
Comprehensive Phenotyping in Inflammatory Bowel Disease: Search for Biomarker Algorithms in the Transkingdom Interactions Context

 , , , and
, , , and
Abstract
1. Introduction
2. Materials and Methods
2.1. Ethics Statement
2.2. Selection of Participants and Environmental Data
2.3. General Diagnosis
2.4. Biochemical Measurements
2.5. Blood and Stool RNA Extraction
2.6. miRNAs Identification
2.7. Stool Samples Collection and DNA Extraction
2.8. 16S Bacterial rRNA Fragment NGS
2.9. Sequence Analysis and Comparison of Microbial Communities
2.10. Logistic Regression Model
2.11. Weighted Correlation Network Analysis
2.12. Data Accession
3. Results
3.1. Characteristics of the Studied Population
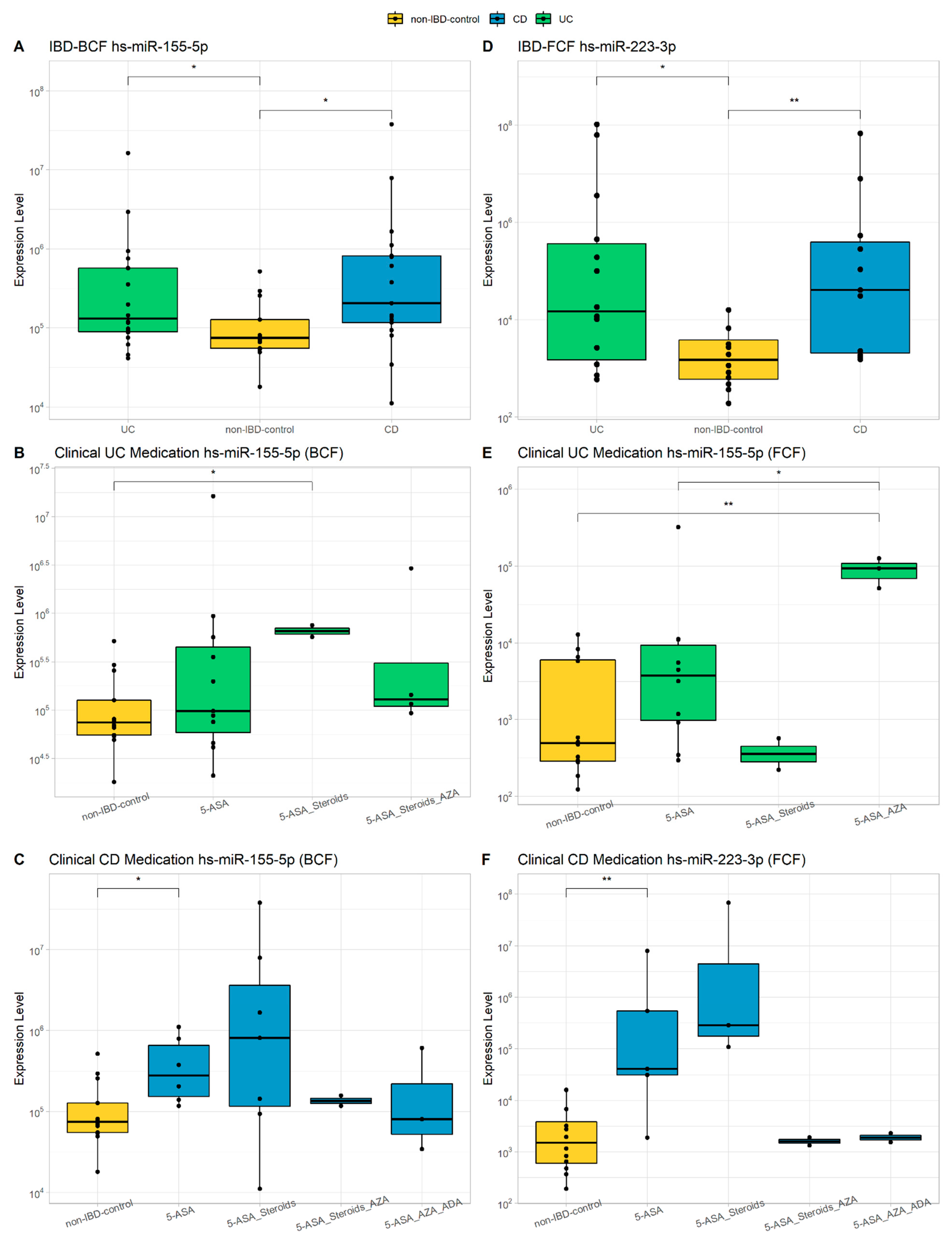
3.2. miRNAs Characterization
3.3. Microbial Composition
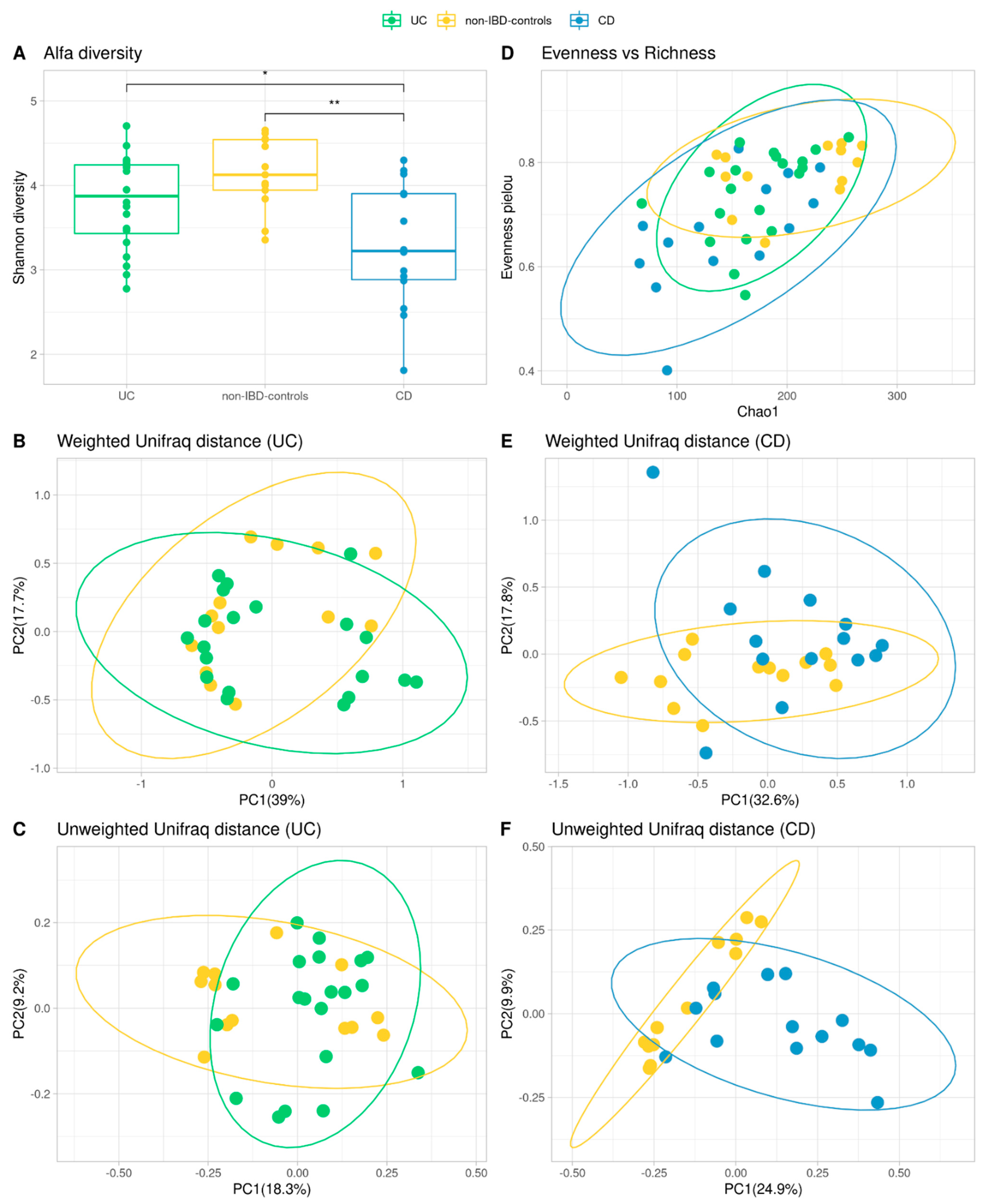
3.4. Alpha and Beta Diversity
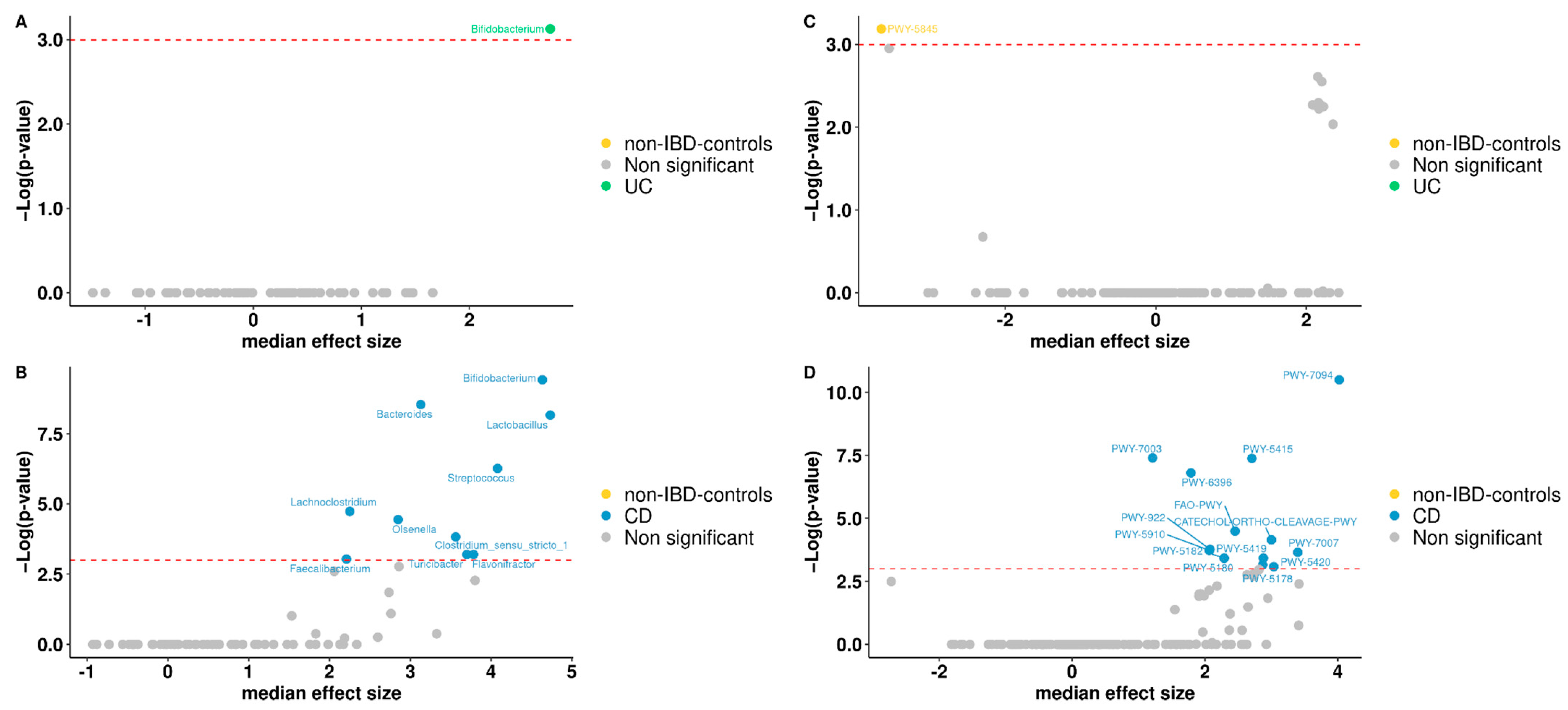
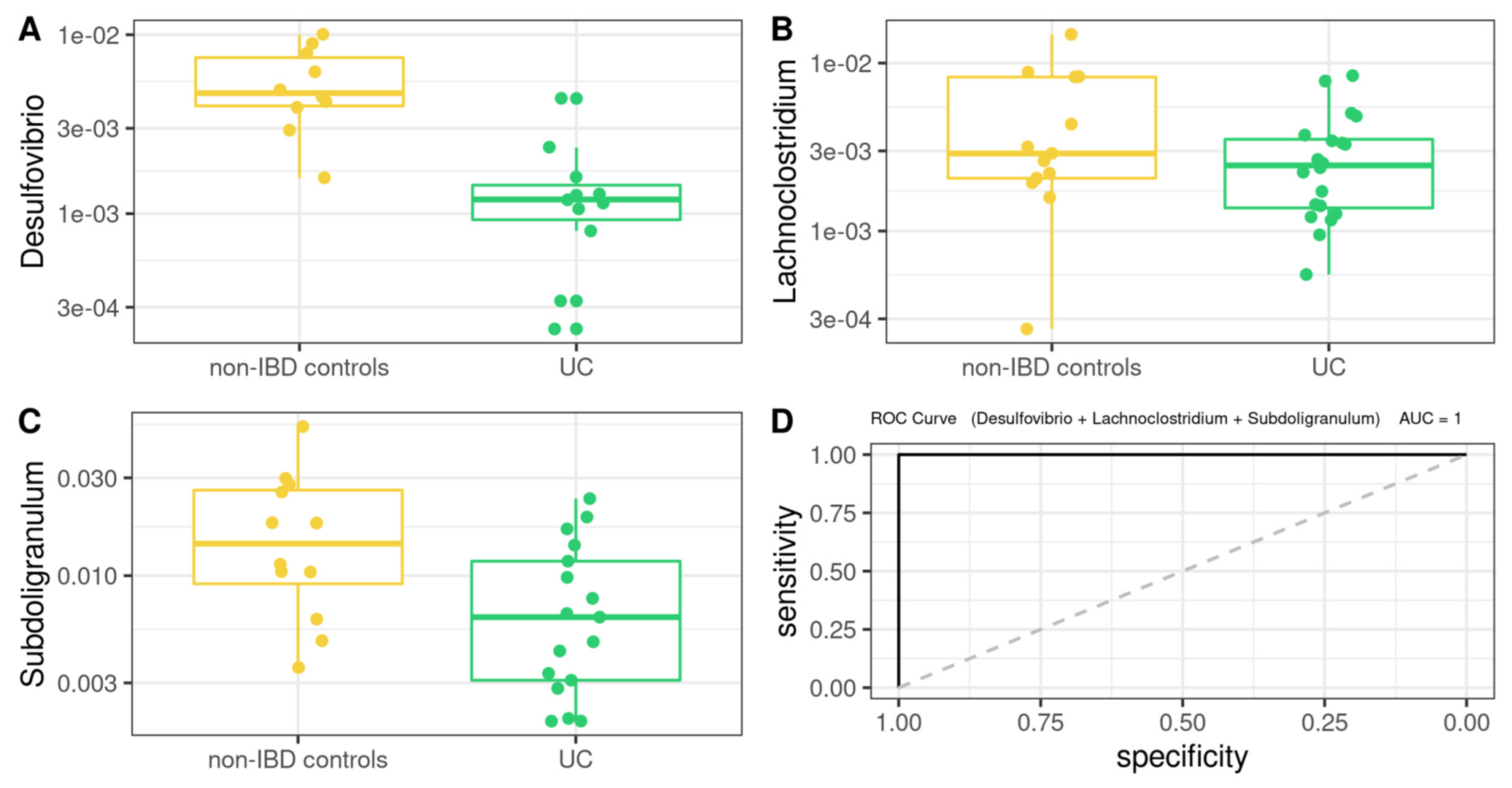
3.5. Differentially Abundant Taxa between Patients and Controls
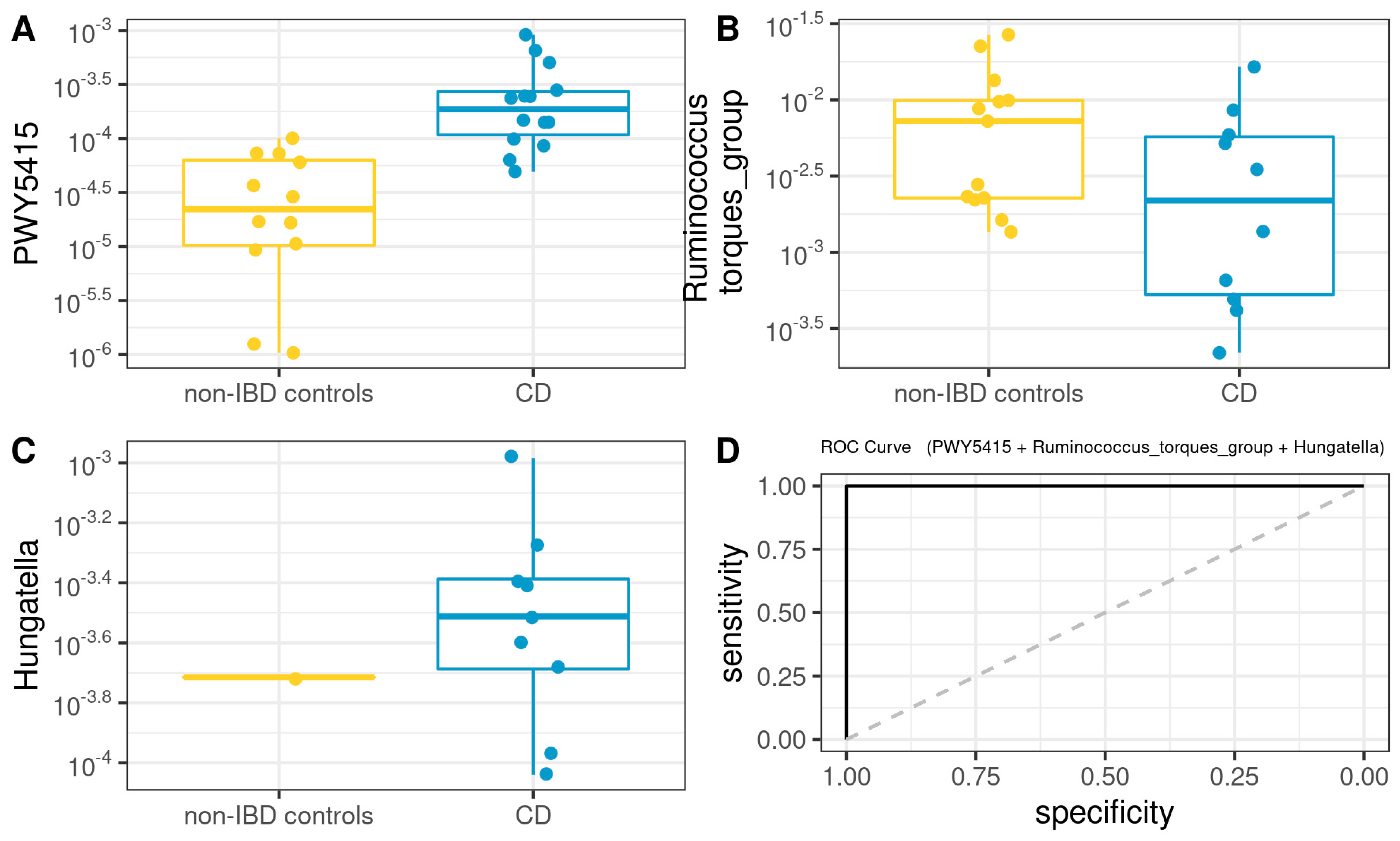
3.6. Functional Analysis
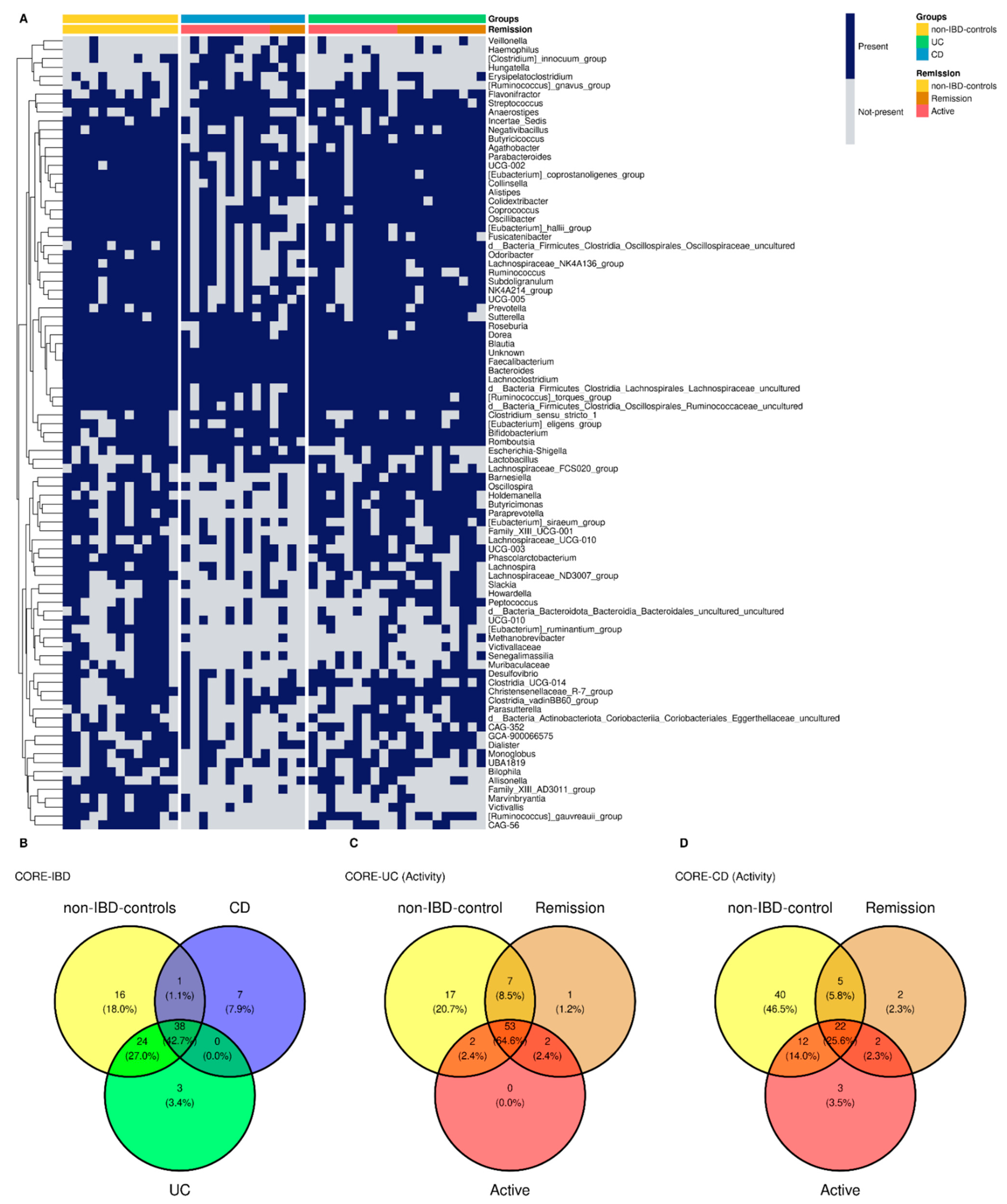
3.7. Common Core Microbiota
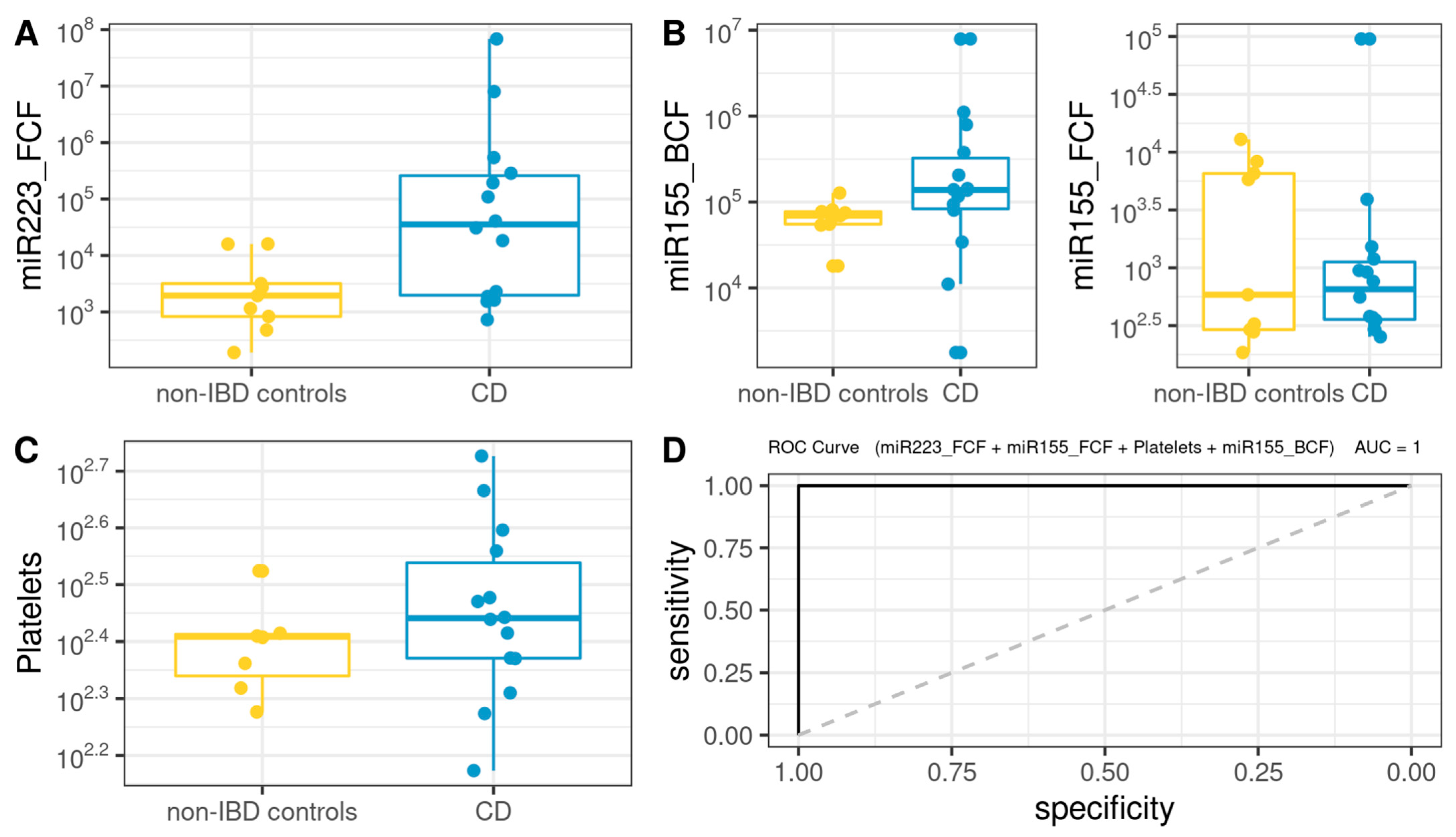
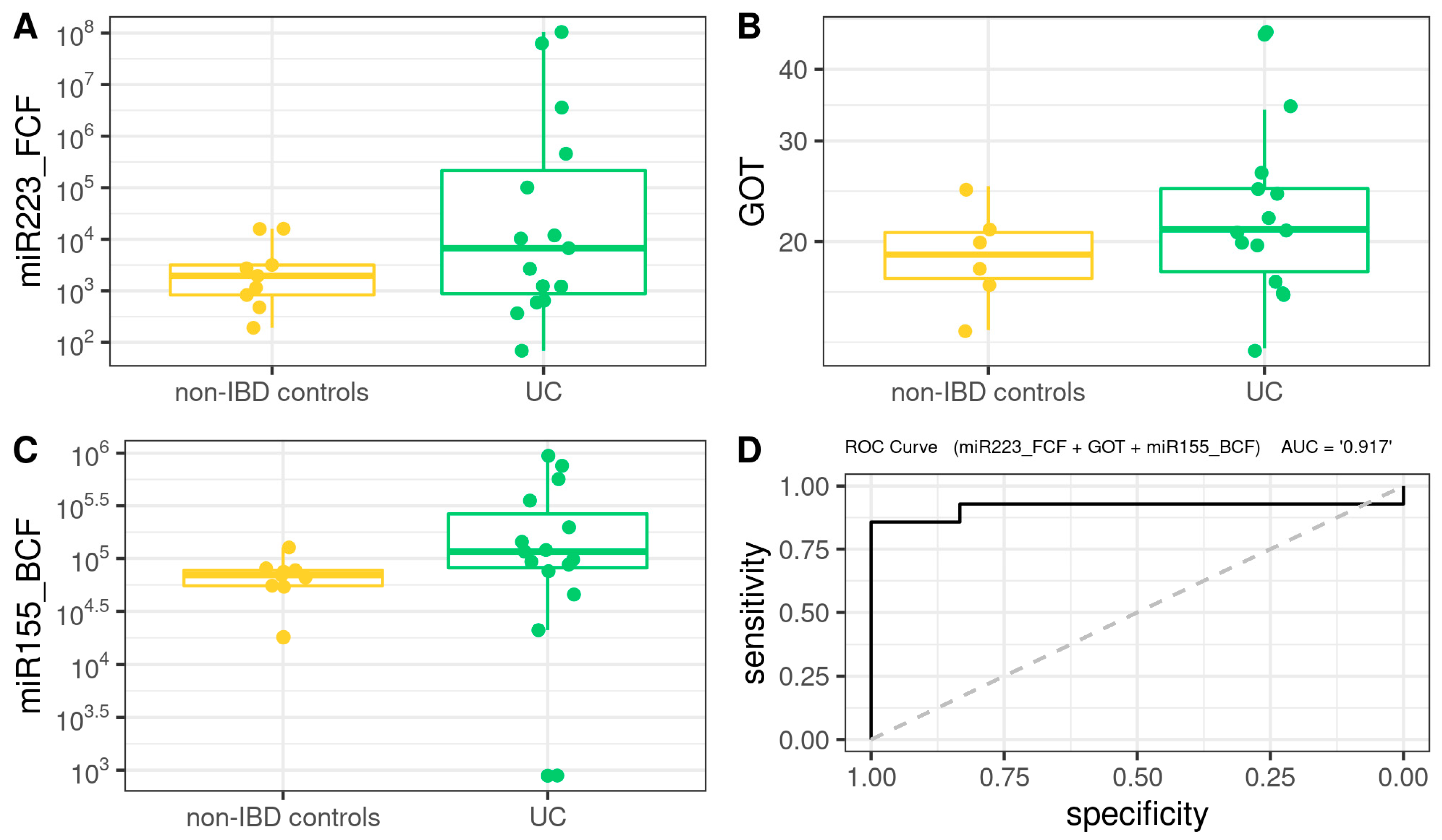
3.8. Comprehensive Phenotyping Algorithms
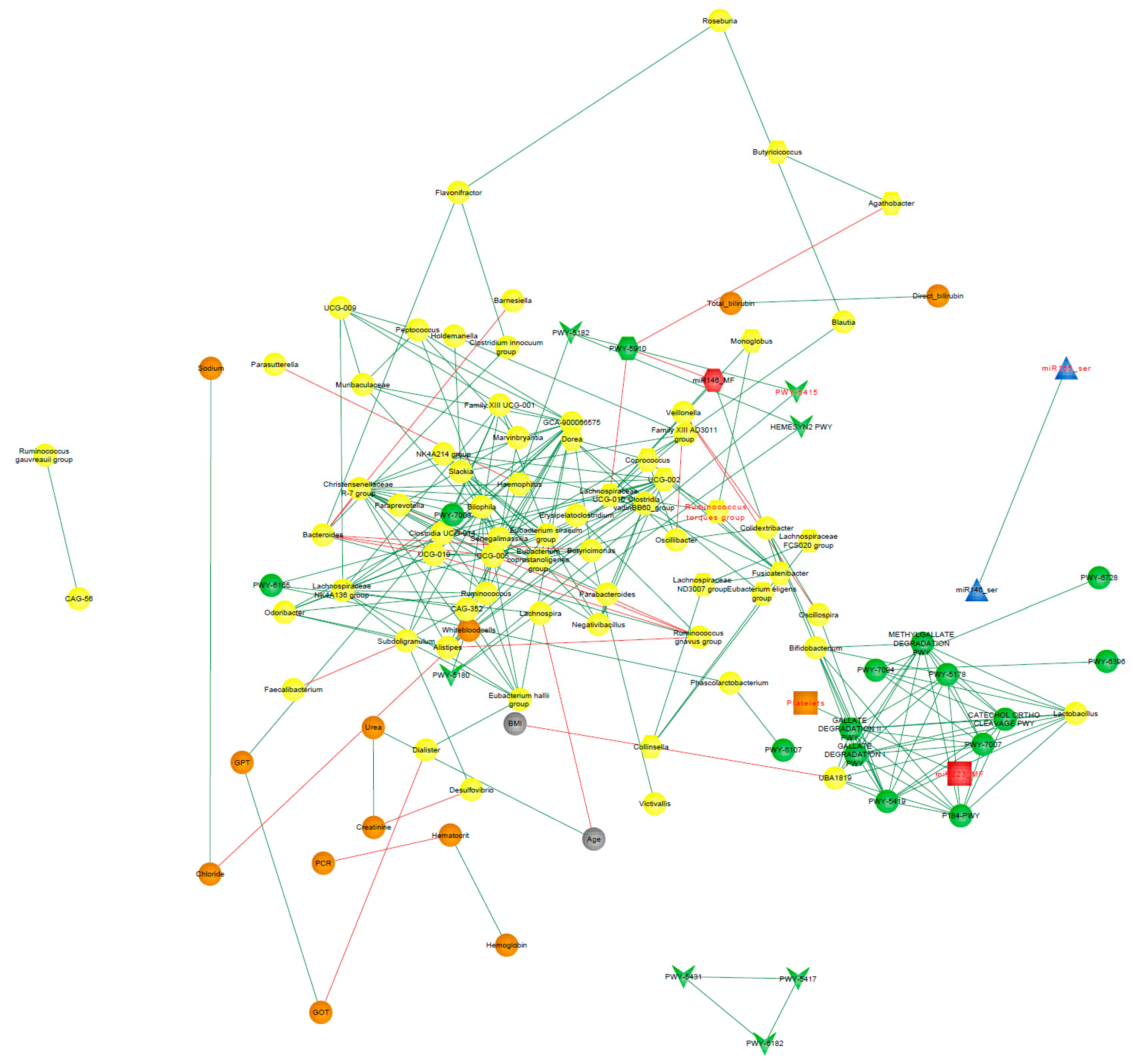
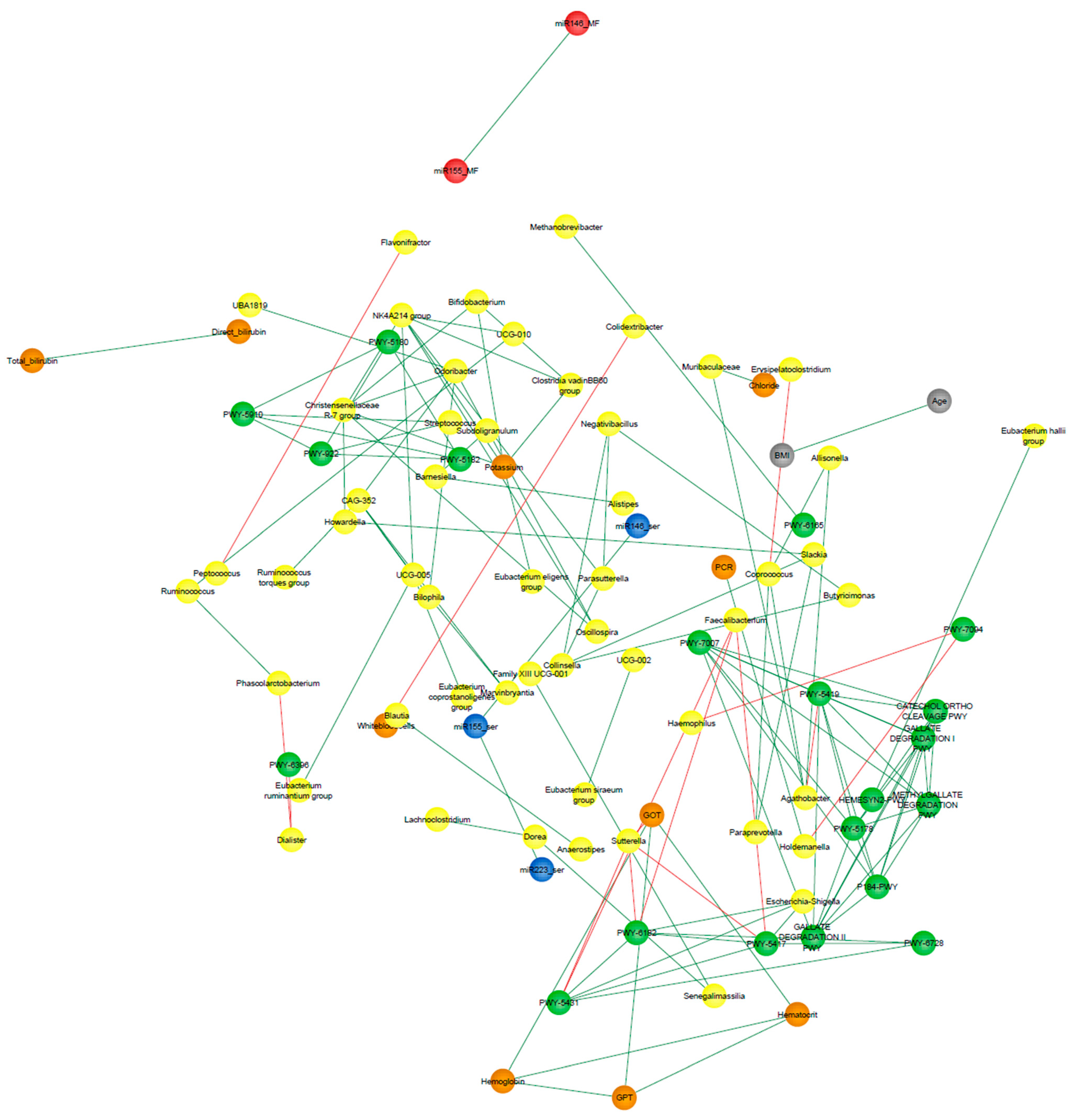
3.9. Correlation Network Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Abraham, C.; Cho, J.H. Inflammatory bowel disease. N. Engl. J. Med. 2009, 361, 2066–2078. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, G.G.; Ng, S.C. Understanding and Preventing the Global Increase of Inflammatory Bowel Disease. Gastroenterology 2017, 152, 313–321.e2. [Google Scholar] [CrossRef] [PubMed]
- Magro, F.; Gionchetti, P.; Eliakim, R.; Ardizzone, S.; Armuzzi, A.; Barreiro-de Acosta, M.; Burisch, J.; Gecse, K.B.; Hart, A.L.; Hindryckx, P.; et al. Third European Evidence-based Consensus on Diagnosis and Management of Ulcerative Colitis. Part 1: Definitions, Diagnosis, Extra-intestinal Manifestations, Pregnancy, Cancer Surveillance, Surgery, and Ileo-anal Pouch Disorders. J. Crohn’s Colitis 2017, 11, 649–670. [Google Scholar] [CrossRef]
- Kaur, A.; Goggolidou, P. Ulcerative colitis: Understanding its cellular pathology could provide insights into novel therapies. J. Inflamm. 2020, 17, 15. [Google Scholar] [CrossRef] [PubMed]
- Valdes, A.M.; Walter, J.; Segal, E.; Spector, T.D. Role of the gut microbiota in nutrition and health. BMJ 2018, 361, k2179. [Google Scholar] [CrossRef]
- Ji, Y.; Li, X.; Zhu, Y.; Li, N.; Zhang, N.; Niu, M. Faecal microRNA as a biomarker of the activity and prognosis of inflammatory bowel diseases. Biochem. Biophys. Res. Commun. 2018, 503, 2443–2450. [Google Scholar] [CrossRef]
- Béres, N.J.; Szabó, D.; Kocsis, D.; Szűcs, D.; Kiss, Z.; Müller, K.E.; Lendvai, G.; Kiss, A.; Arató, A.; Sziksz, E.; et al. Role of Altered Expression of miR-146a, miR-155, and miR-122 in Pediatric Patients with Inflammatory Bowel Disease. Inflamm. Bowel Dis. 2016, 22, 327–335. [Google Scholar] [CrossRef]
- Zhou, G.; Zhou, Y.; Chen, X. New Insight into Inter-kingdom Communication: Horizontal Transfer of Mobile Small RNAs. Front. Microbiol. 2017, 8, 768. [Google Scholar] [CrossRef]
- Shen, Z.-H.; Zhu, C.-X.; Quan, Y.-S.; Yang, Z.-Y.; Wu, S.; Luo, W.-W.; Tan, B.; Wang, X.-Y. Relationship between intestinal microbiota and ulcerative colitis: Mechanisms and clinical application of probiotics and fecal microbiota transplantation. World J. Gastroenterol. 2018, 24, 5–14. [Google Scholar] [CrossRef]
- Medina, D.A.; Li, T.; Thomson, P.; Artacho, A.; Pérez-Brocal, V.; Moya, A. Cross-Regional View of Functional and Taxonomic Microbiota Composition in Obesity and Post-obesity Treatment Shows Country Specific Microbial Contribution. Front. Microbiol. 2019, 10, 2346. [Google Scholar] [CrossRef]
- Fujio-Vejar, S.; Vasquez, Y.; Morales, P.; Magne, F.; Vera-Wolf, P.; Ugalde, J.A.; Navarrete, P.; Gotteland, M. The Gut Microbiota of Healthy Chilean Subjects Reveals a High Abundance of the Phylum Verrucomicrobia. Front. Microbiol. 2017, 8, 1221. [Google Scholar] [CrossRef] [PubMed]
- Muñoz, M.; Restrepo-Montoya, D.; Kumar, N.; Iraola, G.; Camargo, M.; Díaz-Arévalo, D.; Roa-Molina, N.S.; Tellez, M.A.; Herrera, G.; Ríos-Chaparro, D.I.; et al. Integrated genomic epidemiology and phenotypic profiling of Clostridium difficile across intra-hospital and community populations in Colombia. Sci. Rep. 2019, 9, 11293. [Google Scholar] [CrossRef] [PubMed]
- Magne, F.; O’Ryan, M.L.; Vidal, R.; Farfan, M. The human gut microbiome of Latin America populations: A landscape to be discovered. Curr. Opin. Infect. Dis. 2016, 29, 528–537. [Google Scholar] [CrossRef] [PubMed]
- Belforte, F.S.; Fernandez, N.; Monzón, F.T.; Rosso, A.; Quesada, S.; Cimolai, M.C.; Millán, A.; Cerrone, G.E.; Frechtel, G.D.; Burcelin, R.; et al. Getting to Know the Gut Microbial Diversity of Metropolitan Buenos Aires Inhabitants. Front. Microbiol. 2019, 10, 965. [Google Scholar] [CrossRef]
- Aguilera, P.; Mascardi, M.F.; Belforte, F.S.; Rosso, A.D.; Quesada, S.; Llovet, I.; Iraola, G.; Trinks, J.; Penas-Steinhardt, A. A Two-Time Point Analysis of Gut Microbiota in the General Population of Buenos Aires and Its Variation Due to Preventive and Compulsory Social Isolation during the COVID-19 Pandemic. Front. Microbiol. 2022, 13, 803121. [Google Scholar] [CrossRef]
- Dei-Cas, I.; Giliberto, F.; Luce, L.; Dopazo, H.; Penas-Steinhardt, A. Metagenomic analysis of gut microbiota in non-treated plaque psoriasis patients stratified by disease severity: Development of a new Psoriasis-Microbiome Index. Sci. Rep. 2020, 10, 12754. [Google Scholar] [CrossRef]
- La Rosa, P.S.; Brooks, J.P.; Deych, E.; Boone, E.L.; Edwards, D.J.; Wang, Q.; Sodergren, E.; Weinstock, G.; Shannon, W.D. Hypothesis testing and power calculations for taxonomic-based human microbiome data. PLoS ONE 2012, 7, e52078. [Google Scholar] [CrossRef]
- Truelove, S. Cortisone in Ulcerative Colitis Preliminary Report on a Therapeutic Trial. Digestion 1954, 81, 86–90. [Google Scholar] [CrossRef]
- D’Haens, G.; Sandborn, W.J.; Feagan, B.G.; Geboes, K.; Hanauer, S.B.; Irvine, E.J.; Lémann, M.; Marteau, P.; Rutgeerts, P.; Schölmerich, J.; et al. A Review of Activity Indices and Efficacy End Points for Clinical Trials of Medical Therapy in Adults with Ulcerative Colitis. Gastroenterology 2007, 132, 763–786. [Google Scholar] [CrossRef]
- Schroeder, K.W.; Tremaine, W.J.; Ilstrup, D.M. Coated oral 5-aminosalicylic acid therapy for mildly to moderately active ulcerative colitis. A randomized study. N. Engl. J. Med. 1987, 317, 1625–1629. [Google Scholar] [CrossRef]
- Best, W.R.; Becktel, J.M.; Singleton, J.W.; Kern, F., Jr. Development of a Crohn’s disease activity index. National Cooperative Crohn’s Disease Study. Gastroenterology 1976, 70, 439–444. [Google Scholar] [CrossRef]
- Daperno, M.; D’Haens, G.; Van Assche, G.; Baert, F.; Bulois, P.; Maunoury, V.; Sostegni, R.; Rocca, R.; Pera, A.; Gevers, A. Development and validation of a new, simplified endoscopic activity score for Crohn’s disease: The SES-CD. Gastrointest. Endosc. 2004, 60, 505–512. [Google Scholar] [CrossRef]
- Kramer, M.F. Stem-loop RT-qPCR for miRNAs. Curr. Protoc. Mol. Biol. 2011, 95, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Gonzalez Peña, A.; Goodrich, J.K.; Gordon, J.I.; et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef] [PubMed]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef] [PubMed]
- Weinstein, M.M.; Prem, A.; Jin, M.; Tang, S.; Bhasin, J.M. FIGARO: An efficient and objective tool for optimizing microbiome rRNA gene trimming parameters. bioRxiv 2019, 610394. [Google Scholar] [CrossRef]
- Laurence, M.; Hatzis, C.; Brash, D.E. Common contaminants in next-generation sequencing that hinder discovery of low-abundance microbes. PLoS ONE 2014, 9, e97876. [Google Scholar] [CrossRef]
- Salter, S.J.; Cox, M.J.; Turek, E.M.; Calus, S.T.; Cookson, W.O.; Moffatt, M.F.; Turner, P.; Parkhill, J.; Loman, N.J.; Walker, A.W. Reagent and laboratory contamination can critically impact sequence-based microbiome analyses. BMC Biol. 2014, 12, 87. [Google Scholar] [CrossRef]
- Mirarab, S.; Nguyen, N.; Warnow, T. SEPP: SATé-enabled phylogenetic placement. Pac. Symp. Biocomput. 2012, 247–258. [Google Scholar] [CrossRef]
- Janssen, S.; McDonald, D.; Gonzalez, A.; Navas-Molina, J.A.; Jiang, L.; Xu, Z.Z.; Winker, K.; Kado, D.M.; Orwoll, E.; Manary, M.; et al. Phylogenetic Placement of Exact Amplicon Sequences Improves Associations with Clinical Information. mSystems 2018, 3, e00021-18. [Google Scholar] [CrossRef]
- Robeson, M.S.; O’Rourke, D.R.; Kaehler, B.D.; Ziemski, M.; Dillon, M.R.; Foster, J.T.; Bokulich, N.A. RESCRIPt: Reproducible sequence taxonomy reference database management for the masses. bioRxiv 2020. [Google Scholar] [CrossRef]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2013, 41, D590–D596. [Google Scholar] [CrossRef] [PubMed]
- Anderson, M.J. A new method for non-parametric multivariate analysis of variance. Austral Ecol. 2001, 26, 32–46. [Google Scholar]
- Caspi, R.; Billington, R.; Ferrer, L.; Foerster, H.; Fulcher, C.A.; Keseler, I.M.; Kothari, A.; Krummenacker, M.; Latendresse, M.; Mueller, L.A.; et al. The MetaCyc database of metabolic pathways and enzymes and the BioCyc collection of pathway/genome databases. Nucleic Acids Res. 2016, 44, D471–D480. [Google Scholar] [CrossRef] [PubMed]
- Douglas, G.M.; Maffei, V.J.; Zaneveld, J.R.; Yurgel, S.N.; Brown, J.R.; Taylor, C.M.; Huttenhower, C.; Langille, M.G.I. PICRUSt2 for prediction of metagenome functions. Nat. Biotechnol. 2020, 38, 685–688. [Google Scholar] [CrossRef] [PubMed]
- Mandal, S.; Van Treuren, W.; White, R.A.; Eggesbø, M.; Knight, R.; Peddada, S.D. Analysis of composition of microbiomes: A novel method for studying microbial composition. Microb. Ecol. Health Dis. 2015, 26, 27663. [Google Scholar] [CrossRef] [PubMed]
- McMurdie, P.J.; Holmes, S. phyloseq: An R package for reproducible interactive analysis and graphics of microbiome census data. PLoS ONE 2013, 8, e61217. [Google Scholar] [CrossRef]
- Lahti, L.; Shetty, S. Microbiome R Package (2012–2019). Available online: https://www.bioconductor.org/packages/release/bioc/html/microbiome.html (accessed on 29 September 2022).
- Harrell, F.E., Jr.; Dupont, C. Hmisc: Harrell Miscellaneous. R Package Version 4.6-0. 2021. Available online: https://CRAN.R-project.org/package=Hmisc/index.html (accessed on 11 August 2022).
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Biswas, A.; Patgiri, R.; Biswas, B. Principles of Social Networking: The New Horizon and Emerging Challenges; Springer Nature: Berlin/Heidelberg, Germany, 2021. [Google Scholar]
- Layeghifard, M.; Hwang, D.M.; Guttman, D.S. Disentangling Interactions in the Microbiome: A Network Perspective. Trends Microbiol. 2017, 25, 217–228. [Google Scholar] [CrossRef]
- Clauset, A.; Newman, M.E.J.; Moore, C. Finding Community Structure in Very Large Networks. Available online: https://www.arxiv.org/abs/cond-mat/0408187 (accessed on 29 September 2022).
- Wu, Y.; Li, Q.; Zhang, R.; Dai, X.; Chen, W.; Xing, D. Circulating microRNAs: Biomarkers of disease. Clin. Chim. Acta 2021, 516, 46–54. [Google Scholar] [CrossRef]
- Galvão-Lima, L.J.; Morais, A.H.F.; Valentim, R.A.M.; Barreto, E.J.S.S. miRNAs as biomarkers for early cancer detection and their application in the development of new diagnostic tools. Biomed. Eng. Online 2021, 20, 21. [Google Scholar] [CrossRef] [PubMed]
- Okuda, Y.; Shimura, T.; Iwasaki, H.; Fukusada, S.; Nishigaki, R.; Kitagawa, M.; Katano, T.; Okamoto, Y.; Yamada, T.; Horike, S.-I.; et al. Urinary microRNA biomarkers for detecting the presence of esophageal cancer. Sci. Rep. 2021, 11, 8508. [Google Scholar] [CrossRef] [PubMed]
- Masi, L.; Capobianco, I.; Magrì, C.; Marafini, I.; Petito, V.; Scaldaferri, F. MicroRNAs as Innovative Biomarkers for Inflammatory Bowel Disease and Prediction of Colorectal Cancer. Int. J. Mol. Sci. 2022, 23, 7991. [Google Scholar] [CrossRef] [PubMed]
- Mi, S.; Zhang, J.; Zhang, W.; Huang, R.S. Circulating microRNAs as biomarkers for inflammatory diseases. Microrna 2013, 2, 63–71. [Google Scholar] [CrossRef]
- Shannon, C.E. A Mathematical Theory of Communication. Bell Syst. Tech. J. 1948, 27, 623–656. [Google Scholar] [CrossRef]
- Selvaratnam, S.; Gullino, S.; Shim, L.; Lee, E.; Lee, A.; Paramsothy, S.; Leong, R.W. Epidemiology of inflammatory bowel disease in South America: A systematic review. World J. Gastroenterol. 2019, 25, 6866–6875. [Google Scholar] [CrossRef]
- Iskandar, H.N.; Ciorba, M.A. Biomarkers in inflammatory bowel disease: Current practices and recent advances. Transl Res. 2012, 159, 313–325. [Google Scholar] [CrossRef]
- Paraskevi, A.; Theodoropoulos, G.; Papaconstantinou, I.; Mantzaris, G.; Nikiteas, N.; Gazouli, M. Circulating MicroRNA in inflammatory bowel disease. J. Crohns Colitis. 2012, 6, 900–904. [Google Scholar] [CrossRef]
- Pathak, P.H.; Feng, X.; Hu, P.; Mohapatra, P. Visible Light Communication, Networking, and Sensing: A Survey, Potential and Challenges. IEEE Commun. Surv. Tutor. 2015, 17, 2047–2077. [Google Scholar] [CrossRef]
- Ichii, O.; Horino, T. MicroRNAs associated with the development of kidney diseases in humans and animals. J. Toxicol. Pathol. 2018, 31, 23–34. [Google Scholar] [CrossRef]
- Pathak, S.; Grillo, A.R.; Scarpa, M.; Brun, P.; D’Incà, R.; Nai, L.; Banerjee, A.; Cavallo, D.; Barzon, L.; Palù, G.; et al. MiR-155 Modulates the Inflammatory Phenotype of Intestinal Myofibroblasts by Targeting SOCS1 in Ulcerative Colitis. Exp. Mol. Med. 2015, 47, e164. [Google Scholar] [CrossRef] [PubMed]
- Szűcs, D.; Béres, N.J.; Rokonay, R.; Boros, K.; Borka, K.; Kiss, Z.; Arató, A.; Szabó, A.J.; Vannay, Á.; Sziksz, E.; et al. Increased Duodenal Expression of miR-146a and -155 in Pediatric Crohn’s Disease. World J. Gastroenterol. 2016, 22, 6027–6035. [Google Scholar] [CrossRef] [PubMed]
- Yuan, X.; Berg, N.; Lee, J.W.; Le, T.-T.; Neudecker, V.; Jing, N.; Eltzschig, H. MicroRNA miR-223 as Regulator of Innate Immunity. J. Leukoc. Biol. 2018, 104, 515–524. [Google Scholar] [CrossRef] [PubMed]
- Carpenter, S.; Carlson, T.; Dellacasagrande, J.; Garcia, A.; Gibbons, S.; Hertzog, P.; Lyons, A.; Lin, L.-L.; Lynch, M.; Monie, T.; et al. TRIL, a Functional Component of the TLR4 Signaling Complex, Highly Expressed in Brain. J. Immunol. 2009, 183, 3989–3995. [Google Scholar] [CrossRef]
- Wang, H.; Chao, K.; Ng, S.C.; Bai, A.H.; Yu, Q.; Yu, J.; Li, M.; Cui, Y.; Chen, M.; Hu, J.-F.; et al. Pro-Inflammatory miR-223 Mediates the Cross-Talk between the IL23 Pathway and the Intestinal Barrier in Inflammatory Bowel Disease. Genome Biol. 2016, 17, 58. [Google Scholar] [CrossRef]
- Landgraf, P.; Rusu, M.; Sheridan, R.; Sewer, A.; Iovino, N.; Aravin, A.; Pfeffer, S.; Rice, A.; Kamphorst, A.O.; Landthaler, M.; et al. A Mammalian microRNA Expression Atlas Based on Small RNA Library Sequencing. Cell 2007, 129, 1401–1414. [Google Scholar] [CrossRef]
- Bajer, L.; Kverka, M.; Kostovcik, M.; Macinga, P.; Dvorak, J.; Stehlikova, Z.; Brezina, J.; Wohl, P.; Spicak, J.; Drastich, P. Distinct Gut Microbiota Profiles in Patients with Primary Sclerosing Cholangitis and Ulcerative Colitis. World J. Gastroenterol. 2017, 23, 4548–4558. [Google Scholar] [CrossRef]
- Chen, G.-L.; Zhang, Y.; Wang, W.-Y.; Ji, X.-L.; Meng, F.; Xu, P.-S.; Yang, N.-M.; Ye, F.-Q.; Bo, X.-C. Partners of Patients with Ulcerative Colitis Exhibit a Biologically Relevant Dysbiosis in Fecal Microbial Metacommunities. World J. Gastroenterol. 2017, 23, 4624–4631. [Google Scholar] [CrossRef]
- Soen, Y. Environmental Disruption of Host-Microbe Co-Adaptation as a Potential Driving Force in Evolution. Front. Genet. 2014, 5, 168. [Google Scholar] [CrossRef]
- Wang, W.; Chen, L.; Zhou, R.; Wang, X.; Song, L.; Huang, S.; Wang, G.; Xia, B. Increased Proportions of Bifidobacterium and the Lactobacillus Group and Loss of Butyrate-Producing Bacteria in Inflammatory Bowel Disease. J. Clin. Microbiol. 2014, 52, 398–406. [Google Scholar] [CrossRef]
- Fedorak, R.N. Probiotics in the Management of Ulcerative Colitis. Gastroenterol. Hepatol. 2010, 6, 688–690. [Google Scholar]
- Linares, D.M.; Gómez, C.; Renes, E.; Fresno, J.M.; Tornadijo, M.E.; Ross, R.P.; Stanton, C. Lactic Acid Bacteria and Bifidobacteria with Potential to Design Natural Biofunctional Health-Promoting Dairy Foods. Front. Microbiol. 2017, 8, 846. [Google Scholar] [CrossRef]
- Singh, S.; Bhatia, R.; Khare, P.; Sharma, S.; Rajarammohan, S.; Bishnoi, M.; Bhadada, S.K.; Sharma, S.S.; Kaur, J.; Kondepudi, K.K. Anti-Inflammatory Bifidobacterium Strains Prevent Dextran Sodium Sulfate Induced Colitis and Associated Gut Microbial Dysbiosis in Mice. Sci. Rep. 2020, 10, 18597. [Google Scholar] [CrossRef]
- Kuwabara, A.; Tanaka, K.; Tsugawa, N.; Nakase, H.; Tsuji, H.; Shide, K.; Kamao, M.; Chiba, T.; Inagaki, N.; Okano, T.; et al. High Prevalence of Vitamin K and D Deficiency and Decreased BMD in Inflammatory Bowel Disease. Osteoporos. Int. 2009, 20, 935–942. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, X.; Tian, Z.; Li, N.; Mao, R.; Li, X.; Zhao, M.; Xiong, S.; Zeng, Z.; Feng, R.; Chen, M. Gut Microbiota Profiles and Microbial-Based Therapies in Post-Operative Crohn’s Disease: A Systematic Review. Front. Med. 2020, 7, 615858. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; He, Y.; Liu, L.; Zhou, W.; Wang, P.; Hu, H.; Nie, Y.; Chen, Y. Alterations in Gut Microbial Communities Across Anatomical Locations in Inflammatory Bowel Diseases. Front. Nutr. 2021, 8, 615064. [Google Scholar] [CrossRef]
- Alam, M.T.; Amos, G.C.A.; Murphy, A.R.J.; Murch, S.; Wellington, E.M.H.; Arasaradnam, R.P. Microbial Imbalance in Inflammatory Bowel Disease Patients at Different Taxonomic Levels. Gut Pathog. 2020, 12, 1. [Google Scholar] [CrossRef]
- Amos, G.C.A.; Sergaki, C.; Logan, A.; Iriarte, R.; Bannaga, A.; Chandrapalan, S.; Wellington, E.M.H.; Rijpkema, S.; Arasaradnam, R.P. Exploring How Microbiome Signatures Change across Inflammatory Bowel Disease Conditions and Disease Locations. Sci. Rep. 2021, 11, 18699. [Google Scholar] [CrossRef]
- Liu, Y.; Hou, Y.; Wang, G.; Zheng, X.; Hao, H. Gut Microbial Metabolites of Aromatic Amino Acids as Signals in Host-Microbe Interplay. Trends Endocrinol. Metab. 2020, 31, 818–834. [Google Scholar] [CrossRef]
- Dubinsky, V.; Reshef, L.; Rabinowitz, K.; Yadgar, K.; Godny, L.; Zonensain, K.; Wasserberg, N.; Dotan, I.; Gophna, U. Dysbiosis in Metabolic Genes of the Gut Microbiomes of Patients with an Ileo-Anal Pouch Resembles That Observed in Crohn’s Disease. mSystems 2021, 6. [Google Scholar] [CrossRef]
- Santoru, M.L.; Piras, C.; Murgia, A.; Palmas, V.; Camboni, T.; Liggi, S.; Ibba, I.; Lai, M.A.; Orrù, S.; Blois, S.; et al. Cross Sectional Evaluation of the Gut-Microbiome Metabolome Axis in an Italian Cohort of IBD Patients. Sci. Rep. 2017, 7. [Google Scholar] [CrossRef] [PubMed]
- Jansson, J.; Willing, B.; Lucio, M.; Fekete, A.; Dicksved, J.; Halfvarson, J.; Tysk, C.; Schmitt-Kopplin, P. Metabolomics Reveals Metabolic Biomarkers of Crohn’s Disease. PLoS One 2009, 4, e6386. [Google Scholar] [CrossRef] [PubMed]
- Shade, A.; Handelsman, J. Beyond the Venn Diagram: The Hunt for a Core Microbiome. Environ. Microbiol. 2012, 14, 4–12. [Google Scholar] [CrossRef] [PubMed]
- Neu, A.T.; Allen, E.E.; Roy, K. Defining and Quantifying the Core Microbiome: Challenges and Prospects. Proc. Natl. Acad. Sci. USA 2021, 118, e2104429118. [Google Scholar] [CrossRef]
- Kushkevych, I.; Dordević, D.; Kollár, P. Analysis of Physiological Parameters of Desulfovibrio Strains from Individuals with Colitis. Open Life Sci. 2019, 13, 481–488. [Google Scholar] [CrossRef]
- Palm, N.W.; de Zoete, M.R.; Cullen, T.W.; Barry, N.A.; Stefanowski, J.; Hao, L.; Degnan, P.H.; Hu, J.; Peter, I.; Zhang, W.; et al. Immunoglobulin A Coating Identifies Colitogenic Bacteria in Inflammatory Bowel Disease. Cell 2014, 158, 1000–1010. [Google Scholar] [CrossRef]
- Joossens, M.; Huys, G.; Cnockaert, M.; De Preter, V.; Verbeke, K.; Rutgeerts, P.; Vandamme, P.; Vermeire, S. Dysbiosis of the Faecal Microbiota in Patients with Crohn’s Disease and Their Unaffected Relatives. Gut 2011, 60, 631–637. [Google Scholar] [CrossRef]
- Shapiro, J.M.; de Zoete, M.R.; Palm, N.W.; Laenen, Y.; Bright, R.; Mallette, M.; Bu, K.; Bielecka, A.A.; Xu, F.; Hurtado-Lorenzo, A.; et al. Immunoglobulin A Targets a Unique Subset of the Microbiota in Inflammatory Bowel Disease. Cell Host Microbe 2021, 29, 83–93.e3. [Google Scholar] [CrossRef]
- Qiu, Z.; Yang, H.; Rong, L.; Ding, W.; Chen, J.; Zhong, L. Targeted Metagenome Based Analyses Show Gut Microbial Diversity of Inflammatory Bowel Disease Patients. Indian J. Microbiol. 2017, 57, 307–315. [Google Scholar] [CrossRef]
- Jones, C.M.A.; Connors, J.; Dunn, K.A.; Bielawski, J.P.; Comeau, A.M.; Langille, M.G.I.; Van Limbergen, J. Bacterial Taxa and Functions Are Predictive of Sustained Remission Following Exclusive Enteral Nutrition in Pediatric Crohn’s Disease. Inflamm. Bowel Dis. 2020, 26, 1026–1037. [Google Scholar] [CrossRef]
- Qin, J.; Li, R.; Raes, J.; Arumugam, M.; Burgdorf, K.S.; Manichanh, C.; Nielsen, T.; Pons, N.; Levenez, F.; Yamada, T.; et al. A Human Gut Microbial Gene Catalogue Established by Metagenomic Sequencing. Nature 2010, 464, 59–65. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Xu, P.; Zhang, Z.; Zhou, X.; Chen, C.; Lu, C. Platelets Can Reflect the Severity of Crohn’s Disease without the Effect of Anemia. Clinics 2020, 75, e1596. [Google Scholar] [CrossRef] [PubMed]
- Garraud, O. Platelets as Immune Cells in Physiology and Immunopathology; Frontiers Media SA: Lausanne, Switzerland, 2016. [Google Scholar]
- Cognasse, F.; Lafarge, S.; Chavarin, P.; Acquart, S.; Garraud, O. Lipopolysaccharide Induces sCD40L Release through Human Platelets TLR4, but Not TLR2 and TLR9. Intensive Care Med. 2007, 33, 382–384. [Google Scholar] [CrossRef] [PubMed]
- Smyth, S.S.; McEver, R.P.; Weyrich, A.S.; Morrell, C.N.; Hoffman, M.R.; Arepally, G.M.; French, P.A.; Dauerman, H.L.; Becker, R.C. Platelet functions beyond hemostasis. J. Thromb. Haemost. 2009, 7, 1759–1766. [Google Scholar] [CrossRef] [PubMed]
- Banchereau, J.; Steinman, R.M. Dendritic Cells and the Control of Immunity. Nature 1998, 392, 245–252. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.-F.; Niu, G.-C.; Li, C.-Y.; Guo, J.-B.; Song, J.; Li, H.; Zhang, X.-L. Mechanism of Ulcerative Colitis-Aggravated Liver Fibrosis: The Activation of Hepatic Stellate Cells and TLR4 Signaling through Gut-Liver Axis. Front. Physiol. 2021, 12, 695019. [Google Scholar] [CrossRef]
- Ni, J.; Wu, G.D.; Albenberg, L.; Tomov, V.T. Gut Microbiota and IBD: Causation or Correlation? Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 573–584. [Google Scholar] [CrossRef]
- Mazzini, F.N.; Cook, F.; Gounarides, J.; Marciano, S.; Haddad, L.; Tamaroff, A.J.; Casciato, P.; Narvaez, A.; Mascardi, M.F.; Anders, M.; et al. Plasma and Stool Metabolomics to Identify Microbiota Derived-Biomarkers of Metabolic Dysfunction-Associated Fatty Liver Disease: Effect of PNPLA3 Genotype. Metabolomics 2021, 17, 58. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Groups | UC | CD | Non-IBD Controls | p | |
|---|---|---|---|---|---|
| General descriptions | Overall subjects | n = 20 | n = 14 | n = 13 | - |
| Female % | 60 | 57 | 69 | ns | |
| Male % | 40 | 43 | 31 | ns | |
| Mean age, years ± (SD) | 46.65 ± (17.80) | 44.42 ± (16.61) | 52.84 ± (17.94) | ns | |
| BMI ± (SD) | 26.60 ± (4.09) | 25.01 ± (3.84) | 28.40 ± (5.44) | ns |
| Groups | UC | CD | |
|---|---|---|---|
| mean ± (SD) | |||
| Biochemical data | Hemoglobin, g/dL | 13.74 ± (1.41) | 12.61 ± (1.92) |
| us-CRP, mg/dL | 0.35 ± (0.26) | 1.37 ± (2.48) | |
| Albumin, g/dL | 4.55 ± (0.34) | 4.39± (0.25) | |
| Platelets, ×103/mL | 238.11 ± (58.02) | 297.86 ± (107.15) | |
| FPG, mg/dL | 92.43 ± (13.36) | 97.35 ± (15.98) | |
| Creatinine, mg/dL | 0.77 ± (0.16) | 0.82 ± (0.22) | |
| Triglycerides, mg/dL | 92.47 ± (49.33) | 107.22 ± (43.28) | |
| Total cholesterol, mg/dL | 177.77 ± (43.04) | 216.16 ± (53.09) | |
| LDL-C, mg/dL | 109.07 ± (42.50) | 136.89 ± (41.31) | |
| HDL-C, mg/dL | 56.87 ± (13.44) | 61.50 ± (22.45) | |
| GOT (UI/I) | 23.89 ± (8.49) | 23.89 ± (8.49) | |
| GPT (UI/I) | 22.14 ± (10.88) | 23.14 ± (10.88) | |
| No. (%) | |||
| IBD therapy | 5-ASA | 20 (100.00) | 14 (100.00) |
| Steroids | 1 (5.00) | 5 (35.71) | |
| AZA | 4 (20.00) | 3 (21.42) | |
| Rectal budesonide | 5 (25.00) | 4 (28.57) | |
| ADA | 0 (0.00) | 2 (14.28) | |
| Lesion localization UC | E1 proctitis | 1 (5.00) | - |
| E2 left-sided colitis | 8 (40.00) | - | |
| E3 extensive | 11 (55.00) | - | |
| Lesion localization CD | L1 ileal | - | 0 (0.00) |
| L2 colon | - | 8 (57.14) | |
| L3 ileocolonic | - | 4 (28.57) | |
| L2-L4 upper GIT | - | 1 (7.14) | |
| L3-L4 upper GIT | - | 1 (7.14) | |
| General activity according to medical criteria | General remission | 10 (50.00) | 4 (28.57) |
| General active | 10 (50.00) | 10 (71.42) | |
| Clinical activity (Truelove & Witts and CDAI) | Remission | 15 (75.00) | 10 (71.42) |
| Mild | 5 (25.00) | 4 (28.57) | |
| Moderate | 0 (0.00) | 0 (0.00) | |
| Severe | 0 (0.00) | 0 (0.00) | |
| Endoscopic score (Mayo score and SES-CD) | Normal | 10 (50.00) | 4 (28.57) |
| Mild | 7 (35.00) | 4 (28.57) | |
| Moderate | 1 (5.00) | 3 (21.42) | |
| Severe | 2 (10.00) | 3 (21.42) | |
| Histology activity | Quiescent | 10 (50.00) | 3 (21.42) |
| Inflammatory infiltrate | 10 (50.00) | 11 (78.57) | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rosso, A.D.; Aguilera, P.; Quesada, S.; Mascardi, F.; Mascuka, S.N.; Cimolai, M.C.; Cerezo, J.; Spiazzi, R.; Conlon, C.; Milano, C.; et al. Comprehensive Phenotyping in Inflammatory Bowel Disease: Search for Biomarker Algorithms in the Transkingdom Interactions Context. Microorganisms 2022, 10, 2190. https://doi.org/10.3390/microorganisms10112190
Rosso AD, Aguilera P, Quesada S, Mascardi F, Mascuka SN, Cimolai MC, Cerezo J, Spiazzi R, Conlon C, Milano C, et al. Comprehensive Phenotyping in Inflammatory Bowel Disease: Search for Biomarker Algorithms in the Transkingdom Interactions Context. Microorganisms. 2022; 10(11):2190. https://doi.org/10.3390/microorganisms10112190
Chicago/Turabian StyleRosso, Ayelén D., Pablo Aguilera, Sofía Quesada, Florencia Mascardi, Sebastian N. Mascuka, María C. Cimolai, Jimena Cerezo, Renata Spiazzi, Carolina Conlon, Claudia Milano, and et al. 2022. "Comprehensive Phenotyping in Inflammatory Bowel Disease: Search for Biomarker Algorithms in the Transkingdom Interactions Context" Microorganisms 10, no. 11: 2190. https://doi.org/10.3390/microorganisms10112190
APA StyleRosso, A. D., Aguilera, P., Quesada, S., Mascardi, F., Mascuka, S. N., Cimolai, M. C., Cerezo, J., Spiazzi, R., Conlon, C., Milano, C., Iraola, G. M., Penas-Steinhardt, A., & Belforte, F. S. (2022). Comprehensive Phenotyping in Inflammatory Bowel Disease: Search for Biomarker Algorithms in the Transkingdom Interactions Context. Microorganisms, 10(11), 2190. https://doi.org/10.3390/microorganisms10112190