
Direct Detection of Antibacterial-Producing Soil Isolates Utilizing a Novel High-Throughput Screening Assay


Abstract
1. Introduction
2. Materials and Methods
2.1. Bacterial Strains
2.2. Bacterial Growth
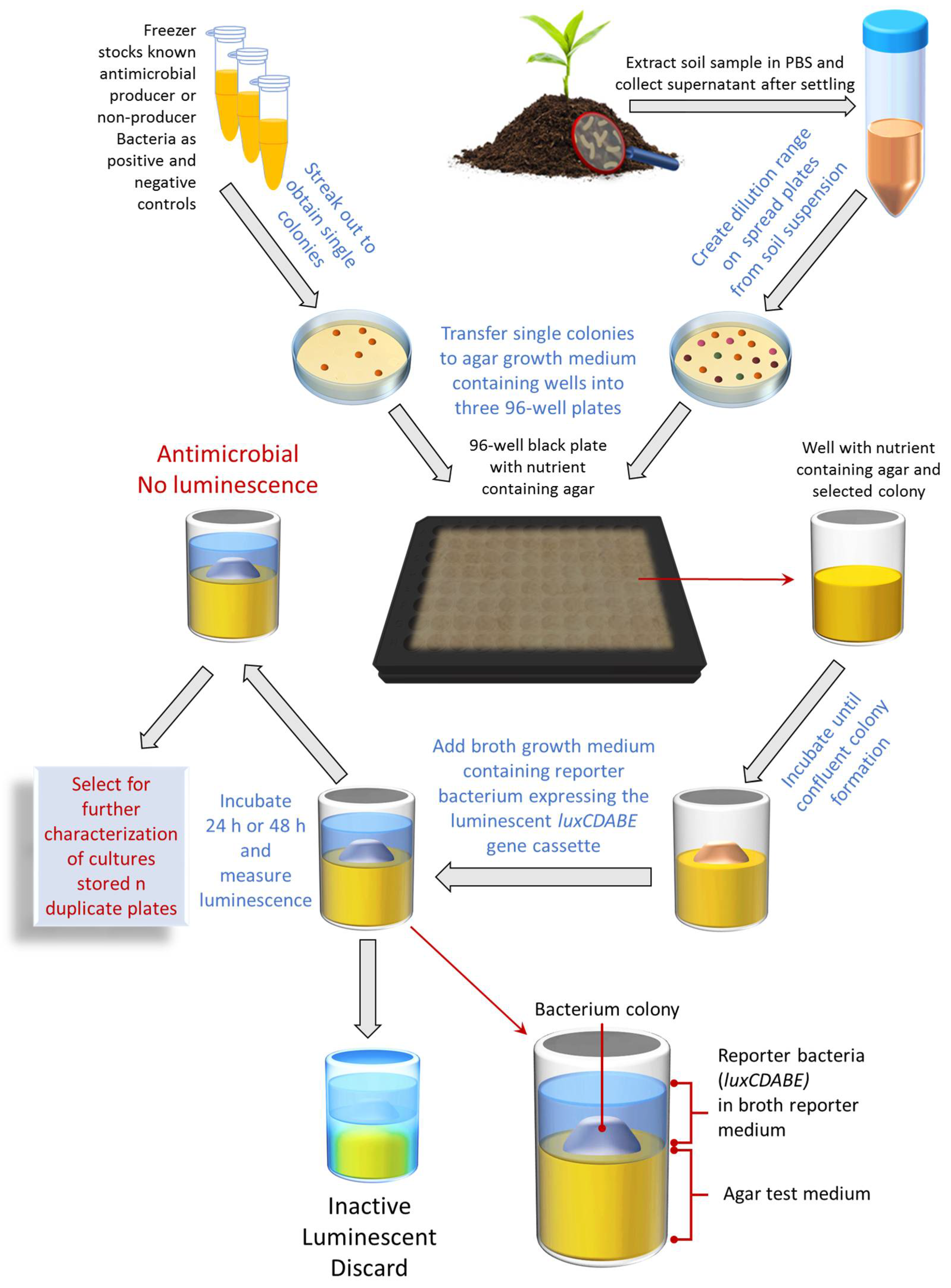
2.3. Bioluminescent Simultaneous Antagonism Assay
2.4. Mass Spectrometry of Test Culture Extracts
2.5. Test Bacterium Activity
2.6. BLSA Assay pH Resistance
2.7. Assay Quality Statistics
2.8. Limit of Detection
2.9. Soil Sample Screening
3. Results
3.1. BLSA Assay Performance against Known Test Bacteria
3.2. pH Stability
3.3. Assay Quality and Robustness
3.4. Screening of a Soil Sample
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Smith, P.W.; Watkins, K.; Hewlett, A. Infection control through the ages. Am. J. Infect. Contr. 2012, 40, 35–42. [Google Scholar] [CrossRef] [PubMed]
- Dodds, D.R. Antibiotic resistance: A current epilogue. Biochem. Pharmacol. 2017, 134, 139–146. [Google Scholar] [CrossRef] [PubMed]
- CDC. COVID-19: U.S. Impact on Antimicrobial Resistance, Special Report 2022. Atlanta, GA: U.S. Department of Health and Human Services. Available online: https://www.cdc.gov/drugresistance/covid19.htmlCDC (accessed on 11 October 2022).
- CDC. Antibiotic Resistance Threats in the United States, 2019. Atlanta, GA: U.S. Department of Health and Human Services. Available online: www.cdc.gov/DrugResistance/Biggest-Threats.html. (accessed on 31 January 2022).
- Silberstein, S. Zur frage der salvarsanresistenten lues. Arch. Dermatol. 1924, 147, 116–130. [Google Scholar] [CrossRef]
- Williams, K. The introduction of ‘chemotherapy’ using arsphenamine—The first magic bullet. J. R. Soc. Med. 2009, 102, 343–348. [Google Scholar] [CrossRef]
- Drake, J.A.; Thomson, M.S. A New phase in early syphilis. Br. J. Dermatol. 1932, 44, 297–304. [Google Scholar] [CrossRef]
- Beckh, W.; Kulchar, G.V. Treatment-resistant syphilis: An evaluation of the causative factors in eighteen cases. Arch. Dermatol. Syphilol. 1939, 40, 1–12. [Google Scholar] [CrossRef]
- Barber, M.; Rozwadowska-Dowzenko, M. Infection by penicillin-resistant staphylococci. The Lancet 1948, 252, 641–644. [Google Scholar] [CrossRef]
- Hutchings, M.I.; Truman, A.W.; Wilkinson, B. Antibiotics: Past, Present and Future. Curr. Opin. Microbiol. 2019, 51, 72–80. [Google Scholar] [CrossRef]
- Waksman, S.A. Microbial Antagonisms and Antibiotic Substances, 1st ed.; The Commonwealth Fund: New York, NY, USA, 1945. [Google Scholar]
- Ribeiro da Cunha, B.; Fonseca, L.P.; Calado, C.R.C. Antibiotic discovery: Where have we come from, where do we go? Antibiotics 2019, 8, 45. [Google Scholar] [CrossRef]
- Solden, L.; Lloyd, K.; Wrighton, K. The Bright Side of Microbial Dark Matter: Lessons learned from the uncultivated majority. Curr. Opin. Microbiol. 2016, 31, 217–226. [Google Scholar] [CrossRef]
- Vartoukian, S.R.; Palmer, R.M.; Wade, W.G. Strategies for culture of ‘unculturable’ bacteria. FEMS Microbiol. Lett. 2010, 309, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Nichols, D.; Cahoon, N.; Trakhtenberg, E.M.; Pham, L.; Mehta, A.; Belanger, A.; Kanigan, T.; Lewis, K.; Epstein, S.S. Use of Ichip for high-throughput in situ cultivation of “uncultivable” microbial species. Appl. Environ. Microbiol. 2010, 76, 2445–2450. [Google Scholar] [CrossRef] [PubMed]
- Ling, L.L.; Schneider, T.; Peoples, A.J.; Spoering, A.L.; Engels, I.; Conlon, B.P.; Mueller, A.; Schäberle, T.F.; Hughes, D.E.; Epstein, S.; et al. A new antibiotic kills pathogens without detectable resistance. Nature 2015, 517, 455–459. [Google Scholar] [CrossRef] [PubMed]
- Mills, S.D.; Dougherty, T.J. Cell-Based Screening in Antibacterial Discovery. In Antibiotic Discovery and Development; Springer: Berlin, Germany, 2012; pp. 901–929. [Google Scholar]
- Meighen, E.A. Bacterial Bioluminescence: Organization, Regulation, and application of the Lux genes. FASEB J. 1993, 7, 1016–1022. [Google Scholar] [CrossRef]
- Naveh, A.; Potasman, I.; Bassan, H.; Ulitzur, S. A new rapid and sensitive bioluminescence assay for antibiotics that inhibit protein synthesis. J. Appl. Bacteriol. 1984, 56, 457–463. [Google Scholar] [CrossRef]
- Shawar, R.M.; Humble, D.J.; Van Dalfsen, J.M.; Stover, C.K.; Hickey, M.J.; Steele, S.; Mitscher, L.A.; Baker, W. Rapid screening of natural products for antimycobacterial activity by using luciferase-expressing strains of Mycobacterium Bovis BCG and Mycobacterium intracellulare. Antimicrob. Agents Chemother. 1997, 41, 570–574. [Google Scholar] [CrossRef]
- Hilpert, K.; Hancock, R.E.W. Use of luminescent bacteria for rapid screening and characterization of short cationic antimicrobial peptides synthesized on cellulose using peptide array technology. Nat. Protoc. 2007, 2, 1652–1660. [Google Scholar] [CrossRef]
- Travis, S.M.; Anderson, N.N.; Forsyth, W.R.; Espiritu, C.; Conway, B.D.; Greenberg, E.P.; McCray, P.B.; Lehrer, R.I.; Welsh, M.J.; Tack, B.F. Bactericidal activity of mammalian cathelicidin-derived peptides. Infect. Immun. 2000, 68, 2748–2755. [Google Scholar] [CrossRef]
- R Core Team R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria. Available online: https://www.r-project.org/ (accessed on 9 August 2021).
- Wickham, H.; Averick, M.; Bryan, J.; Chang, W.; McGowan, L.D.; François, R.; Grolemund, G.; Hayes, A.; Henry, L.; Hester, J.; et al. Welcome to the Tidyverse. J. Open Source Softw. 2019, 4, 1686. [Google Scholar] [CrossRef]
- Zhang, J.-H.; Chung, T.D.Y.; Oldenburg, K.R. A simple statistical parameter for use in evaluation and validation of high-throughput screening assays. J. Biomol. Screen 1999, 4, 67–73. [Google Scholar] [CrossRef]
- Inglese, J.; Johnson, R.L.; Simeonov, A.; Xia, M.; Zheng, W.; Austin, C.P.; Auld, D.S. High-throughput screening assays for the identification of chemical probes. Nat. Chem. Biol. 2007, 3, 466–479. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, M.; Wickham, H. Tidymodels: A Collection of Packages for Modeling and Machine learning Using Tidyverse Principles. Available online: https://www.tidymodels.org (accessed on 11 October 2022).
- Koga, K.; Harada, T.; Shimizu, H.; Tanaka, K. Bacterial luciferase activity and the intracellular redox pool in Escherichia coli. Mol. Genet. Genomics 2005, 274, 180–188. [Google Scholar] [CrossRef]
- Lema, C.; Varela-Ramirez, A.; Aguilera, R.J. Differential nuclear staining assay for high-throughput screening to identify cytotoxic compounds. Curr. Cell Biochem. 2011, 1, 1–14. [Google Scholar] [PubMed]
- Moran, J.C.; Crank, E.L.; Ghabban, H.A.; Horsburgh, M.J. Deferred growth inhibition assay to quantify the effect of bacteria-derived antimicrobials on competition. J. Vis. Exp. 2016, 54437. [Google Scholar] [CrossRef] [PubMed]
- Sui, Y.; Wu, Z. Alternative statistical parameter for high-throughput screening assay quality assessment. J. Biomol Screen 2007, 12, 229–234. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Bacterium | Strain | Activity a | Antibacterial Compound(s) in Culture Extracts b | |
|---|---|---|---|---|
| E. coli | S. aureus | |||
| Aneurinibacillus migulanus | ATCC9999 | + | + | Gramicidin S |
| Bacillus licheniformis | LB5 | + | + | Bacitracin, Unknown |
| Bacillus subtilis | ATCC21332 | - | +/- | Surfactin |
| Bacillus subtilis | OKB105 | - | + | Surfactin |
| Bacillus subtilis | OKB120 | - | + | Shortened surfactin c |
| Bacillus subtilis | OKB168 | - | - | None |
| Brevibacillus laterosporus | LB4 | + | + | Loloatins and bogorols |
| Brevibacillus parabrevis | ATCC10068 | - | + | Tyrothricin |
| Brevibacillus parabrevis | ATCC8185 | - | + | Tyrothricin |
| Escherichia coli | K12 | - | - | None |
| Micrococcus luteus | NCTC8340 | - | - | None |
| Pseudomonas aeruginosa | ATCC27853 | + | + | Rhamnolipids c |
| Plate | Well | E. coli Xen14 | S. aureus Xen29 | ||
|---|---|---|---|---|---|
| BLSA | SA | BLSA | SA | ||
| 1 | E8 | + | + | + | + |
| 1 | G3 | + | + | + | + |
| 2 | A3 | - | - | + | + |
| 2 | A10 | - | - | + | + |
| 2 | F4 | - | - | + | - |
| 2 | G10 | - | - | + | - |
| 2 | H10 | + | + | + | + |
| 3 | A12 | - | - | + | - |
| 3 | B9 | - | - | + | - |
| 3 | D9 | - | - | + | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Laubscher, W.E.; Rautenbach, M. Direct Detection of Antibacterial-Producing Soil Isolates Utilizing a Novel High-Throughput Screening Assay. Microorganisms 2022, 10, 2235. https://doi.org/10.3390/microorganisms10112235
Laubscher WE, Rautenbach M. Direct Detection of Antibacterial-Producing Soil Isolates Utilizing a Novel High-Throughput Screening Assay. Microorganisms. 2022; 10(11):2235. https://doi.org/10.3390/microorganisms10112235
Chicago/Turabian StyleLaubscher, Wikus Ernst, and Marina Rautenbach. 2022. "Direct Detection of Antibacterial-Producing Soil Isolates Utilizing a Novel High-Throughput Screening Assay" Microorganisms 10, no. 11: 2235. https://doi.org/10.3390/microorganisms10112235
APA StyleLaubscher, W. E., & Rautenbach, M. (2022). Direct Detection of Antibacterial-Producing Soil Isolates Utilizing a Novel High-Throughput Screening Assay. Microorganisms, 10(11), 2235. https://doi.org/10.3390/microorganisms10112235