
Deciphering Rhizosphere Microbiome Assembly of Castanea henryi in Plantation and Natural Forest

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Collection
2.2. DNA Extraction and Metagenomic Data Analysis
2.3. Construction of Molecular Ecological Network
2.4. Statistical Analysis
3. Results
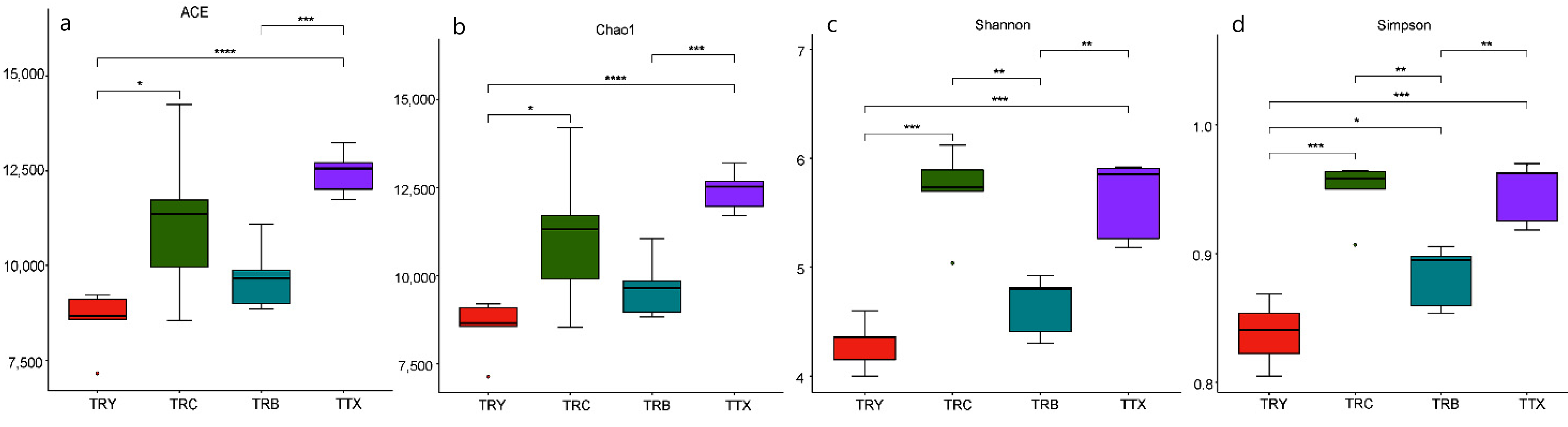
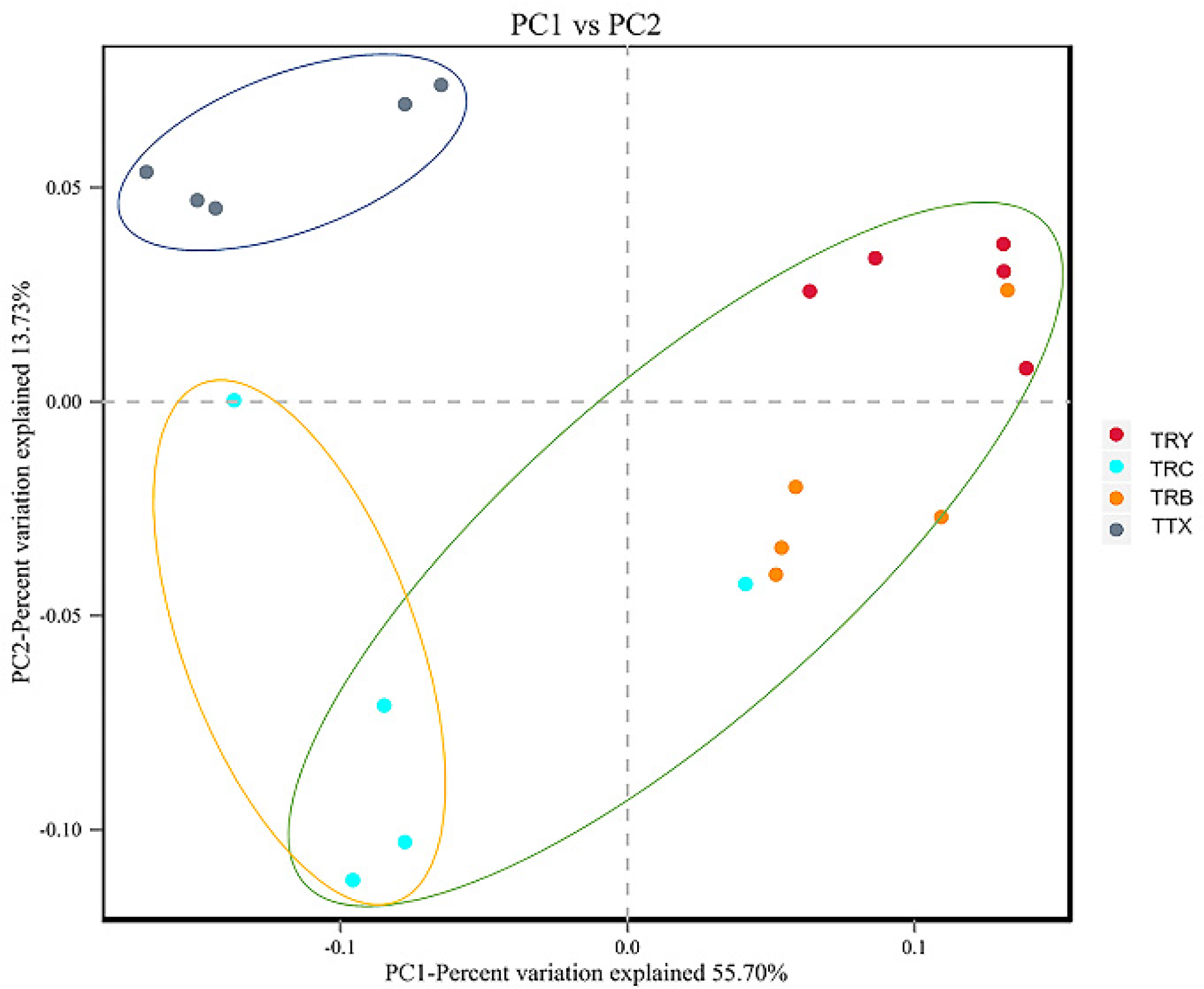
3.1. Diversity of Microbial Communities at Different Forest Types and Cultivated Varieties
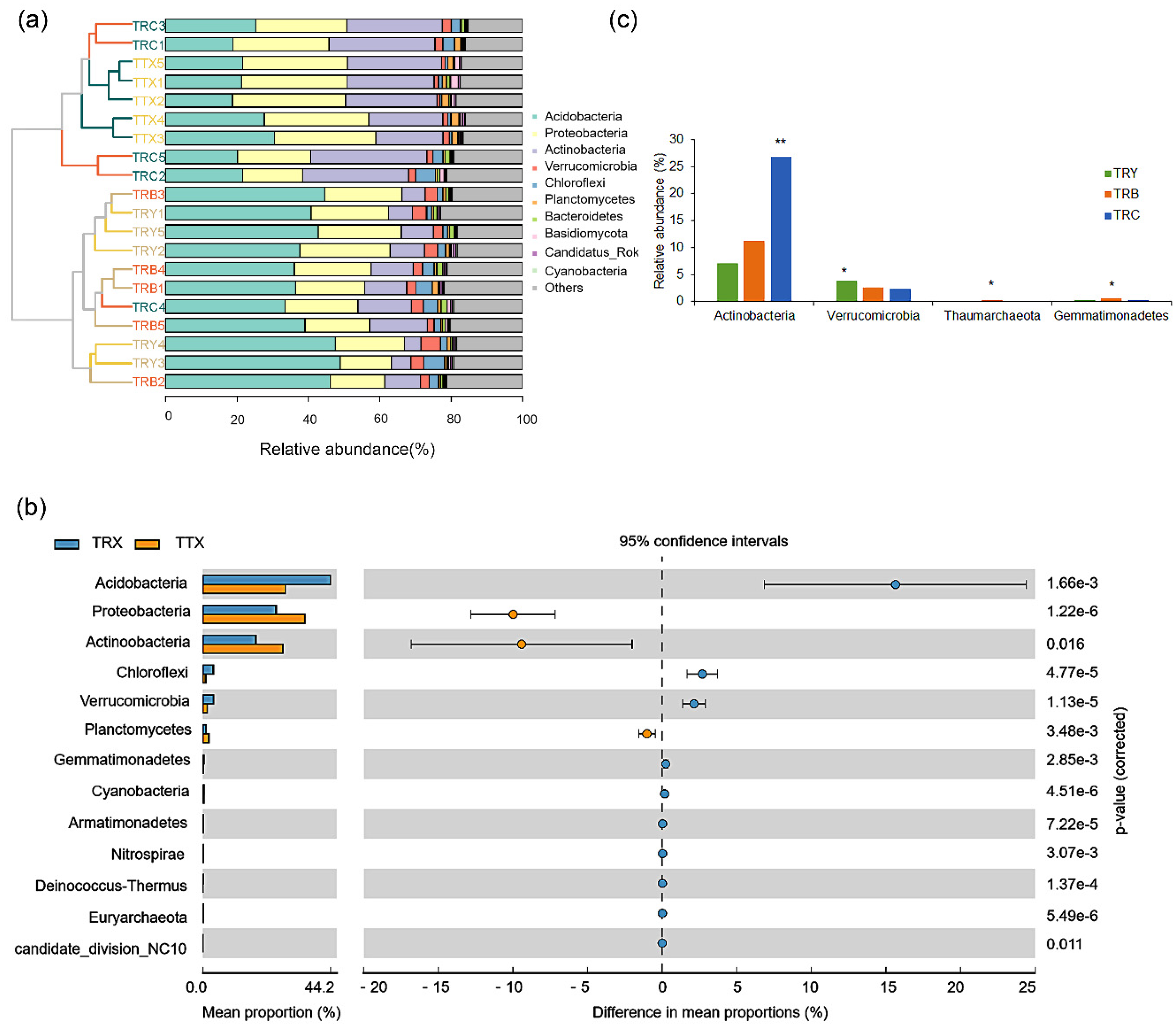
3.2. Specific Differences in Composition of Microbiome at Different Forest Types and Cultivated Varieties
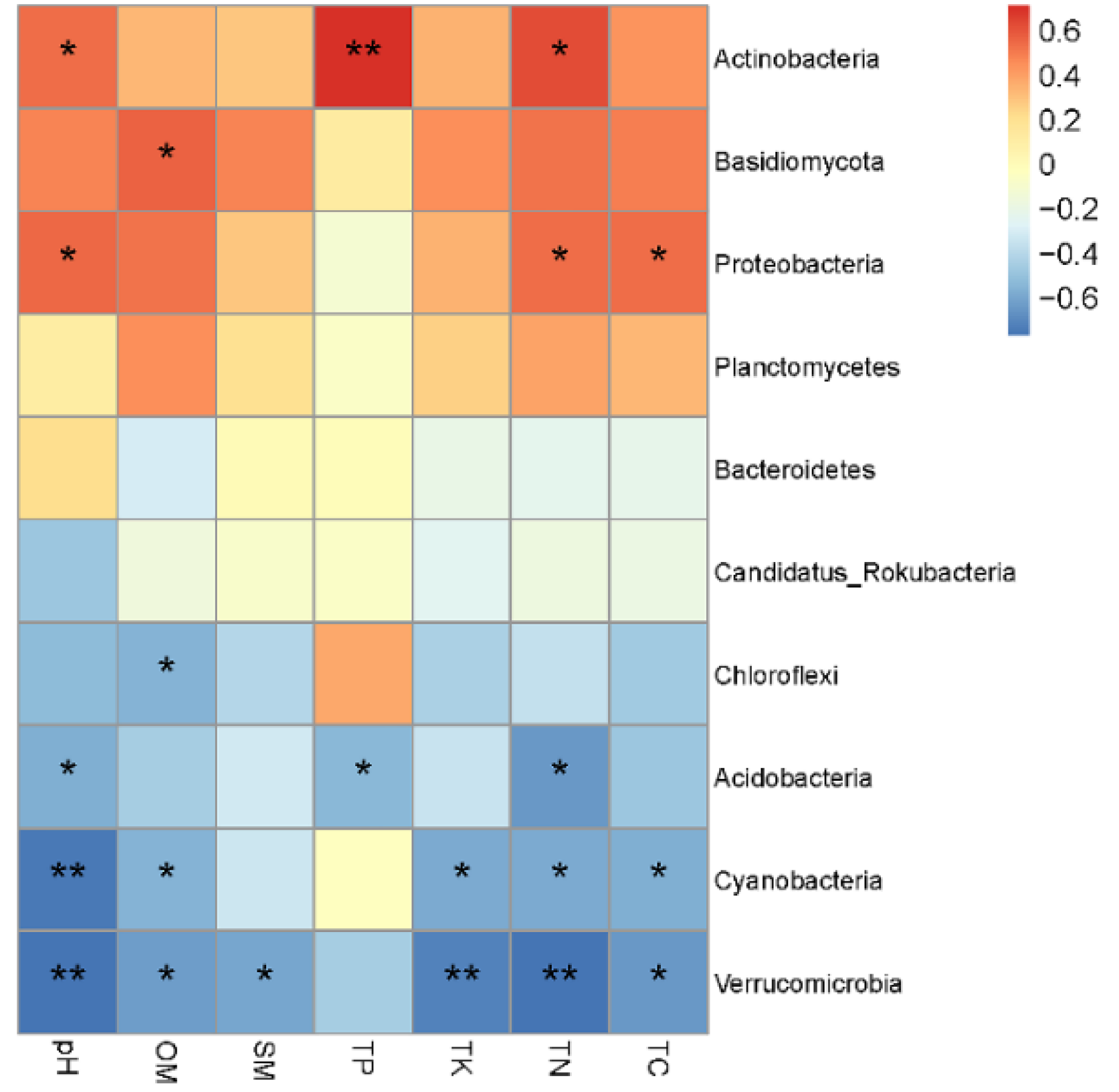
3.3. Relationship between Microbial Communities and Soil Physicochemical Properties
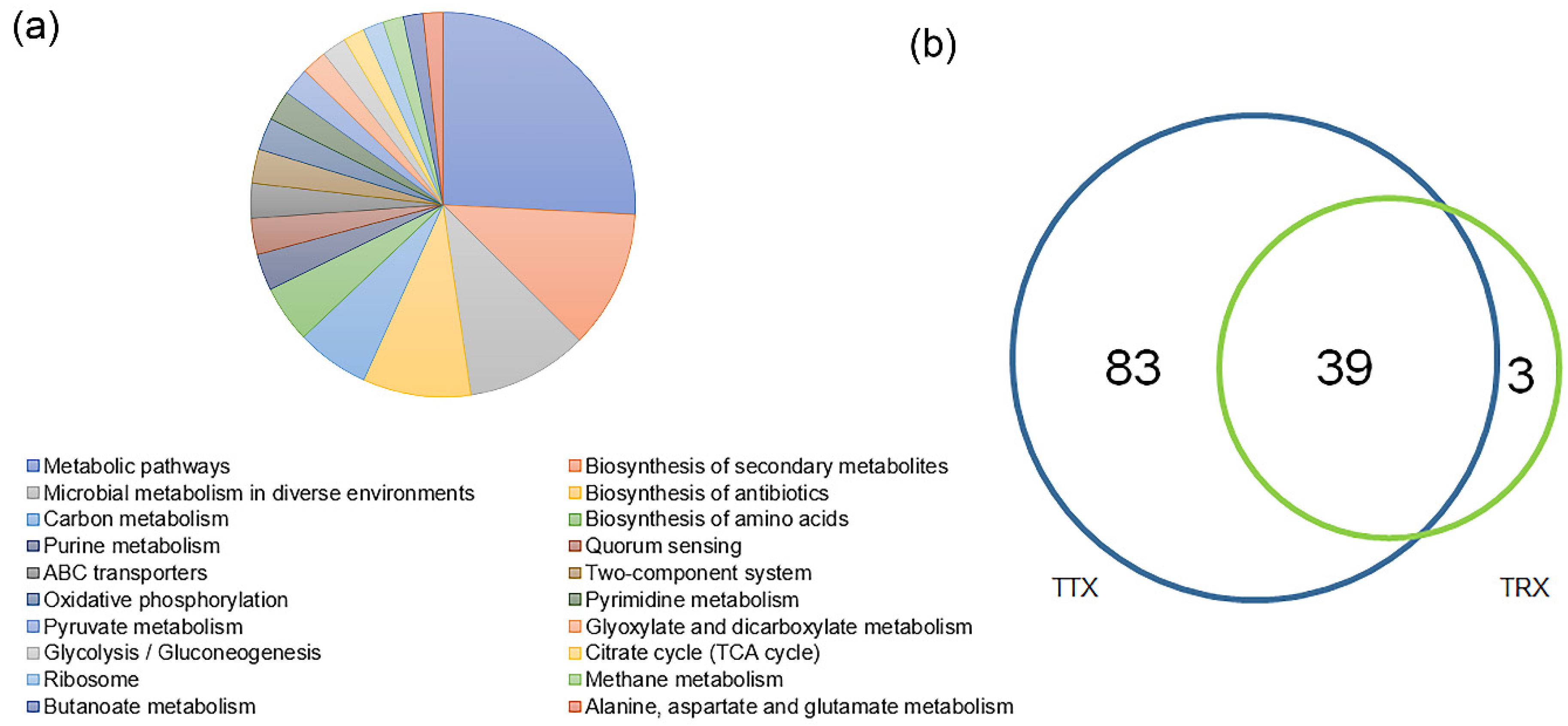
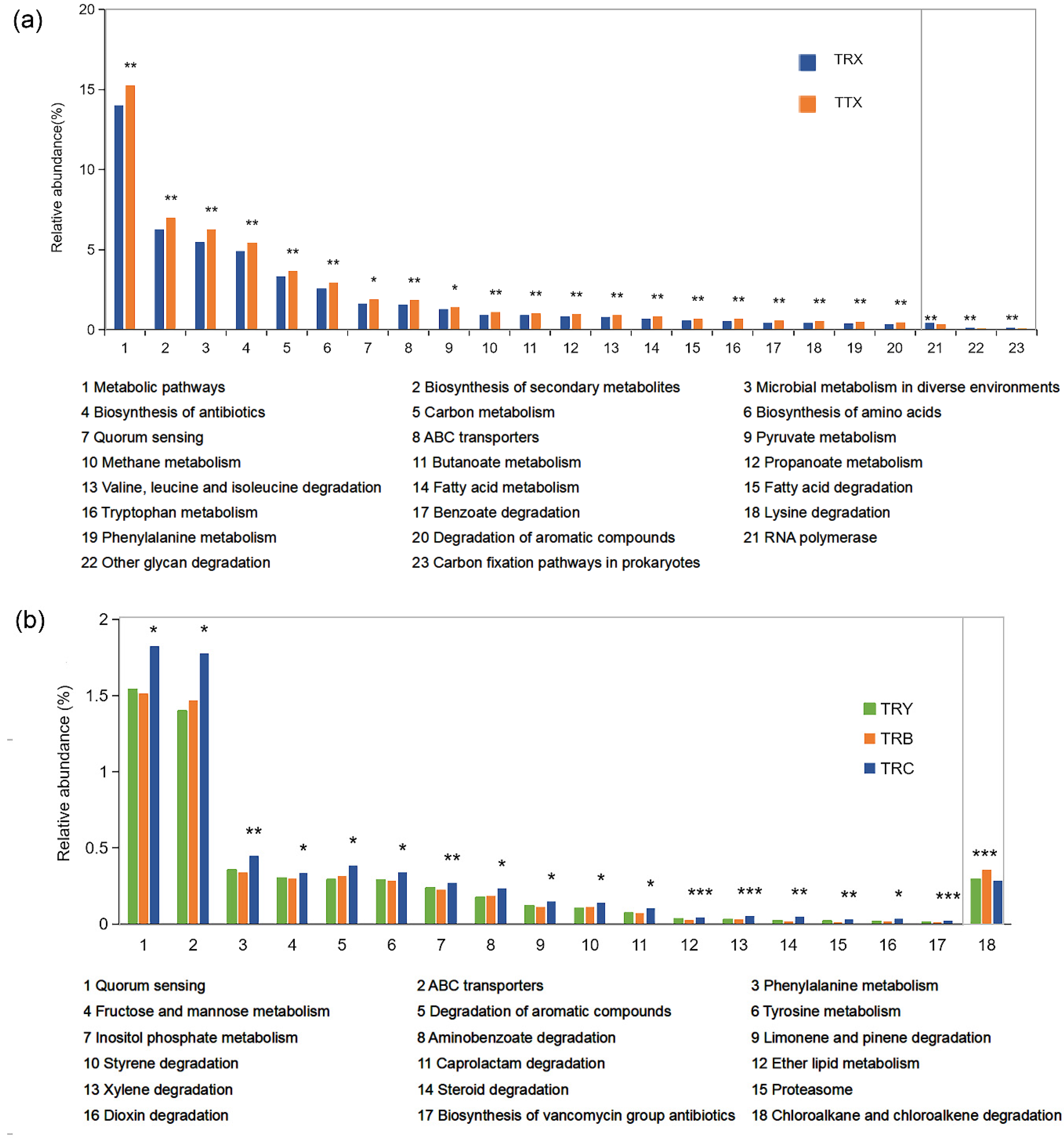
3.4. Specific Differences in Function of Microbiome at Different Forest Types and Different Cultivated Varieties
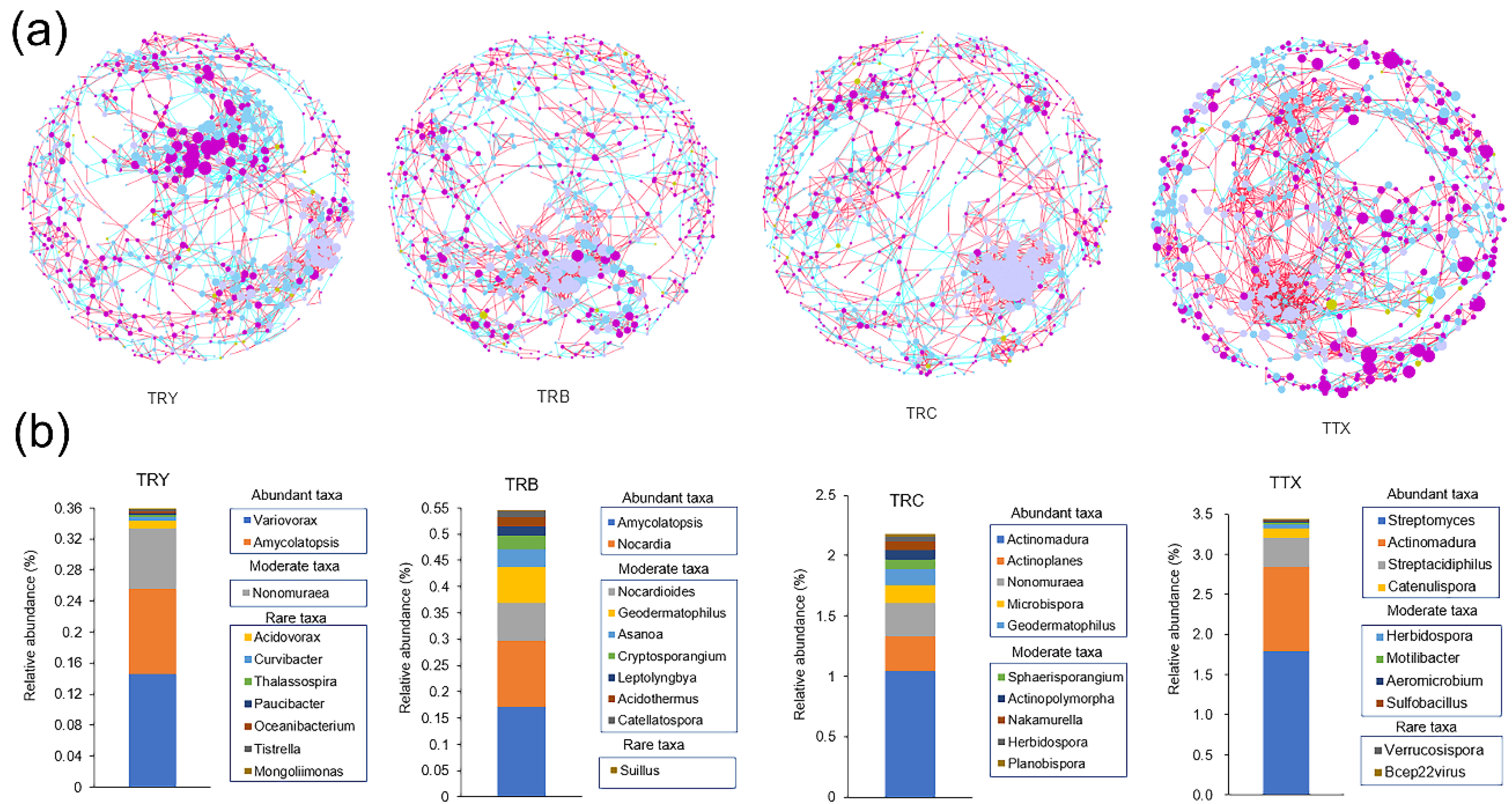
3.5. Molecular Ecological Network Structure of Soil Microbial Community
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bardgett, R.D.; van der Putten, W.H. Belowground biodiversity and ecosystem functioning. Nature 2014, 515, 505–511. [Google Scholar] [CrossRef] [PubMed]
- Mommer, L.; Cotton, T.E.A.; Raaijmakers, J.M.; Termorshuizen, A.J.; van Ruijven, J.; Hendriks, M.; van Rijssel, S.Q.; van de Mortel, J.E.; van der Paauw, J.W.; Schijlen, E.; et al. Lost in diversity: The interactions between soil-borne fungi, biodiversity and plant productivity. New Phytol. 2018, 218, 542–553. [Google Scholar] [CrossRef] [Green Version]
- Wagg, C.; Bender, S.F.; Widmer, F.; van der Heijden, M.G.A. Soil biodiversity and soil community composition determine ecosystem multifunctionality. Proc. Natl. Acad. Sci. USA 2014, 111, 5266–5270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiao, S.; Peng, Z.H.; Qi, J.J.; Gao, J.M.; Wei, G.H. Linking Bacterial-Fungal Relationships to Microbial Diversity and Soil Nutrient Cycling. mSystems 2021, 6, 16. [Google Scholar] [CrossRef]
- Luo, G.; Xue, C.; Jiang, Q.; Xiao, Y.; Zhang, F.; Guo, S.; Shen, Q.; Ling, N. Soil carbon, nitrogen, and phosphorus cycling microbial populations and their resistance to global change depend on soil C:N:P stoichiometry. mSystems 2020, 5, 15. [Google Scholar] [CrossRef]
- Xu, R.; Li, B.; Xiao, E.; Young, L.Y.; Sun, X.; Kong, T.; Dong, Y.; Wang, Q.; Yang, Z.; Chen, L.; et al. Uncovering microbial responses to sharp geochemical gradients in a terrace contaminated by acid mine drainage. Environ. Pollut. 2020, 261, 114226. [Google Scholar] [CrossRef]
- Zhang, M.; Li, Z.; Haggblom, M.M.; Young, L.; He, Z.; Li, F.; Xu, R.; Sun, X.; Sun, W. Characterization of Nitrate-Dependent As(III)-Oxidizing Communities in Arsenic-Contaminated Soil and Investigation of Their Metabolic Potentials by the Combination of DNA-Stable Isotope Probing and Metagenomics. Environ. Sci. Technol. 2020, 54, 7366–7377. [Google Scholar] [CrossRef] [PubMed]
- Aranda, E.; Scervino, J.M.; Godoy, P.; Reina, R.; Ocampo, J.A.; Wittich, R.M.; Garcia-Romera, I. Role of arbuscular mycorrhizal fungus Rhizophagus custos in the dissipation of PAHs under root-organ culture conditions. Environ. Pollut. 2013, 181, 182–189. [Google Scholar] [CrossRef] [PubMed]
- Pellitier, P.T.; Ibanez, I.; Zak, D.R.; Argiroff, W.A.; Acharya, K. Ectomycorrhizal access to organic nitrogen mediates CO2 fertilization response in a dominant temperate tree. Nat. Commun. 2021, 12, 10. [Google Scholar] [CrossRef] [PubMed]
- Lu, T.; Ke, M.; Lavoie, M.; Jin, Y.; Fan, X.; Zhang, Z.; Fu, Z.; Sun, L.; Gillings, M.; Penuelas, J.; et al. Rhizosphere microorganisms can influence the timing of plant flowering. Microbiome 2018, 6, 1–12. [Google Scholar] [CrossRef]
- de Vries, F.T.; Griffiths, R.I.; Bailey, M.; Craig, H.; Girlanda, M.; Gweon, H.S.; Hallin, S.; Kaisermann, A.; Keith, A.M.; Kretzschmar, M.; et al. Soil bacterial networks are less stable under drought than fungal networks. Nat. Commun. 2018, 9, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Vries, F.T.; Griffiths, R.I.; Knight, C.G.; Nicolitch, O.; Williams, A. Harnessing rhizosphere microbiomes for drought-resilient crop production. Science 2020, 368, 270–274. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.Y.; Chen, H.Y.; Ding, G.J.; Li, K.F.; Ren, Q.F. Identification of candidate genes conferring tolerance to aluminum stress in Pinus massoniana inoculated with ectomycorrhizal fungus. BMC Plant Biol. 2020, 20, 13. [Google Scholar] [CrossRef]
- Cong, J.; Yang, Y.; Liu, X.; Lu, H.; Liu, X.; Zhou, J.; Li, D.; Yin, H.; Ding, J.; Zhang, Y. Analyses of soil microbial community compositions and functional genes reveal potential consequences of natural forest succession. Sci. Rep. 2015, 5, 10007. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.; He, C.; Xu, G.; Mo, L.; Gu, D.; Yao, Y.; Huang, Y. Soil microbial properties under different vegetation types in Shiwan Mountain, Guangxi Province. Chin. J. Appl. Ecol. 2015, 34, 352–359. [Google Scholar]
- Ai, C.; Zhang, S.Q.; Zhang, X.; Guo, D.D.; Zhou, W.; Huang, S.M. Distinct responses of soil bacterial and fungal communities to changes in fertilization regime and crop rotation. Geoderma 2018, 319, 156–166. [Google Scholar] [CrossRef]
- Li, B.-B.; Roley, S.S.; Duncan, D.S.; Guo, J.; Quensen, J.F.; Yu, H.-Q.; Tiedje, J.M. Long-term excess nitrogen fertilizer increases sensitivity of soil microbial community to seasonal change revealed by ecological network and metagenome analyses. Soil Biol. Biochem. 2021, 160, 108349. [Google Scholar] [CrossRef]
- Dang, Q.; Tan, W.; Zhao, X.; Li, D.; Li, Y.; Yang, T.; Li, R.; Zu, G.; Xi, B. Linking the response of soil microbial community structure in soils to long-term wastewater irrigation and soil depth. Sci. Total Environ. 2019, 688, 26–36. [Google Scholar] [CrossRef] [PubMed]
- Fu, B.; Li, Z.; Gao, X.; Wu, L.; Lan, J.; Peng, W. Effects of subsurface drip irrigation on alfalfa (Medicago sativa L.) growth and soil microbial community structures in arid and semi-arid areas of northern China. Appl. Soil Ecol. 2021, 159, 103859. [Google Scholar] [CrossRef]
- Jesus, E.D.; Marsh, T.L.; Tiedje, J.M.; Moreira, F.M.D. Changes in land use alter the structure of bacterial communities in Western Amazon soils. ISME J. 2009, 3, 1004–1011. [Google Scholar] [CrossRef]
- Vitali, F.; Mastromei, G.; Senatore, G.; Caroppo, C.; Casalone, E. Long lasting effects of the conversion from natural forest to poplar plantation on soil microbial communities. Microbiol. Res. 2016, 182, 89–98. [Google Scholar] [CrossRef] [PubMed]
- Monkai, J.; Goldberg, S.D.; Hyde, K.D.; Harrison, R.D.; Mortimer, P.E.; Xu, J.C. Natural forests maintain a greater soil microbial diversity than that in rubber plantations in Southwest China. Agric. Ecosyst. Environ. 2018, 265, 190–197. [Google Scholar] [CrossRef]
- Lejon, D.P.H.; Chaussod, R.; Ranger, J.; Ranjard, L. Microbial community structure and density under different tree species in an acid forest soil (Morvan, France). Microb. Ecol. 2005, 50, 614–625. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.Q.; Guo, Z.Y.; Chen, C.R.; Jia, Z.J. Soil microbial community structure and diversity are largely influenced by soil pH and nutrient quality in 78-year-old tree plantations. Biogeosciences 2017, 14, 2101–2111. [Google Scholar] [CrossRef] [Green Version]
- Peay, K.G.; Baraloto, C.; Fine, P.V.A. Strong coupling of plant and fungal community structure across western Amazonian rainforests. ISME J. 2013, 7, 1852–1861. [Google Scholar] [CrossRef]
- Urbanova, M.; Snajdr, J.; Baldrian, P. Composition of fungal and bacterial communities in forest litter and soil is largely determined by dominant trees. Soil Biol. Biochem. 2015, 84, 53–64. [Google Scholar] [CrossRef]
- Sims, A.; Zhang, Y.; Galaraj, S.; Brown, P.B.; Hu, Z. Toward the development of microbial indicators for wetland assessment. Water Res. 2013, 47, 1711–1725. [Google Scholar] [CrossRef]
- Vinhal-Freitas, I.C.; Correa, G.F.; Wendling, B.; Bobul’ska, L.; Ferreira, A.S. Soil textural class plays a major role in evaluating the effects of land use on soil quality indicators. Ecol. Indic. 2017, 74, 182–190. [Google Scholar] [CrossRef]
- Chen, W.; He, L.; Tian, S.; Masabni, J.; Xiong, H.; Zou, F.; Yuan, D. Factors involved in the success ofCastanea henryistem cuttings in different cutting mediums and cutting selection periods. J. For. Res. 2021, 32, 1627–1639. [Google Scholar] [CrossRef]
- Fan, X.; Yuan, D.; Tang, J.; Tian, X.; Zhang, L.; Zou, F.; Tan, X. Sporogenesis and gametogenesis in Chinese chinquapin (Castanea henryi (Skam) Rehder & Wilson) and their systematic implications. Trees-Struct. Funct. 2015, 29, 1713–1723. [Google Scholar]
- Ma, H.; Jiang, X.; Gong, B.; Wu, K.; Zhao, X. Advance of Researches on Castanea henryi and of Further Development in China. J. Zhejiang For. Sci. Technol. 2013, 33, 62–67. [Google Scholar]
- Felton, A.; Lindbladh, M.; Brunet, J.; Fritz, Ö. Replacing coniferous monocultures with mixed-species production stands: An assessment of the potential benefits for forest biodiversity in northern Europe. For. Ecol. Manag. 2010, 260, 939–947. [Google Scholar] [CrossRef]
- Epron, D.; Mouanda, C.; Mareschal, L.; Koutika, L.S. Impacts of organic residue management on the soil C dynamics in a tropical eucalypt plantation on a nutrient-poor sandy soil after three rotations. Soil Biol. Biochem. 2015, 85, 183–189. [Google Scholar] [CrossRef]
- Guo, L.B.; Gifford, R.M. Soil carbon stocks and land use change: A meta analysis. Glob. Change Biol. 2002, 8, 345–360. [Google Scholar] [CrossRef]
- Yang, Y.; He, Z.; Zou, S.; Yu, X. A study on the soil microbes and biochemistry of rhizospheric and total soil in natural forest and plantation of Castanopsis kauakamii. Acta Ecol. Sin. 1998, 18, 198–202. [Google Scholar]
- Ezirim, C.; Chikezie, P.; Iheanacho, K.; Nwachukwu, N. Comparative Activities of Soil Enzymes from Polluted Sites in Egbema, Imo State, Nigeria. J. Pollut. Eff. Control 2017, 5, 2. [Google Scholar] [CrossRef] [Green Version]
- Lieb, A.M.; Darrouzet-Nardi, A.; Bowman, W.D. Nitrogen deposition decreases acid buffering capacity of alpine soils in the southern Rocky Mountains. Geoderma 2011, 164, 220–224. [Google Scholar] [CrossRef]
- Satapute, P.; Kamble, M.V.; Adhikari, S.S.; Jogaiah, S. Influence of triazole pesticides on tillage soil microbial populations and metabolic changes. Sci. Total Environ. 2019, 651, 2334–2344. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Jiang, L.; Li, L.; Li, A.; Wu, M.; Wan, S. Diversity-dependent stability under mowing and nutrient addition: Evidence from a 7-year grassland experiment. Ecol. Lett. 2012, 15, 619–626. [Google Scholar] [CrossRef]
- Chen, D.; Huang, C. Production status and development countermeasures and ways of Castanea henryi orchard in Songxi, Fujian. Pract. Technol. Inf. Fruit Trees 2017, 8, 40–41. [Google Scholar]
- Hamer, U.; Makeschin, F.; An, S.; Zheng, F. Microbial activity and community structure in degraded soils on the Loess Plateau of China. J. Plant Nutr. Soil Sci. 2009, 172, 118–126. [Google Scholar] [CrossRef]
- Ma, X.; Zhao, C.; Gao, Y.; Liu, B.; Wang, T.; Yuan, T.; Hale, L.R.; Van Nostrand, J.D.; Wan, S.; Zhou, J.; et al. Divergent taxonomic and functional responses of microbial communities to field simulation of aeolian soil erosion and deposition. Mol. Ecol. 2017, 26, 4186–4196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qiu, L.; Zhang, Q.; Zhu, H.; Reich, P.B.; Banerjee, S.; van der Heijden, M.G.A.; Sadowsky, M.J.; Ishii, S.; Jia, X.; Shao, M.; et al. Erosion reduces soil microbial diversity, network complexity and multifunctionality. ISME J. 2021, 15, 2474–2489. [Google Scholar] [CrossRef]
- Chen, H.; He, F. The effect of plant growth substances and rare-earth elements on the yield of Castanea henryi. For. Res. 2004, 17, 83–88. [Google Scholar]
- Liu, B.; Lin, R.; Jiang, Y.; Jiang, S.; Xiong, Y.; Lian, H.; Zeng, Q.; Liu, X.; Liu, Z.; Chen, S. Transcriptome Analysis and Identification of Genes Associated with Starch Metabolism in Castanea henryi Seed (Fagaceae). Int. J. Mol. Sci. 2020, 21, 1431. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Xu, J.; Riera, N.; Jin, T.; Li, J.; Wang, N. Huanglongbing impairs the rhizosphere-to-rhizoplane enrichment process of the citrus root-associated microbiome. Microbiome 2017, 5, 1–17. [Google Scholar] [CrossRef]
- Yanai, J.; Okada, T.; Yamada, H. Elemental composition of agricultural soils in Japan in relation to soil type, land use and region. Soil Sci. Plant Nutr. 2012, 58, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Rao, B.K.R.; Bailey, J.; Wingwafi, R.W. Comparison of Three Digestion Methods for Total Soil Potassium Estimation in Soils of Papua New Guinea Derived from Varying Parent Materials. Commun. Soil Sci. Plant Anal. 2011, 42, 1259–1265. [Google Scholar] [CrossRef]
- Zhou, J.; Bruns, M.A.; Tiedje, J.M. DNA recovery from soils of diverse composition. Appl. Environ. Microbiol. 1996, 62, 316–322. [Google Scholar] [CrossRef] [Green Version]
- Ju, F.; Zhang, T. Bacterial assembly and temporal dynamics in activated sludge of a full-scale municipal wastewater treatment plant. ISME J. 2015, 9, 683–695. [Google Scholar] [CrossRef] [PubMed]
- Galand, P.E.; Casamayor, E.O.; Kirchman, D.L.; Lovejoy, C. Ecology of the rare microbial biosphere of the Arctic Ocean. Proc. Natl. Acad. Sci. USA 2009, 106, 22427–22432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiong, C.; He, J.-Z.; Singh, B.K.; Zhu, Y.-G.; Wang, J.-T.; Li, P.-P.; Zhang, Q.-B.; Han, L.-L.; Shen, J.-P.; Ge, A.-H.; et al. Rare taxa maintain the stability of crop mycobiomes and ecosystem functions. Environ. Microbiol. 2021, 23, 1907–1924. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y.; Xiao, X.; Nuccio, E.E.; Yuan, M.; Zhang, N.; Xue, K.; Cohan, F.M.; Zhou, J.; Sun, B. Differentiation strategies of soil rare and abundant microbial taxa in response to changing climatic regimes. Environ. Microbiol. 2020, 22, 1327–1340. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Chen, Y.; Castro-Izaguirre, N.; Baruffol, M.; Brezzi, M.; Lang, A.; Li, Y.; Hardtle, W.; Oheimb, G.; Yang, X.; et al. Impacts of species richness on productivity in a large-scale subtropical forest experiment. Science 2018, 362, 80–83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, B.K.; Quince, C.; Macdonald, C.A.; Khachane, A.; Thomas, N.; Abu Al-Soud, W.; Sorensen, S.J.; He, Z.L.; White, D.; Sinclair, A.; et al. Loss of microbial diversity in soils is coincident with reductions in some specialized functions. Environ. Microbiol. 2014, 16, 2408–2420. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Yang, Y.; Xie, J.; Guo, J.; Gao, R.; Qian, W. Conversion of a natural broad-leafed evergreen forest into pure plantation forests in a subtropical area: Effects on carbon storage. Ann. For. Sci. 2005, 62, 659–668. [Google Scholar] [CrossRef] [Green Version]
- Allen, K.; Corre, M.D.; Tjoa, A.; Veldkamp, E. Soil Nitrogen-Cycling Responses to Conversion of Lowland Forests to Oil Palm and Rubber Plantations in Sumatra, Indonesia. PLoS ONE 2015, 10, 21. [Google Scholar] [CrossRef] [Green Version]
- Liang, Q.; Chen, H.; Gong, Y.; Fan, M.; Yang, H.; Lal, R.; Kuzyakov, Y. Effects of 15 years of manure and inorganic fertilizers on soil organic carbon fractions in a wheat-maize system in the North China Plain. Nutr. Cycl. Agroecosyst. 2012, 92, 21–33. [Google Scholar] [CrossRef]
- Miao, Y.; Stewart, B.A.; Zhang, F. Long-term experiments for sustainable nutrient management in China. A review. Agron. Sustain. Dev. 2011, 31, 397–414. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Cong, J.; Lu, H.; Li, G.; Xue, Y.; Deng, Y.; Li, H.; Zhou, J.; Li, D. Soil bacterial diversity patterns and drivers along an elevational gradient on Shennongjia Mountain, China. Microb. Biotechnol. 2015, 8, 739–746. [Google Scholar] [CrossRef] [Green Version]
- Fierer, N.; Jackson, R.B. The diversity and biogeography of soil bacterial communities. Proc. Natl. Acad. Sci. USA 2006, 103, 626–631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, K.; Weisenhorn, P.; Gilbert, J.A.; Shi, Y.; Bai, Y.; Chu, H. Soil pH correlates with the co-occurrence and assemblage process of diazotrophic communities in rhizosphere and bulk soils of wheat fields. Soil Biol. Biochem. 2018, 121, 185–192. [Google Scholar] [CrossRef]
- Zhang, P.; Ren, C.; Sun, H.; Min, L. Sorption, desorption and degradation of neonicotinoids in four agricultural soils and their effects on soil microorganisms. Sci. Total Environ. 2018, 615, 59–69. [Google Scholar] [CrossRef] [PubMed]
- Hu, L.; Robert, C.A.M.; Cadot, S.; Zhang, X.; Ye, M.; Li, B.; Manzo, D.; Chervet, N.; Steinger, T.; van der Heijden, M.G.A.; et al. Root exudate metabolites drive plant-soil feedbacks on growth and defense by shaping the rhizosphere microbiota. Nat. Commun. 2018, 9, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Yaryura, P.M.; Leon, M.; Correa, O.S.; Kerber, N.L.; Pucheu, N.L.; Garcia, A.F. Assessment of the role of chemotaxis and biofilm formation as requirements for colonization of roots and seeds of soybean plants by Bacillus amyloliquefaciens BNM339. Curr. Microbiol. 2008, 56, 625–632. [Google Scholar] [CrossRef]
- Wu, K.; Yuan, S.; Xun, G.; Shi, W.; Pan, B.; Guan, H.; Shen, B.; Shen, Q. Root exudates from two tobacco cultivars affect colonization of Ralstonia solanacearum and the disease index. Eur. J. Plant Pathol. 2015, 141, 667–677. [Google Scholar] [CrossRef]
- Kuzmicheva, Y.V.; Shaposhnikov, A.I.; Petrova, S.N.; Makarova, N.M.; Tychinskaya, I.L.; Puhalsky, J.V.; Parahin, N.V.; Tikhonovich, I.A.; Belimov, A.A. Variety specific relationships between effects of rhizobacteria on root exudation, growth and nutrient uptake of soybean. Plant Soil 2017, 419, 83–96. [Google Scholar] [CrossRef]
- Zechmeister-Boltenstern, S.; Keiblinger, K.M.; Mooshammer, M.; Penuelas, J.; Richter, A.; Sardans, J.; Wanek, W. The application of ecological stoichiometry to plant-microbial-soil organic matter transformations. Ecol. Monogr. 2015, 85, 133–155. [Google Scholar] [CrossRef] [Green Version]
- Kopecky, J.; Kyselkova, M.; Omelka, M.; Cermak, L.; Novotna, J.; Grundmann, G.L.; Moenne-Loccoz, Y.; Sagova-Mareckova, M. Actinobacterial community dominated by a distinct clade in acidic soil of a waterlogged deciduous forest. FEMS Microbiol. Ecol. 2011, 78, 386–394. [Google Scholar] [CrossRef] [Green Version]
- Fukuda, M. Cometabolism of PCBs and DDE by the aromatic degradation system of a Rhodococcus bacterium. J. Biotechnol. 2010, 150, S44–S44. [Google Scholar] [CrossRef]
- Jose, P.A.; Maharshi, A.; Jha, B. Actinobacteria in natural products research: Progress and prospects. Microbiol. Res. 2021, 246, 14. [Google Scholar] [CrossRef] [PubMed]
- Bull, A.T.; Goodfellow, M. Dark, rare and inspirational microbial matter in the extremobiosphere: 16,000 m of bioprospecting campaigns. Microbiology 2019, 165, 1252–1264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonzalez-Pimentel, J.L.; Miller, A.Z.; Jurado, V.; Laiz, L.; Pereir, M.F.C.; Saiz-Jimenez, C. Yellow coloured mats from lava tubes of La Palma (Canary Islands, Spain) are dominated by metabolically active Actinobacteria. Sci. Rep. 2018, 8, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Sun, M.; Zhang, H.; Xu, N.; Sun, G. Use of mulberry-soybean intercropping in salt-alkali soil impacts the diversity of the soil bacterial community. Microb. Biotechnol. 2016, 9, 293–304. [Google Scholar] [CrossRef]
- Fierer, N.; Lauber, C.L.; Ramirez, K.S.; Zaneveld, J.; Bradford, M.A.; Knight, R. Comparative metagenomic, phylogenetic and physiological analyses of soil microbial communities across nitrogen gradients. ISME J. 2012, 6, 1007–1017. [Google Scholar] [CrossRef] [Green Version]
- Barns, S.M.; Cain, E.C.; Sommerville, L.; Kuske, C.R. Acidobactetia phylum sequences in uranium-contaminated subsurface sediments greatly expand the known diversity within the phylum. Appl. Environ. Microbiol. 2007, 73, 3113–3116. [Google Scholar] [CrossRef] [Green Version]
- Griffiths, R.I.; Thomson, B.C.; James, P.; Bell, T.; Bailey, M.; Whiteley, A.S. The bacterial biogeography of British soils. Environ. Microbiol. 2011, 13, 1642–1654. [Google Scholar] [CrossRef]
- Rawat, S.R.; Mannisto, M.K.; Bromberg, Y.; Haggblom, M.M. Comparative genomic and physiological analysis provides insights into the role of Acidobacteria in organic carbon utilization in Arctic tundra soils. FEMS Microbiol. Ecol. 2012, 82, 341–355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, S.; Nuccio, E.E.; Shi, Z.; He, Z.; Zhou, J.; Firestone, M.K. The interconnected rhizosphere: High network complexity dominates rhizosphere assemblages. Ecol. Lett. 2016, 19, 926–936. [Google Scholar] [CrossRef] [Green Version]
- Faust, K.; Raes, J. Microbial interactions: From networks to models. Nat. Rev. Microbiol. 2012, 10, 538–550. [Google Scholar] [CrossRef]
- Matchado, M.S.; Lauber, M.; Reitmeier, S.; Kacprowski, T.; Baumbach, J.; Haller, D.; List, M. Network analysis methods for studying microbial communities: A mini review. Comp. Struct. Biotechnol. J. 2021, 19, 2687–2698. [Google Scholar] [CrossRef]
- Foster, E.A.; Franks, D.W.; Morrell, L.J.; Balcomb, K.C.; Parsons, K.M.; van Ginneken, A.; Croft, D.P. Social network correlates of food availability in an endangered population of killer whales, Orcinus orca. Anim. Behav. 2012, 83, 731–736. [Google Scholar] [CrossRef] [Green Version]
- Henzi, S.P.; Lusseau, D.; Weingrill, T.; van Schaik, C.P.; Barrett, L. Cyclicity in the structure of female baboon social networks. Behav. Ecol. Sociobiol. 2009, 63, 1015–1021. [Google Scholar] [CrossRef]
- Bauer, M.A.; Kainz, K.; Carmona-Gutierrez, D.; Madeo, F. Microbial wars: Competition in ecological niches and within the microbiome. Microb. Cell 2018, 5, 215–219. [Google Scholar] [CrossRef] [PubMed]
- Lu, L.; Yin, S.; Liu, X.; Zhang, W.; Gu, T.; Shen, Q.; Qiu, H. Fungal networks in yield-invigorating and -debilitating soils induced by prolonged potato monoculture. Soil Biol. Biochem. 2013, 65, 186–194. [Google Scholar] [CrossRef] [Green Version]
- van der Heijden, M.G.A.; Hartmann, M. Networking in the Plant Microbiome. PLoS Biol. 2016, 14, 9. [Google Scholar] [CrossRef]
- Xue, Y.; Chen, H.; Yang, J.; Liu, M.; Huang, B.; Yang, J. Distinct patterns and processes of abundant and rare eukaryotic plankton communities following a reservoir cyanobacterial bloom. ISME J. 2018, 12, 2263–2277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Banerjee, S.; Schlaeppi, K.; van der Heijden, M.G.A. Keystone taxa as drivers of microbiome structure and functioning. Nat. Rev. Microbiol. 2018, 16, 567–576. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | TRY | TRB | TRC | TTX |
|---|---|---|---|---|
| pH | 4.26 ± 0.40 b | 4.55 ± 0.12 ab | 4.41 ± 0.13 ab | 4.82 ± 0.13 a |
| OM (g/kg) | 29.99 ± 4.25 b | 30.80 ± 4.43 b | 32.75 ± 3.86 b | 54.18 ± 7.25 a |
| TP (g/kg) | 0.10 ± 0.03 c | 0.24 ± 0.02 a | 0.20 ± 0.04 ab | 0.18 ± 0.03 b |
| TK (g/kg) | 6.19 ± 2.44 b | 8.37 ± 1.05 b | 8.39 ± 1.73 b | 22.94 ± 5.69 a |
| TC (g/kg) | 15.73 ± 3.08 b | 17.61 ± 2.08 b | 16.76 ± 1.83 b | 29.02 ± 2.85 a |
| TN (g/kg) | 1.31 ± 0.13 b | 1.60 ± 0.16 b | 1.45 ± 0.13 b | 2.03 ± 0.14 a |
| SM (g/kg) | 202.66 ± 22.75 b | 215.11 ± 32.36 b | 259.59 ± 35.88 b | 283.71 ± 36.15 a |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cheng, Y.; Zhou, L.; Liang, T.; Man, J.; Wang, Y.; Li, Y.; Chen, H.; Zhang, T. Deciphering Rhizosphere Microbiome Assembly of Castanea henryi in Plantation and Natural Forest. Microorganisms 2022, 10, 42. https://doi.org/10.3390/microorganisms10010042
Cheng Y, Zhou L, Liang T, Man J, Wang Y, Li Y, Chen H, Zhang T. Deciphering Rhizosphere Microbiome Assembly of Castanea henryi in Plantation and Natural Forest. Microorganisms. 2022; 10(1):42. https://doi.org/10.3390/microorganisms10010042
Chicago/Turabian StyleCheng, Yuanyuan, Lexin Zhou, Tian Liang, Jiayin Man, Yinghao Wang, Yu Li, Hui Chen, and Taoxiang Zhang. 2022. "Deciphering Rhizosphere Microbiome Assembly of Castanea henryi in Plantation and Natural Forest" Microorganisms 10, no. 1: 42. https://doi.org/10.3390/microorganisms10010042
APA StyleCheng, Y., Zhou, L., Liang, T., Man, J., Wang, Y., Li, Y., Chen, H., & Zhang, T. (2022). Deciphering Rhizosphere Microbiome Assembly of Castanea henryi in Plantation and Natural Forest. Microorganisms, 10(1), 42. https://doi.org/10.3390/microorganisms10010042