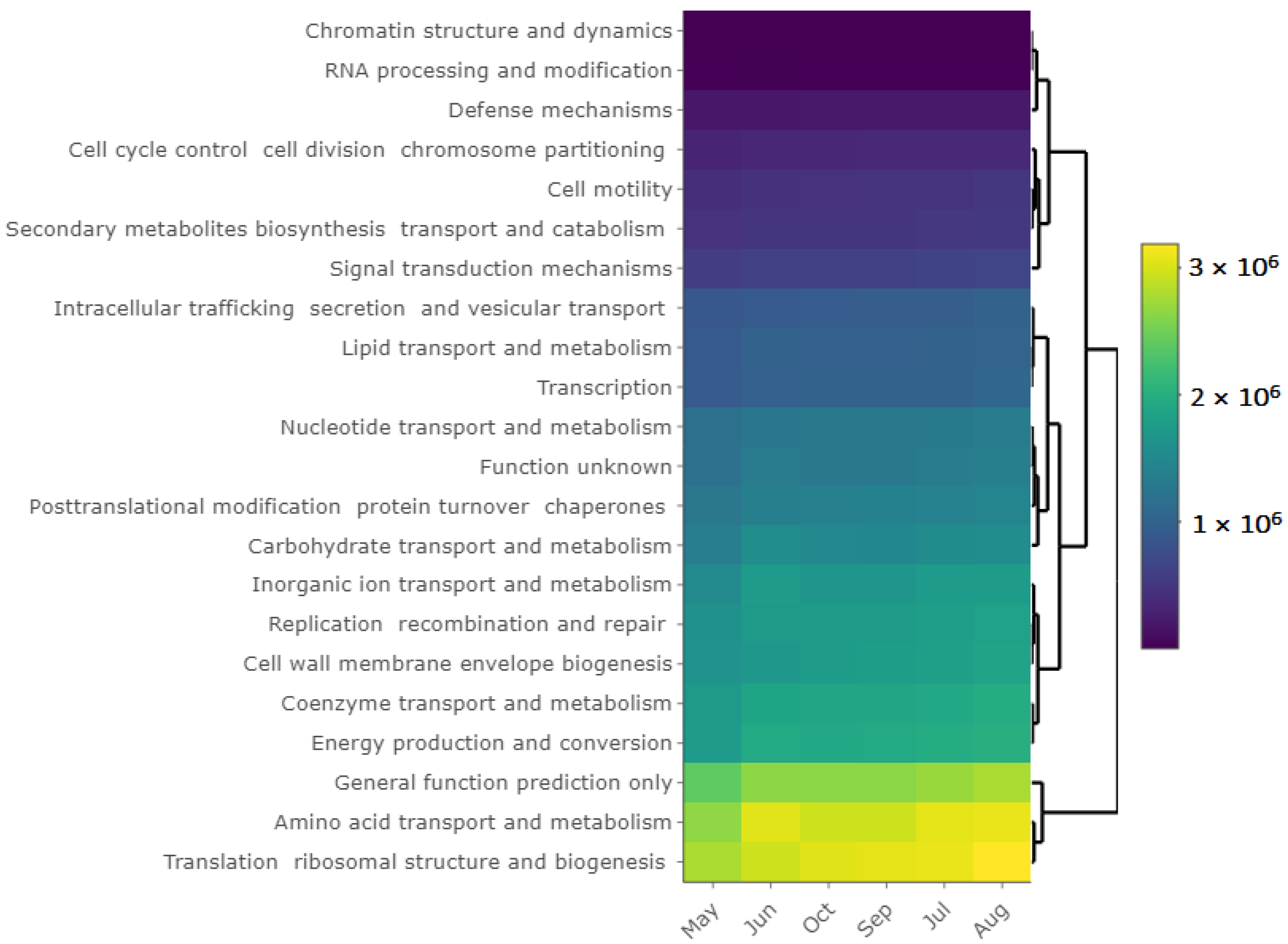
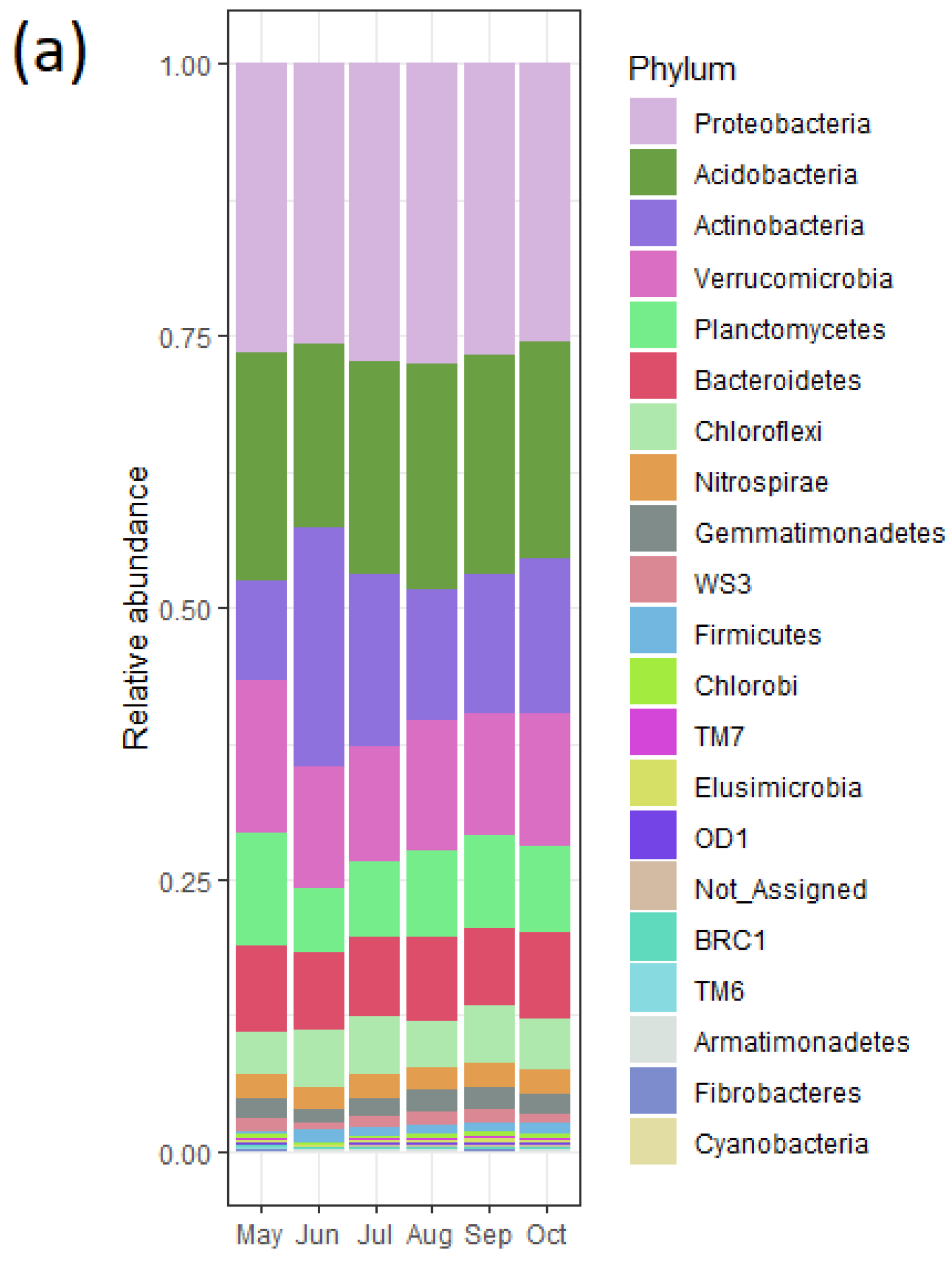
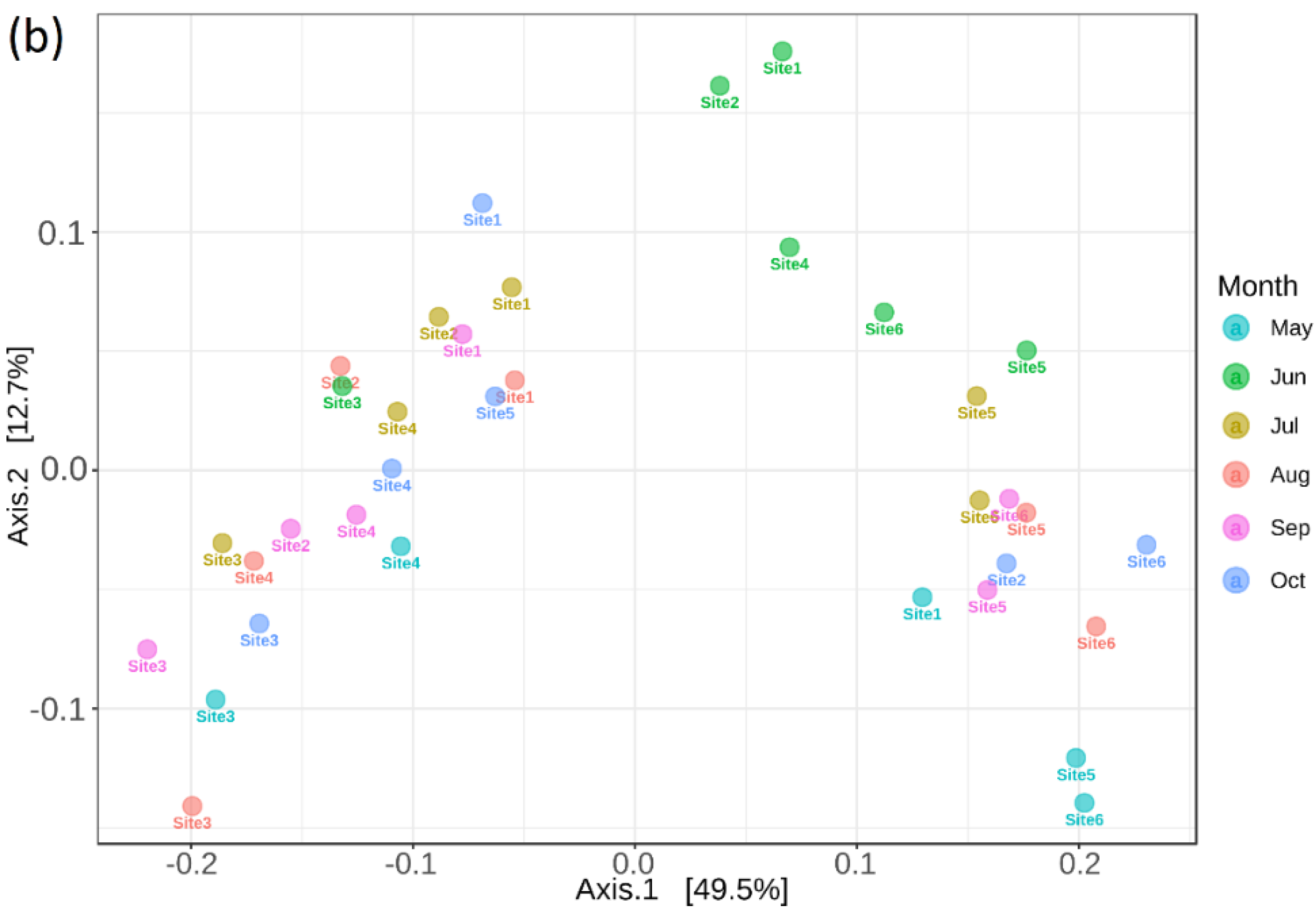
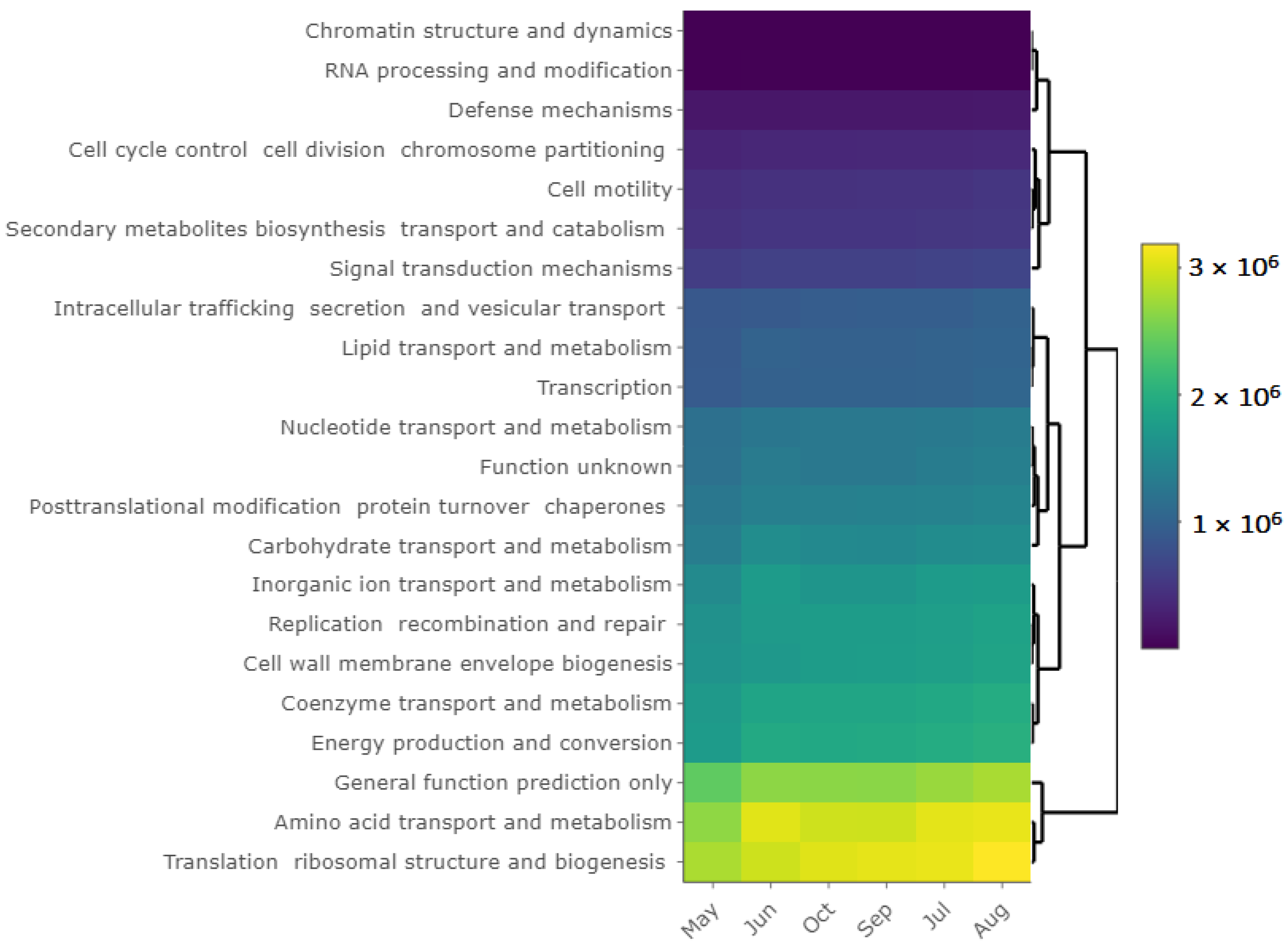
3.6. Bacterial Community and Functional Potential Were Unaffected by the Seasons
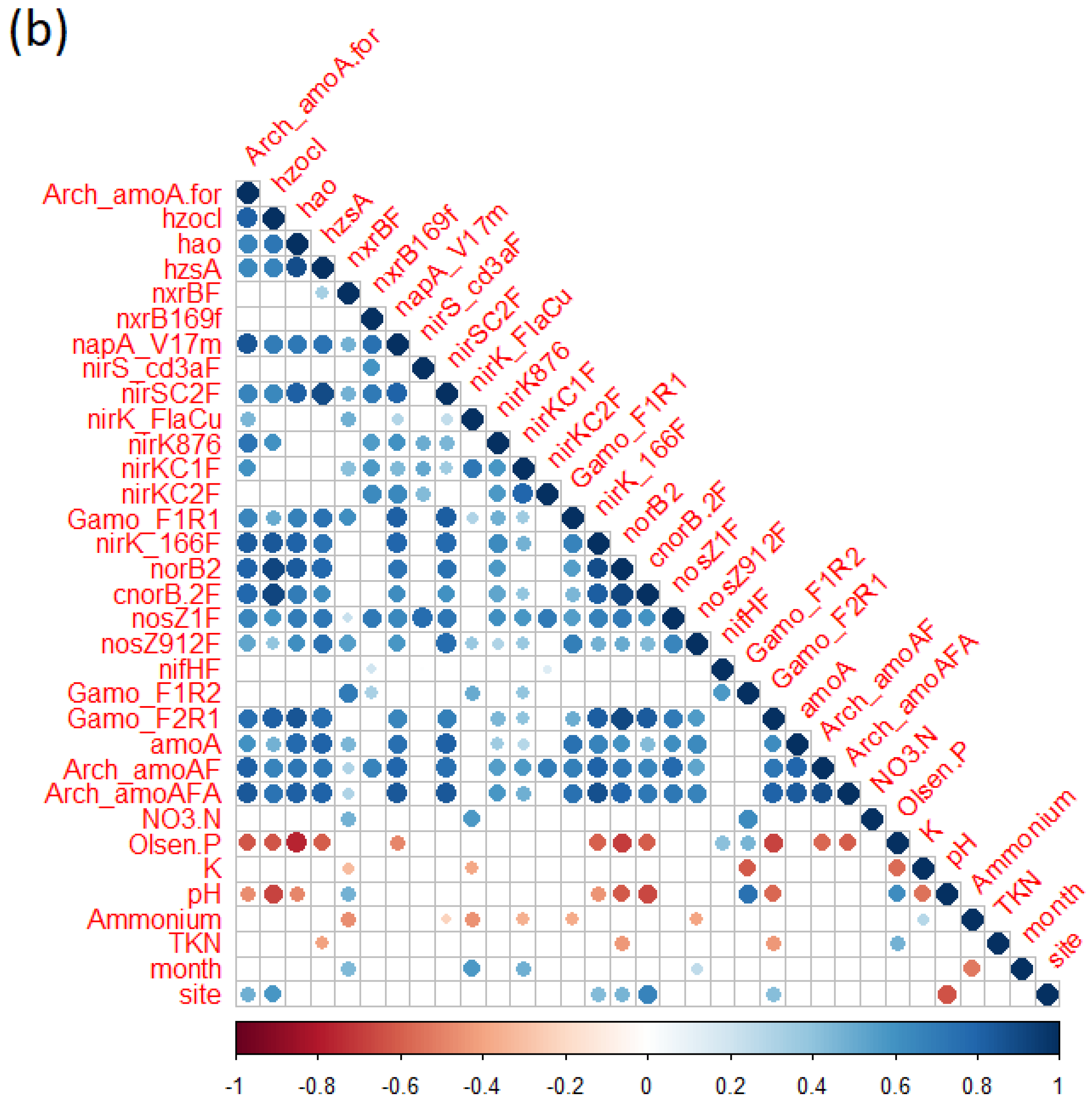
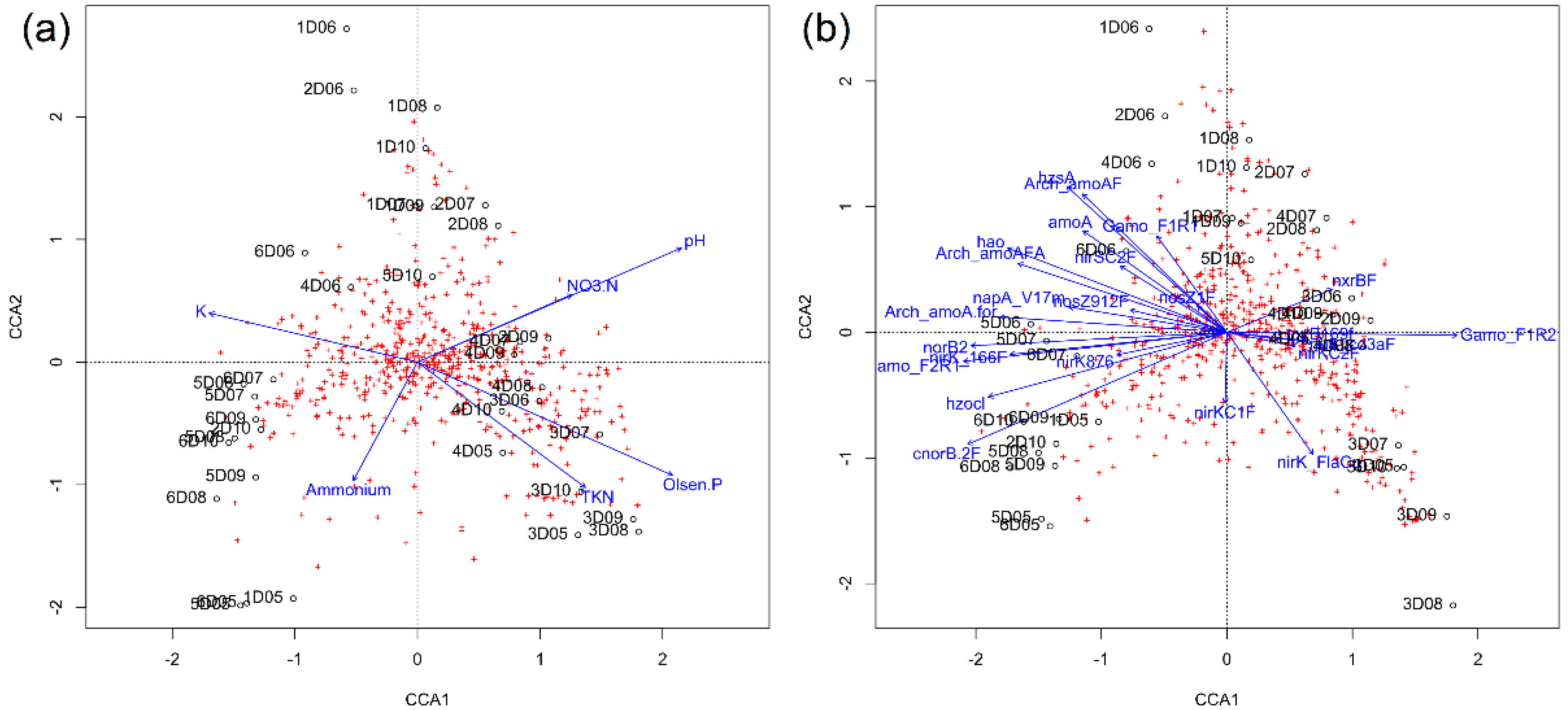
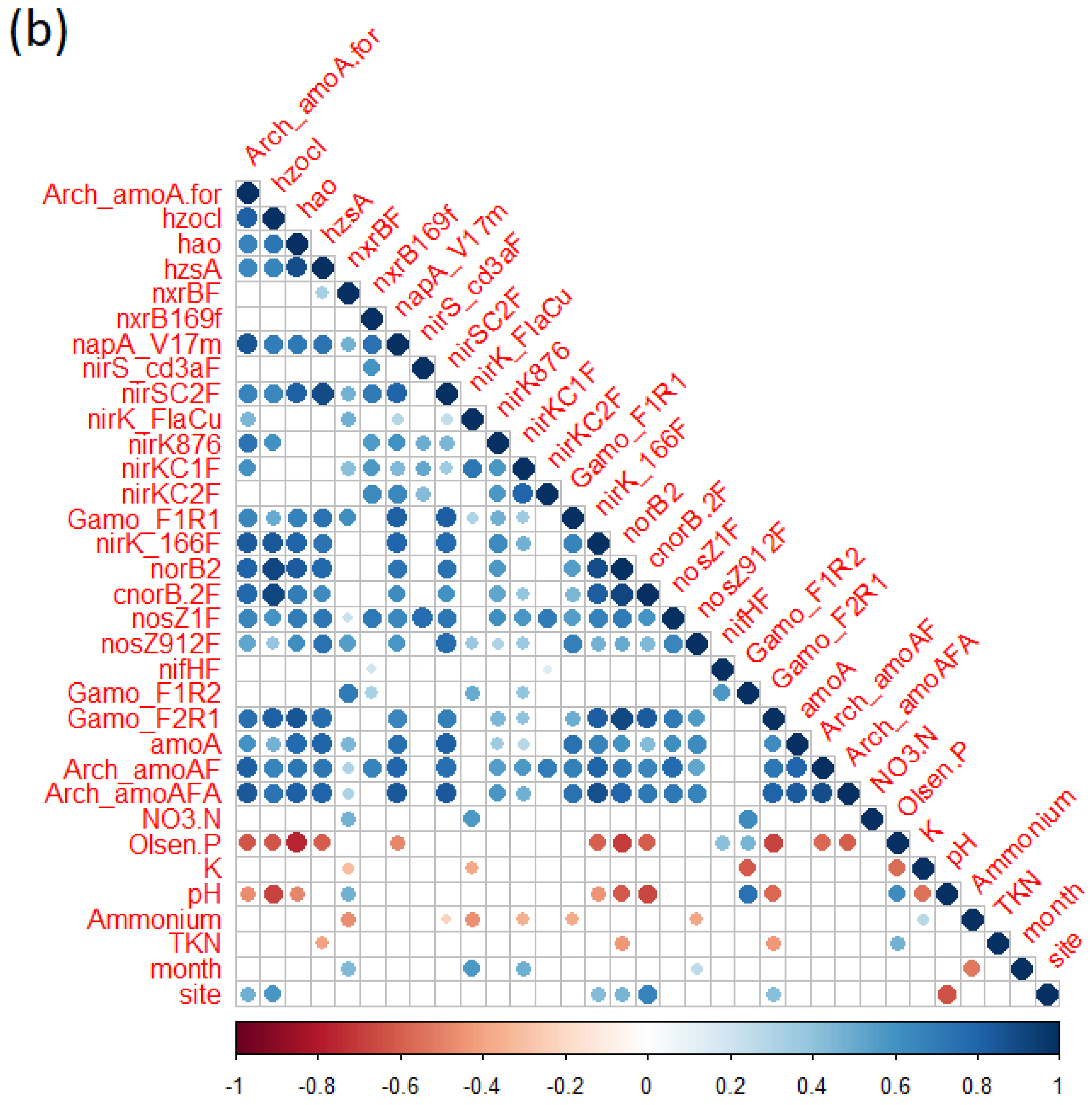
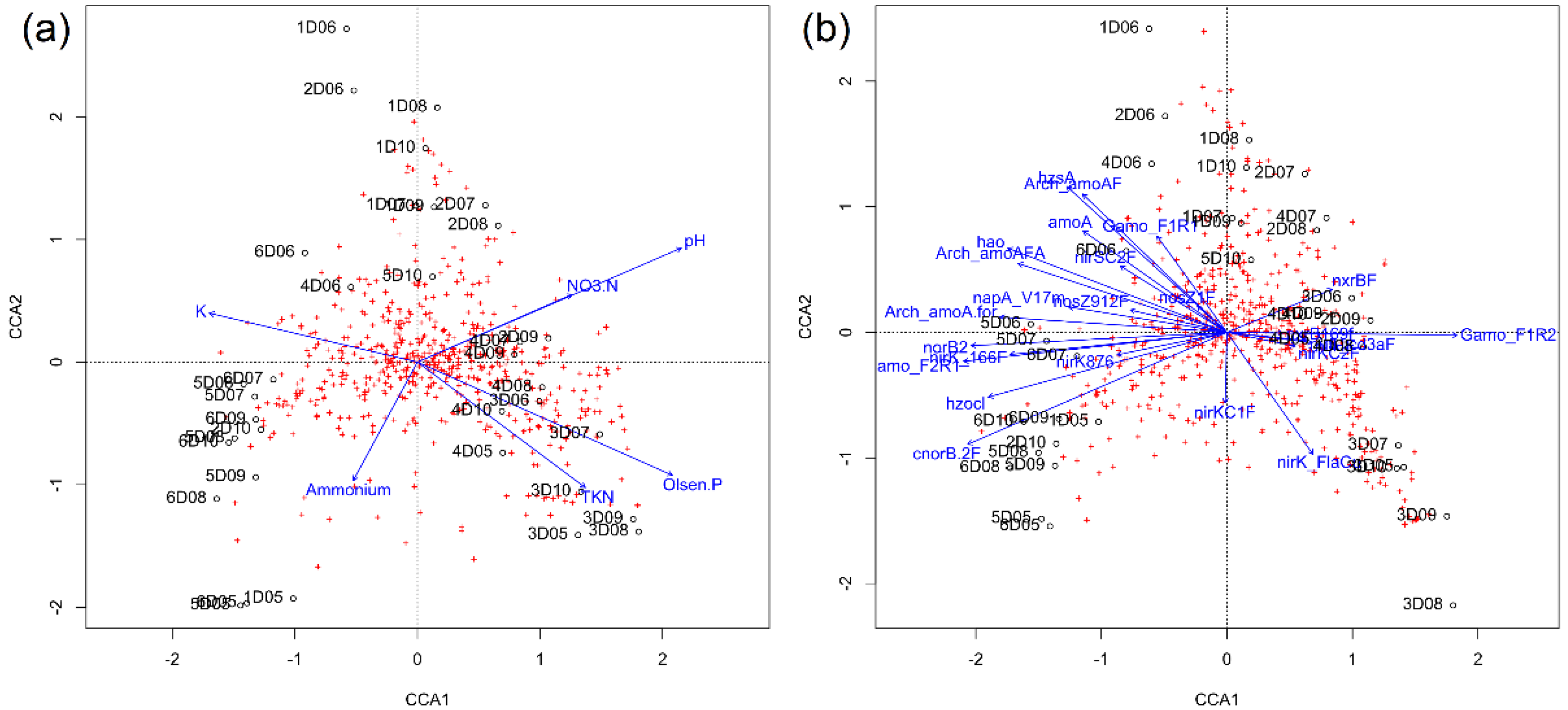
3.7. Bacterial Diversity, N Cycle Gene Expression, and Soil Chemistry
4. Discussion
4.1. N Cycle Genes and Transcript Quantities in a Heterogenous Grassland Are Stable
4.2. Nitrogen Cycle Genes and Soil Physio-Chemical Characteristics Are Strongly Correlated
4.3. Bacterial Communities and Functional Capabilities Are Synchronously Stable
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Monti, A.; Zegada-Lizarazu, W.; Zanetti, F.; Casler, M. Nitrogen Fertilization Management of Switchgrass, Miscanthus and Giant Reed: A Review. Adv. Agron. 2019, 153, 87. [Google Scholar]
- Kaiser, K.; Wemheuer, B.; Korolkow, V.; Wemheuer, F.; Nacke, H.; Schöning, I.; Schrumpf, M.; Daniel, R. Driving forces of soil bacterial community structure, diversity, and function in temperate grasslands and forests. Sci. Rep. 2016, 6, 33696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Samson, F.; Knopf, F. Prairie conservation in north america. BioScience 1994, 44, 418–421. [Google Scholar] [CrossRef] [Green Version]
- Wagle, P.; Gowda, P.H. Tallgrass prairie responses to management practices and disturbances: A review. Agronomy 2018, 8, 300. [Google Scholar] [CrossRef]
- Schnitzer, S.A.; Klironomos, J.N.; HilleRisLambers, J.; Kinkel, L.L.; Reich, P.B.; Xiao, K.; Rillig, M.C.; Sikes, B.A.; Callaway, R.M.; Mangan, S.A.; et al. Soil microbes drive the classic plant diversity-productivity pattern. Ecology 2011, 92, 296–303. [Google Scholar] [CrossRef]
- Entry, J.A.; Mills, D.; Mathee, K.; Jayachandran, K.; Sojka, R.; Narasimhan, G. Influence of irrigated agriculture on soil microbial diversity. Appl. Soil Ecol. 2008, 40, 146–154. [Google Scholar] [CrossRef]
- Tsiafouli, M.A.; Thébault, E.; Sgardelis, S.P.; De Ruiter, P.C.; Van Der Putten, W.H.; Birkhofer, K.; Hemerik, L.; De Vries, F.T.; Bardgett, R.D.; Brady, M.V. Intensive agriculture reduces soil biodiversity across Europe. Glob. Chang. Biol. 2015, 21, 973–985. [Google Scholar] [CrossRef]
- Black, H.; Parekh, N.; Chaplow, J.; Monson, F.; Watkins, J.; Creamer, R.; Potter, E.; Poskitt, J.; Rowland, P.; Ainsworth, G. Assessing soil biodiversity across Great Britain: National trends in the occurrence of heterotrophic bacteria and invertebrates in soil. J. Environ. Manag. 2003, 67, 255–266. [Google Scholar] [CrossRef] [Green Version]
- Haichar, F.E.; Marol, C.; Berge, O.; Rangel-Castro, J.I.; Prosser, J.I.; Balesdent, J.; Heulin, T.; Achouak, W. Plant host habitat and root exudates shape soil bacterial community structure. ISME J. 2008, 2, 1221–1230. [Google Scholar] [CrossRef]
- Coskun, D.; Britto, D.T.; Shi, W.; Kronzucker, H.J. How plant root exudates shape the nitrogen cycle. Trends Plant Sci. 2017, 22, 661–673. [Google Scholar] [CrossRef]
- Zhao, Q.; Thompson, A.M.; Callister, S.J.; Tfaily, M.M.; Bell, S.L.; Hobbie, S.E.; Hofmockel, K.S. Dynamics of organic matter molecular composition under aerobic decomposition and their response to the nitrogen addition in grassland soils. Sci. Total Environ. 2022, 806, 150514. [Google Scholar] [CrossRef] [PubMed]
- Tavi, N.M.; Martikainen, P.J.; Lokko, K.; Kontro, M.; Wild, B.; Richter, A.; Biasi, C. Linking microbial community structure and allocation of plant-derived carbon in an organic agricultural soil using (CO2)-C-13 pulse-chase labelling combined with C-13-PLFA profiling. Soil Biol. Biochem. 2013, 58, 207–215. [Google Scholar] [CrossRef]
- Shen, X.; Yang, F.; Xiao, C.; Zhou, Y. Increased contribution of root exudates to soil carbon input during grassland degradation. Soil Biol. Biochem. 2020, 146, 107817. [Google Scholar] [CrossRef]
- Preece, C.; Peñuelas, J. A return to the wild: Root exudates and food security. Trends Plant Sci. 2020, 25, 14–21. [Google Scholar] [CrossRef]
- Farrell, M.; Hill, P.W.; Farrar, J.; Bardgett, R.D.; Jones, D.L. Seasonal variation in soluble soil carbon and nitrogen across a grassland productivity gradient. Soil Biol. Biochem. 2011, 43, 835–844. [Google Scholar] [CrossRef]
- Gransee, A.; Wittenmayer, L. Qualitative and quantitative analysis of water-soluble root exudates in relation to plant species and development. J. Plant Nutr. Soil Sci. 2000, 163, 381–385. [Google Scholar] [CrossRef]
- Hu, L.; Robert, C.A.; Cadot, S.; Zhang, X.; Ye, M.; Li, B.; Manzo, D.; Chervet, N.; Steinger, T.; Van Der Heijden, M.G. Root exudate metabolites drive plant-soil feedbacks on growth and defense by shaping the rhizosphere microbiota. Nat. Commun. 2018, 9, 2738. [Google Scholar] [CrossRef] [Green Version]
- Barber, N.A.; Chantos-Davidson, K.M.; Amel Peralta, R.; Sherwood, J.P.; Swingley, W.D. Soil microbial community composition in tallgrass prairie restorations converge with remnants across a 27-year chronosequence. Environ. Microbiol. 2017, 19, 3118–3131. [Google Scholar] [CrossRef]
- Ryden, J. Denitrification loss from a grassland soil in the field receiving different rates of nitrogen as ammonium nitrate. J. Soil Sci. 1983, 34, 355–365. [Google Scholar] [CrossRef]
- Stein, L.Y.; Klotz, M.G. The nitrogen cycle. Curr. Biol. 2016, 26, R94–R98. [Google Scholar] [CrossRef] [Green Version]
- Coolon, J.D.; Jones, K.L.; Todd, T.C.; Blair, J.M.; Herman, M.A. Long-term nitrogen amendment alters the diversity and assemblage of soil bacterial communities in tallgrass prairie. PLoS ONE 2013, 8, e67884. [Google Scholar] [CrossRef] [PubMed]
- Regan, K.; Stempfhuber, B.; Schloter, M.; Rasche, F.; Prati, D.; Philippot, L.; Boeddinghaus, R.S.; Kandeler, E.; Marhan, S. Spatial and temporal dynamics of nitrogen fixing, nitrifying and denitrifying microbes in an unfertilized grassland soil. Soil Biol. Biochem. 2017, 109, 214–226. [Google Scholar] [CrossRef]
- Pereira e Silva, M.C.; Semenov, A.V.; van Elsas, J.D.; Salles, J.F. Seasonal variations in the diversity and abundance of diazotrophic communities across soils. FEMS Microbiol. Ecol. 2011, 77, 57–68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langarica-Fuentes, A.; Manrubia, M.; Giles, M.E.; Mitchell, S.; Daniell, T.J. Effect of model root exudate on denitrifier community dynamics and activity at different water-filled pore space levels in a fertilised soil. Soil Biol. Biochem. 2018, 120, 70–79. [Google Scholar] [CrossRef] [Green Version]
- Chang, J.; Clay, D.E.; Smart, A.J.; Clay, S.A. Estimating annual root decomposition in grassland systems. Rangel. Ecol. Manag. 2016, 69, 288–291. [Google Scholar] [CrossRef]
- Oshiki, M.; Segawa, T.; Ishii, S. Nitrogen Cycle Evaluation (NiCE) Chip for Simultaneous Analysis of Multiple N Cycle-Associated Genes. Appl. Environ. Microbiol. 2018, 84, e02615-17. [Google Scholar] [CrossRef] [Green Version]
- Douglas, G.M.; Maffei, V.J.; Zaneveld, J.R.; Yurgel, S.N.; Brown, J.R.; Taylor, C.M.; Huttenhower, C.; Langille, M.G.I. PICRUSt2 for prediction of metagenome functions. Nat. Biotechnol. 2020, 38, 685–688. [Google Scholar] [CrossRef]
- Jacobs, M.B. Micro-Kjeldahl Method for Biologicals. J. Am. Pharm. Assoc. 1951, 40, 151–153. [Google Scholar] [CrossRef]
- Olsen, S.R. Estimation of Available Phosphorus in Soils by Extraction with Sodium Bicarbonate; US Department of Agriculture: Washington, DC, USA, 1954.
- Weisburg, W.G.; Barns, S.M.; Pelletier, D.A.; Lane, D.J. 16S ribosomal DNA amplification for phylogenetic study. J. Bacteriol. 1991, 173, 697–703. [Google Scholar] [CrossRef] [Green Version]
- Muyzer, G.; De Waal, E.C.; Uitterlinden, A.G. Profiling of complex microbial populations by denaturing gradient gel electrophoresis analysis of polymerase chain reaction-amplified genes coding for 16S rRNA. Appl. Environ. Microbiol. 1993, 59, 695–700. [Google Scholar] [CrossRef] [Green Version]
- Jang, J.; Xiong, X.; Liu, C.; Yoo, K.; Ishii, S. Invasive Earthworms Alter Forest Soil Microbiomes and Nitrogen Cycling. bioRxiv 2021. [Google Scholar] [CrossRef]
- Ishii, S.; Segawa, T.; Okabe, S. Simultaneous quantification of multiple food-and waterborne pathogens by use of microfluidic quantitative PCR. Appl. Environ. Microbiol. 2013, 79, 2891–2898. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef] [PubMed]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef] [Green Version]
- Bokulich, N.A.; Kaehler, B.D.; Rideout, J.R.; Dillon, M.; Bolyen, E.; Knight, R.; Huttley, G.A.; Caporaso, J.G. Optimizing taxonomic classification of marker-gene amplicon sequences with QIIME 2’s q2-feature-classifier plugin. Microbiome 2018, 6, 90. [Google Scholar] [CrossRef]
- McDonald, D.; Price, M.N.; Goodrich, J.; Nawrocki, E.P.; DeSantis, T.Z.; Probst, A.; Andersen, G.L.; Knight, R.; Hugenholtz, P. An improved Greengenes taxonomy with explicit ranks for ecological and evolutionary analyses of bacteria and archaea. ISME J. 2012, 6, 610–618. [Google Scholar] [CrossRef]
- Oren, A.; Garrity, G.M. Valid publication of the names of forty-two phyla of prokaryotes. Int. J. Syst. Evol. Microbiol. 2021, 71, 005056. [Google Scholar] [CrossRef]
- Team, R. RStudio: Integrated Development for R; RStudio, PBC: Boston, MA, USA, 2020. [Google Scholar]
- Team, R.C. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2021. [Google Scholar]
- Wickham, H.; Francois, R.; Henry, L.; Müller, K. dplyr: A Grammar of Data Manipulation. R Package Version 0.4; R Foundation for Statistical Computing: Vienna, Austria, 2015; Volume 3. [Google Scholar]
- Wickham, H. Ggplot2: Elegant Graphics for Data Analysis; Springer: Berlin/Heidelberg, Germany, 2016. [Google Scholar]
- De Mendiburu, F.; Simon, R. Agricolae-Ten Years of an Open Source Statistical Tool for Experiments in Breeding, Agriculture and Biology. PeerJ PrePrints 2015, 3, e1404v1. [Google Scholar]
- Signorell, A.; Aho, K.; Alfons, A.; Anderegg, N.; Aragon, T.; Arppe, A.; Baddeley, A.; Barton, K.; Bolker, B.; Borchers, H. DescTools: Tools for descriptive statistics. R Package Version 0.99. 34; R Foundation for Statistical Computing: Vienna, Austria, 2020. [Google Scholar]
- Revelle, W.R. psych: Procedures for Personality and Psychological Research; Northwestern University: Evanston, IL, USA, 2017. [Google Scholar]
- Kassambara, A.; Kassambara, M.A. Package ‘ggpubr’. 2020. Available online: https://cran.r-project.org/web/packages/ggpubr/index.html (accessed on 13 March 2021).
- Oksanen, J.; Kindt, R.; Legendre, P.; O’Hara, B.; Stevens, M.H.H.; Oksanen, M.J.; Suggests, M. The vegan package. Community Ecol. Package 2007, 10, 719. [Google Scholar]
- Wickham, H.; Averick, M.; Bryan, J.; Chang, W.; McGowan, L.D.A.; François, R.; Grolemund, G.; Hayes, A.; Henry, L.; Hester, J. Welcome to the Tidyverse. J. Open Source Softw. 2019, 4, 1686. [Google Scholar] [CrossRef]
- Fox, J.; Weisberg, S. An R Companion to Applied Regression; Sage Publications: New York, NY, USA, 2018. [Google Scholar]
- Dhariwal, A.; Chong, J.; Habib, S.; King, I.L.; Agellon, L.B.; Xia, J. MicrobiomeAnalyst: A web-based tool for comprehensive statistical, visual and meta-analysis of microbiome data. Nucleic Acids Res. 2017, 45, W180–W188. [Google Scholar] [CrossRef] [PubMed]
- Chong, J.; Liu, P.; Zhou, G.; Xia, J. Using MicrobiomeAnalyst for comprehensive statistical, functional, and meta-analysis of microbiome data. Nat. Protoc. 2020, 15, 799–821. [Google Scholar] [CrossRef]
- Větrovský, T.; Baldrian, P. The variability of the 16S rRNA gene in bacterial genomes and its consequences for bacterial community analyses. PLoS ONE 2013, 8, e57923. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parker, S.S.; Schimel, J.P. Soil nitrogen availability and transformations differ between the summer and the growing season in a California grassland. Appl. Soil Ecol. 2011, 48, 185–192. [Google Scholar] [CrossRef]
- Denton, C.S.; Bardgett, R.D.; Cook, R.; Hobbs, P.J. Low amounts of root herbivory positively influence the rhizosphere microbial community in a temperate grassland soil. Soil Biol. Biochem. 1998, 31, 155–165. [Google Scholar] [CrossRef]
- Nelson, M.B.; Berlemont, R.; Martiny, A.C.; Martiny, J.B. Nitrogen cycling potential of a grassland litter microbial community. Appl. Environ. Microbiol. 2015, 81, 7012–7022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crane, S.L.; van Dorst, J.; Hose, G.C.; King, C.K.; Ferrari, B.C. Microfluidic qPCR enables high throughput quantification of microbial functional genes but requires strict curation of primers. Front. Environ. Sci. 2018, 6, 145. [Google Scholar] [CrossRef] [Green Version]
- Jang, J.; Ashida, N.; Kai, A.; Isobe, K.; Nishizawa, T.; Otsuka, S.; Yokota, A.; Senoo, K.; Ishii, S. Presence of Cu-Type (NirK) and cd1-Type (NirS) Nitrite Reductase Genes in the Denitrifying Bacterium Bradyrhizobium nitroreducens sp. nov. Microbes Environ. 2018, 33, 326–331. [Google Scholar] [CrossRef] [Green Version]
- Gaby, J.C.; Buckley, D.H. A comprehensive evaluation of PCR primers to amplify the nifH gene of nitrogenase. PLoS ONE 2012, 7, e42149. [Google Scholar] [CrossRef] [Green Version]
- Koirala, A.; Brözel, V.S. Phylogeny of Nitrogenase Structural and Assembly Components Reveals New Insights into the Origin and Distribution of Nitrogen Fixation across Bacteria and Archaea. Microorganisms 2021, 9, 1662. [Google Scholar] [CrossRef]
- Gaby, J.C.; Buckley, D.H. The use of degenerate primers in qPCR analysis of functional genes can cause dramatic quantification bias as revealed by investigation of nifH primer performance. Microb. Ecol. 2017, 74, 701–708. [Google Scholar] [CrossRef] [PubMed]
- Dixon, R.; Kahn, D. Genetic regulation of biological nitrogen fixation. Nat. Rev. Microbiol. 2004, 2, 621–631. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Song, Z.; Zhuang, D.; Wang, J.; Xie, S.; Liu, G. Urea fertilization decreases soil bacterial diversity, but improves microbial biomass, respiration, and N-cycling potential in a semiarid grassland. Biol. Fertil. Soils 2019, 55, 229–242. [Google Scholar] [CrossRef]
- Lin, Y.; Ye, G.; Liu, D.; Ledgard, S.; Luo, J.; Fan, J.; Yuan, J.; Chen, Z.; Ding, W. Long-term application of lime or pig manure rather than plant residues suppressed diazotroph abundance and diversity and altered community structure in an acidic Ultisol. Soil Biol. Biochem. 2018, 123, 218–228. [Google Scholar] [CrossRef]
- Wang, D.; Li, T.; Huang, K.; He, X.; Zhang, X.-X. Roles and correlations of functional bacteria and genes in the start-up of simultaneous anammox and denitrification system for enhanced nitrogen removal. Sci. Total Environ. 2019, 655, 1355–1363. [Google Scholar] [CrossRef]
- Girvan, M.S.; Bullimore, J.; Pretty, J.N.; Osborn, A.M.; Ball, A.S. Soil type is the primary determinant of the composition of the total and active bacterial communities in arable soils. Appl. Environ. Microbiol. 2003, 69, 1800–1809. [Google Scholar] [CrossRef] [Green Version]
- Riffkin, P.A.; Quigley, P.E.; Kearney, G.A.; Cameron, F.J.; Gault, R.R.; Peoples, M.B.; Thies, J.E. Factors associated with biological nitrogen fixation in dairy pastures in south-western Victoria. Aust. J. Agric. Res. 1999, 50, 261–272. [Google Scholar] [CrossRef]
- Kuramae, E.; Gamper, H.; van Veen, J.; Kowalchuk, G. Soil and plant factors driving the community of soil-borne microorganisms across chronosequences of secondary succession of chalk grasslands with a neutral pH. FEMS Microbiol. Ecol. 2011, 77, 285–294. [Google Scholar] [CrossRef] [Green Version]
- Lauber, C.L.; Hamady, M.; Knight, R.; Fierer, N. Pyrosequencing-based assessment of soil pH as a predictor of soil bacterial community structure at the continental scale. Appl. Environ. Microbiol. 2009, 75, 5111–5120. [Google Scholar] [CrossRef] [Green Version]
- Mackelprang, R.; Grube, A.M.; Lamendella, R.; Jesus, E.d.C.; Copeland, A.; Liang, C.; Jackson, R.D.; Rice, C.W.; Kapucija, S.; Parsa, B. Microbial community structure and functional potential in cultivated and native tallgrass prairie soils of the Midwestern United States. Front. Microbiol. 2018, 9, 1775. [Google Scholar] [CrossRef] [Green Version]
- Swab, R.; Lorenz, N.; Byrd, S.; Dick, R. Native vegetation in reclamation: Improving habitat and ecosystem function through using prairie species in mine land reclamation. Ecol. Eng. 2017, 108, 525–536. [Google Scholar] [CrossRef]
- Mao, Y.; Yannarell, A.C.; Mackie, R.I. Changes in N-transforming archaea and bacteria in soil during the establishment of bioenergy crops. PLoS ONE 2011, 6, e24750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ritz, K.; McNicol, J.; Nunan, N.; Grayston, S.; Millard, P.; Atkinson, D.; Gollotte, A.; Habeshaw, D.; Boag, B.; Clegg, C. Spatial structure in soil chemical and microbiological properties in an upland grassland. FEMS Microbiol. Ecol. 2004, 49, 191–205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Weil, M.; Dumack, K.; Zak, D.; Münch, D.; Günther, A.; Jurasinski, G.; Blume-Werry, G.; Kreyling, J.; Urich, T. Eukaryotic rather than prokaryotic microbiomes change over seasons in rewetted fen peatlands. bioRxiv 2021. [Google Scholar] [CrossRef] [PubMed]
- Miksch, S.; Meiners, M.; Meyerdierks, A.; Probandt, D.; Wegener, G.; Titschack, J.; Jensen, M.A.; Ellrott, A.; Amann, R.; Knittel, K. Bacterial communities in temperate and polar coastal sands are seasonally stable. ISME Commun. 2021, 1, 29. [Google Scholar] [CrossRef]
- Toole, D.R.; Zhao, J.; Martens-Habbena, W.; Strauss, S.L. Bacterial functional prediction tools detect but underestimate metabolic diversity compared to shotgun metagenomics in southwest Florida soils. Appl. Soil Ecol. 2021, 168, 104129. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Das, B.K.; Ishii, S.; Antony, L.; Smart, A.J.; Scaria, J.; Brözel, V.S. The Microbial Nitrogen Cycling, Bacterial Community Composition, and Functional Potential in a Natural Grassland Are Stable from Breaking Dormancy to Being Dormant Again. Microorganisms 2022, 10, 923. https://doi.org/10.3390/microorganisms10050923
Das BK, Ishii S, Antony L, Smart AJ, Scaria J, Brözel VS. The Microbial Nitrogen Cycling, Bacterial Community Composition, and Functional Potential in a Natural Grassland Are Stable from Breaking Dormancy to Being Dormant Again. Microorganisms. 2022; 10(5):923. https://doi.org/10.3390/microorganisms10050923
Chicago/Turabian StyleDas, Bikram K., Satoshi Ishii, Linto Antony, Alexander J. Smart, Joy Scaria, and Volker S. Brözel. 2022. "The Microbial Nitrogen Cycling, Bacterial Community Composition, and Functional Potential in a Natural Grassland Are Stable from Breaking Dormancy to Being Dormant Again" Microorganisms 10, no. 5: 923. https://doi.org/10.3390/microorganisms10050923
APA StyleDas, B. K., Ishii, S., Antony, L., Smart, A. J., Scaria, J., & Brözel, V. S. (2022). The Microbial Nitrogen Cycling, Bacterial Community Composition, and Functional Potential in a Natural Grassland Are Stable from Breaking Dormancy to Being Dormant Again. Microorganisms, 10(5), 923. https://doi.org/10.3390/microorganisms10050923