
Complete Genome Sequence, Molecular Characterization and Phylogenetic Relationships of a Novel Tern Atadenovirus

 ,
,  , , ,
, , ,  and
and
Abstract
1. Introduction
2. Materials and Methods
2.1. Sampling
2.2. Sample Preparation and Sequencing
2.3. Assembly and Genome Annotation
2.4. Phylogenetic Analysis
2.5. Comparative Analysis
2.6. Species Delimitation
2.7. Codon-Based Analysis of Positive Selection
2.8. Protein 3D Structure Prediction
3. Results
3.1. Genome of TeAdV-1 and Comparative Analyses
3.2. Evolutionary Relationships of TAdV-1
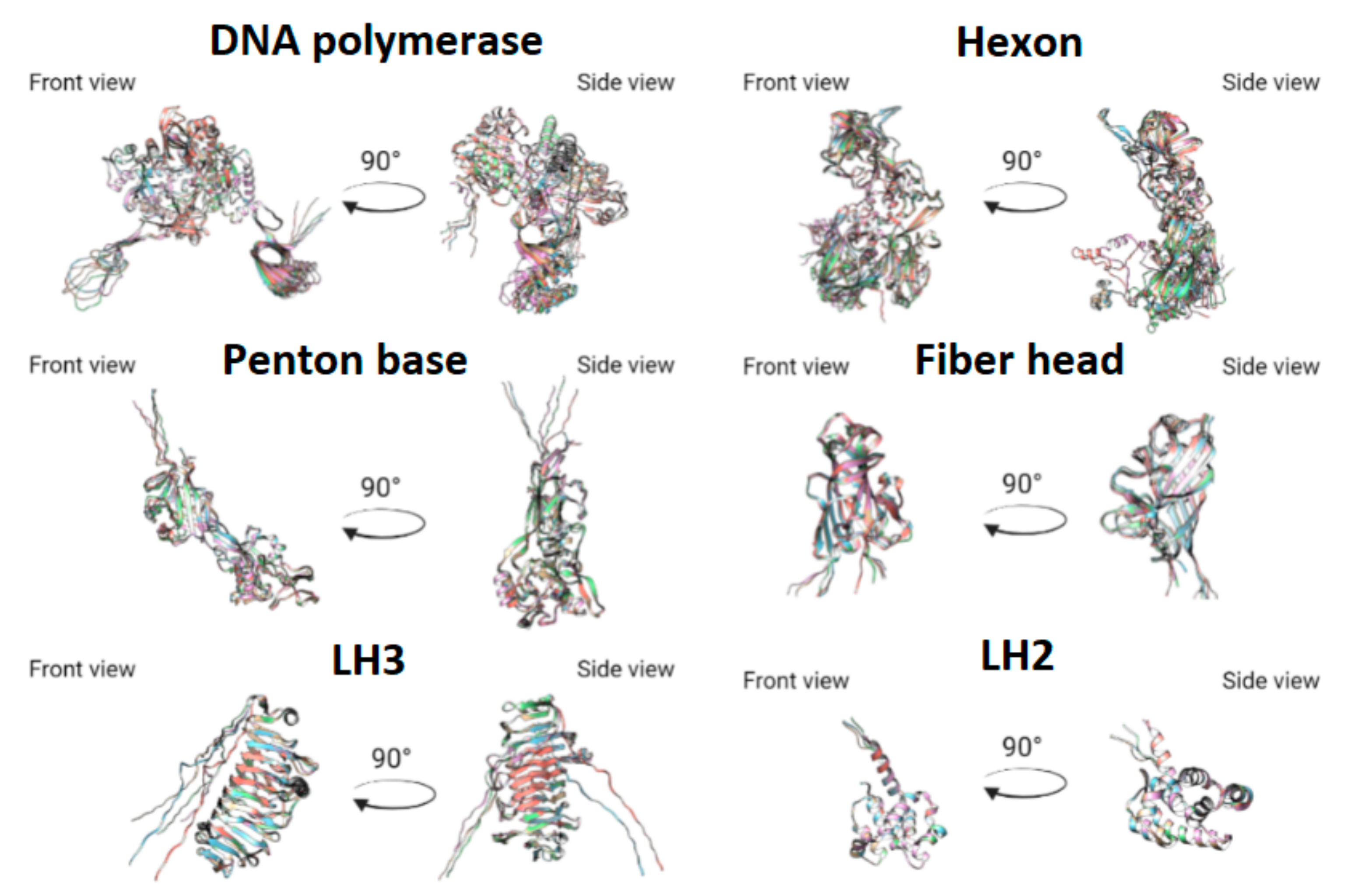
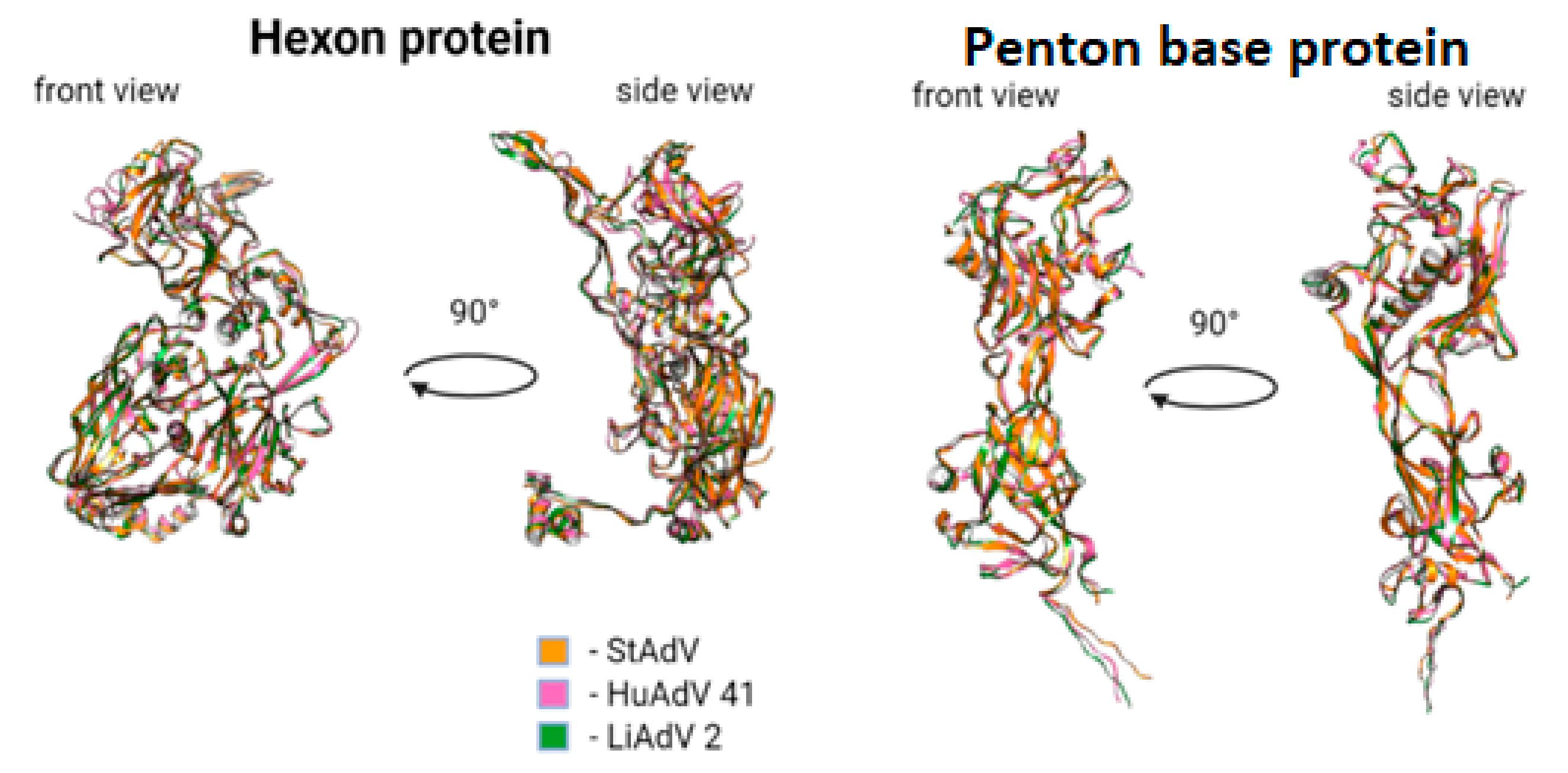
3.3. Protein Prediction
- Absence of alpha-helix, corresponding to Tyr288-Val285 in HAdV-41, lacking in TAdV, lacking in LAdV-2;
- Elongation of alpha-helix Val138-Asn159 (21 aa) in TAdV, which corresponds to Glu173-Ala183 (10 aa) in HAdV-41 and Val138-Gly157 (19 aa) in LAdV-2;
- Presence of beta-sheet-like short structure at Gly208-Asp210 in TAdV, lacking in HAdV-41 and LAdV-2 alike.
- TAdV alpha-helix Thr41-Ser46, presented in HAdV-41 as Asn72-Ala75, lacking in LAdV-2.
- Presence of structure Val233-Leu235 beta-sheet to Tyr236-Ile239 alpha helix, presented in StAdV which is absent in HAdV-41 and LAdV-2.
- Presence of two beta-sheets Glu380-Gly382, Ala400-Ile402, absent in HAdV-41 and LAdV-2;
- Elongation of beta-sheet Gln816-Cys824 (8 aa), corresponding to Val816 –Val823 (7 aa) in LAdV-2 and Ser831-Lys836 (5 aa)
- Presence of beta-sheet Gln229-Leu233, absent in LAdV-2 and HAdV-41.
- Elongation of beta-sheet Ser187-Ile197 (10 aa), corresponding to Arg201-Ile203 (2 aa) in HAdV-41 and absent in LAdV-2
- Presence of alpha-helix Val153-Lys157, absent in HAdV-41 and LAdV-2.
- Elongation of beta-sheet Cys269-Gly273 (4 aa), corresponding to Arg263-Thr265 (2 aa) in LAdV-2 and absent in HAdV-41.
3.4. Detection of Adaptive Evolution Events
3.4.1. Pervasive Positive Selection in the Molecular Evolution of Atadenovirus
3.4.2. Episodic Positive Selection in the Molecular Evolution of TAtV-1
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Harrach, B.; Tarján, Z.L.; Benkő, M. Adenoviruses across the animal kingdom: A walk in the zoo. FEBS Lett. 2019, 593, 3660–3673. [Google Scholar] [CrossRef] [PubMed]
- Lefkowitz, E.J.; Dempsey, D.M.; Hendrickson, R.C.; Orton, R.J.; Siddell, S.G.; Smith, D.B. Virus taxonomy: The database of the International Committee on Taxonomy of Viruses (ICTV). Nucleic Acids Res. 2018, 46, D708–D717. [Google Scholar] [CrossRef] [PubMed]
- Benkő, M.; Aoki, K.; Arnberg, N.; Davison, A.J. ICTV Virus Taxonomy Profile: Adenoviridae. J. Gen. Virol. 2021; in press. ISBN 978-0-12-384684-6. [Google Scholar]
- Davison, A.J.; Benko, M.; Harrach, B. Genetic content and evolution of adenoviruses. J. Gen. Virol. 2003, 84, 2895–2908. [Google Scholar] [CrossRef]
- Gorman, J.J.; Wallis, T.P.; Whelan, D.A.; Shaw, J.; Both, G.W. LH3, a “homologue” of the mastadenoviral E1B 55-kDa protein is a structural protein of atadenoviruses. Virology 2005, 342, 159–166. [Google Scholar] [CrossRef][Green Version]
- Gerlach, H. Viruses. In Avian Medicine: Principles and Application; Ritchie, B.W., Harrison, G.J., Harrison, L.R., Eds.; Wingers Publishing Inc.: Lake Worth, FL, USA, 1994; pp. 862–948. ISBN 978-096369960. [Google Scholar]
- Borkenhagen, L.K.; Fieldhouse, J.K.; Seto, D.; Gray, G.C. Are adenoviruses zoonotic? A systematic review of the evidence. Emerg. Microbes Infect. 2019, 8, 1679–1687. [Google Scholar] [CrossRef]
- Li, K.S.; Guan, Y.; Wang, J.; Smith, G.J.D.; Xu, K.M.; Duan, L.; Rahardjo, A.P.; Puthavathana, P.; Buranathai, C.; Nguyen, T.D.; et al. Genesis of a highly pathogenic and potentially pandemic H5N1 influenza virus in eastern Asia. Nature 2004, 430, 209–213. [Google Scholar] [CrossRef]
- World Health Organization Pandemic H1N1. 2009. Available online: http://apps.who.int/iris/bitstream/handle/10665/78414/9789241503051_eng.pdf?sequence=1 (accessed on 30 November 2021).
- Neumann, G.; Noda, T.; Kawaoka, Y. Emergence and pandemic potential of swine-origin H1N1 influenza virus. Nature 2009, 459, 931–939. [Google Scholar] [CrossRef]
- World Health Organization Statement on the second meeting of the International Health Regulations. 2020. Available online: https://www.who.int/news/item/30-01-2020-statement-on-the-second-meeting-of-the-international-health-regulations-(2005)-emergency-committee-regarding-the-outbreak-of-novel-coronavirus-(2019-ncov) (accessed on 30 November 2021).
- Andersen, K.G.; Rambaut, A.; Lipkin, W.I.; Holmes, E.C.; Garry, R.F. The proximal origin of SARS-CoV-2. Nat. Med. 2020, 26, 450–452. [Google Scholar] [CrossRef]
- Fritzsche McKay, A.; Hoye, B.J. Are Migratory Animals Superspreaders of Infection?: An Introduction to the Symposium. Integr. Comp. Biol. 2016, 56, 260–267. [Google Scholar] [CrossRef]
- Ayginin, A.A.; Pimkina, E.V.; Matsvay, A.D.; Speranskaya, A.S.; Safonova, M.V.; Blinova, E.A.; Artyushin, I.V.; Dedkov, V.G.; Shipulin, G.A.; Khafizov, K. The Study of Viral RNA Diversity in Bird Samples Using De Novo Designed Multiplex Genus-Specific Primer Panels. Adv. Virol. 2018, 2018, 1–10. [Google Scholar] [CrossRef]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A New Genome Assembly Algorithm and Its Applications to Single-Cell Sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef]
- McGinnis, S.; Madden, T.L. BLAST: At the core of a powerful and diverse set of sequence analysis tools. Nucleic Acids Res. 2004, 32, W20–W25. [Google Scholar] [CrossRef]
- Federhen, S. The NCBI Taxonomy database. Nucleic Acids Res. 2012, 40, D136–D143. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinforma. Oxf. Engl. 2009, 25, 1754–1760. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; 1000 Genome Project Data Processing Subgroup. The Sequence Alignment/Map format and SAMtools. Bioinforma. Oxf. Engl. 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- Milne, I.; Stephen, G.; Bayer, M.; Cock, P.J.A.; Pritchard, L.; Cardle, L.; Shaw, P.D.; Marshall, D. Using Tablet for visual exploration of second-generation sequencing data. Brief. Bioinform. 2013, 14, 193–202. [Google Scholar] [CrossRef]
- Kurtz, S.; Phillippy, A.; Delcher, A.L.; Smoot, M.; Shumway, M.; Antonescu, C.; Salzberg, S.L. Versatile and open software for comparing large genomes. Genome Biol. 2004, 5, R12. [Google Scholar] [CrossRef]
- Zhang, K.-Y.; Gao, Y.-Z.; Du, M.-Z.; Liu, S.; Dong, C.; Guo, F.-B. Vgas: A Viral Genome Annotation System. Front. Microbiol. 2019, 10, 184. [Google Scholar] [CrossRef]
- Emms, D.M.; Kelly, S. OrthoFinder: Phylogenetic orthology inference for comparative genomics. Genome Biol. 2019, 20, 238. [Google Scholar] [CrossRef]
- Reese, M.G.; Eeckman, F.H.; Kulp, D.; Haussler, D. Improved Splice Site Detection in Genie. J. Comput. Biol. 1997, 4, 311–323. [Google Scholar] [CrossRef]
- Cock, P.J.A.; Antao, T.; Chang, J.T.; Chapman, B.A.; Cox, C.J.; Dalke, A.; Friedberg, I.; Hamelryck, T.; Kauff, F.; Wilczynski, B.; et al. Biopython: Freely available Python tools for computational molecular biology and bioinformatics. Bioinforma. Oxf. Engl. 2009, 25, 1422–1423. [Google Scholar] [CrossRef]
- Clark, K.; Karsch-Mizrachi, I.; Lipman, D.J.; Ostell, J.; Sayers, E.W. GenBank. Nucleic Acids Res. 2016, 44, D67–D72. [Google Scholar] [CrossRef]
- Larkin, M.A.; Blackshields, G.; Brown, N.P.; Chenna, R.; McGettigan, P.A.; McWilliam, H.; Valentin, F.; Wallace, I.M.; Wilm, A.; Lopez, R.; et al. Clustal W and Clustal X version 2.0. Bioinforma. Oxf. Engl. 2007, 23, 2947–2948. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Castresana, J. Selection of Conserved Blocks from Multiple Alignments for Their Use in Phylogenetic Analysis. Mol. Biol. Evol. 2000, 17, 540–552. [Google Scholar] [CrossRef]
- Lanfear, R.; Frandsen, P.B.; Wright, A.M.; Senfeld, T.; Calcott, B. PartitionFinder 2: New Methods for Selecting Partitioned Models of Evolution for Molecular and Morphological Phylogenetic Analyses. Mol. Biol. Evol. 2017, 34, 772–773. [Google Scholar] [CrossRef]
- Kozlov, A.M.; Darriba, D.; Flouri, T.; Morel, B.; Stamatakis, A. RAxML-NG: A fast, scalable and user-friendly tool for maximum likelihood phylogenetic inference. Bioinformatics 2019, 35, 4453–4455. [Google Scholar] [CrossRef]
- Rambaut, A. FigTree v. 1.4.3. 2018. Available online: http://tree.bio.ed.ac.uk/software/figtree/ (accessed on 30 November 2021).
- Abadi, S.; Azouri, D.; Pupko, T.; Mayrose, I. Model selection may not be a mandatory step for phylogeny reconstruction. Nat. Commun. 2019, 10, 934. [Google Scholar] [CrossRef]
- Spielman, S.J. Relative Model Fit Does Not Predict Topological Accuracy in Single-Gene Protein Phylogenetics. Mol. Biol. Evol. 2020, 37, 2110–2123. [Google Scholar] [CrossRef]
- Letunic, I.; Bork, P. Interactive Tree Of Life (iTOL) v5: An online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 2021, 49, W293–W296. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K. MAFFT: A novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002, 30, 3059–3066. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Kapli, P.; Pavlidis, P.; Stamatakis, A. A general species delimitation method with applications to phylogenetic placements. Bioinformatics 2013, 29, 2869–2876. [Google Scholar] [CrossRef] [PubMed]
- Fujisawa, T.; Barraclough, T.G. Delimiting Species Using Single-Locus Data and the Generalized Mixed Yule Coalescent Approach: A Revised Method and Evaluation on Simulated Data Sets. Syst. Biol. 2013, 62, 707–724. [Google Scholar] [CrossRef]
- Pons, J.; Barraclough, T.G.; Gomez-Zurita, J.; Cardoso, A.; Duran, D.P.; Hazell, S.; Kamoun, S.; Sumlin, W.D.; Vogler, A.P. Sequence-Based Species Delimitation for the DNA Taxonomy of Undescribed Insects. Syst. Biol. 2006, 55, 595–609. [Google Scholar] [CrossRef]
- Fontaneto, D.; Herniou, E.A.; Boschetti, C.; Caprioli, M.; Melone, G.; Ricci, C.; Barraclough, T.G. Independently Evolving Species in Asexual Bdelloid Rotifers. PLoS Biol. 2007, 5, e87. [Google Scholar] [CrossRef]
- Birky, C.W., Jr.; Ricci, C.; Melone, G.; Fontaneto, D. Integrating DNA and morphological taxonomy to describe diversity in poorly studied microscopic animals: New species of the genus Abrochtha Bryce, 1910 (Rotifera: Bdelloidea: Philodinavidae): NEW CRYPTIC ROTIFER SPECIES. Zool. J. Linn. Soc. 2011, 161, 723–734. [Google Scholar] [CrossRef]
- Yule, G.U. II.—A mathematical theory of evolution, based on the conclusions of Dr. J. C. Willis, F.R.S. Philos. Trans. R. Soc. Lond. Ser. B Contain. Pap. Biol. Character 1925, 213, 21–87. [Google Scholar] [CrossRef]
- Hudson, R. Gene genealogies and the coalescent process. Oxf. Surv. Evol. Biol. 1990, 7, 1–44. [Google Scholar]
- Tamura, K.; Battistuzzi, F.U.; Billing-Ross, P.; Murillo, O.; Filipski, A.; Kumar, S. Estimating divergence times in large molecular phylogenies. Proc. Natl. Acad. Sci. USA 2012, 109, 19333–19338. [Google Scholar] [CrossRef]
- Tamura, K.; Tao, Q.; Kumar, S. Theoretical Foundation of the RelTime Method for Estimating Divergence Times from Variable Evolutionary Rates. Mol. Biol. Evol. 2018, 35, 1770–1782. [Google Scholar] [CrossRef]
- Thomas, R.H. Molecular Evolution and Phylogenetics. Heredity 2001, 86, 385. [Google Scholar] [CrossRef]
- Puillandre, N.; Brouillet, S.; Achaz, G. ASAP: Assemble species by automatic partitioning. Mol. Ecol. Resour. 2021, 21, 609–620. [Google Scholar] [CrossRef]
- Boc, A.; Diallo, A.B.; Makarenkov, V. T-REX: A web server for inferring, validating and visualizing phylogenetic trees and networks. Nucleic Acids Res. 2012, 40, W573–W579. [Google Scholar] [CrossRef]
- Birky, C.W.; Adams, J.; Gemmel, M.; Perry, J. Using Population Genetic Theory and DNA Sequences for Species Detection and Identification in Asexual Organisms. PLoS ONE 2010, 5, e10609. [Google Scholar] [CrossRef]
- Kosakovsky Pond, S.L.; Posada, D.; Gravenor, M.B.; Woelk, C.H.; Frost, S.D.W. GARD: A genetic algorithm for recombination detection. Bioinformatics 2006, 22, 3096–3098. [Google Scholar] [CrossRef]
- Pond, S.L.K.; Muse, S.V. HyPhy: Hypothesis Testing Using Phylogenies. In Statistical Methods in Molecular Evolution; Springer: New York, NY, USA, 2005; pp. 125–181. ISBN 978-0-387-22333-9. [Google Scholar]
- Yang, Z. PAML 4: Phylogenetic Analysis by Maximum Likelihood. Mol. Biol. Evol. 2007, 24, 1586–1591. [Google Scholar] [CrossRef]
- Kosakovsky Pond, S.L.; Frost, S.D.W. Not So Different After All: A Comparison of Methods for Detecting Amino Acid Sites Under Selection. Mol. Biol. Evol. 2005, 22, 1208–1222. [Google Scholar] [CrossRef]
- Zhang, J. Evaluation of an Improved Branch-Site Likelihood Method for Detecting Positive Selection at the Molecular Level. Mol. Biol. Evol. 2005, 22, 2472–2479. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera?A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
- Marabini, R.; Condezo, G.N.; Krupovic, M.; Menéndez-Conejero, R.; Gómez-Blanco, J.; San Martín, C. Near-atomic structure of an atadenovirus reveals a conserved capsid-binding motif and intergenera variations in cementing proteins. Sci. Adv. 2021, 7, eabe6008. [Google Scholar] [CrossRef]
- Kundhavai Natchiar, S.; Venkataraman, S.; Mullen, T.-M.; Nemerow, G.R.; Reddy, V.S. Revised Crystal Structure of Human Adenovirus Reveals the Limits on Protein IX Quasi-Equivalence and on Analyzing Large Macromolecular Complexes. J. Mol. Biol. 2018, 430, 4132–4141. [Google Scholar] [CrossRef]
- To, K.K.W.; Tse, H.; Chan, W.-M.; Choi, G.K.Y.; Zhang, A.J.X.; Sridhar, S.; Wong, S.C.Y.; Chan, J.F.W.; Chan, A.S.F.; Woo, P.C.Y.; et al. A Novel Psittacine Adenovirus Identified During an Outbreak of Avian Chlamydiosis and Human Psittacosis: Zoonosis Associated with Virus-Bacterium Coinfection in Birds. PLoS Negl. Trop. Dis. 2014, 8, e3318. [Google Scholar] [CrossRef]
- Harrach, B. Adenoviruses: General Features. In Reference Module in Biomedical Sciences; Elsevier: Amsterdam, The Netherlands, 2014; p. B978012801238302523X. ISBN 978-0-12-801238-3. [Google Scholar]
- Cepko, C.L.; Sharp, P.A. Analysis of Ad5 Hexon and 100K is mutants using conformation-specific monoclonal antibodies. Virology 1983, 129, 137–154. [Google Scholar] [CrossRef]
- Russell, W.C. Adenoviruses: Update on structure and function. J. Gen. Virol. 2009, 90, 1–20. [Google Scholar] [CrossRef]
- Kulanayake, S.; Tikoo, S.K. Adenovirus Core Proteins: Structure and Function. Viruses 2021, 13, 388. [Google Scholar] [CrossRef]
- Parker, E. Adenovirus DNA polymerase: Domain organisation and interaction with preterminal protein. Nucleic Acids Res. 1998, 26, 1240–1247. [Google Scholar] [CrossRef][Green Version]
- Vellinga, J.; Van der Heijdt, S.; Hoeben, R.C. The adenovirus capsid: Major progress in minor proteins. J. Gen. Virol. 2005, 86, 1581–1588. [Google Scholar] [CrossRef]
- Duarte, M.A.; Silva, J.M.F.; Brito, C.R.; Teixeira, D.S.; Melo, F.L.; Ribeiro, B.M.; Nagata, T.; Campos, F.S. Faecal Virome Analysis of Wild Animals from Brazil. Viruses 2019, 11, 803. [Google Scholar] [CrossRef]
- Shriner, D.; Nickle, D.C.; Jensen, M.A.; Mullins, J.I. Potential impact of recombination on sitewise approaches for detecting positive natural selection. Genet. Res. 2003, 81, 115–121. [Google Scholar] [CrossRef] [PubMed]
- Vrati, S.; Brookes, D.E.; Strike, P.; Khatri, A.; Boyle, D.B.; Both, G.W. Unique Genome Arrangement of an Ovine Adenovirus: Identification of New Proteins and Proteinase Cleavage Sites. Virology 1996, 220, 186–199. [Google Scholar] [CrossRef] [PubMed]
- Pénzes, J.J.; Szirovicza, L.; Harrach, B. The complete genome sequence of bearded dragon adenovirus 1 harbors three genes encoding proteins of the C-type lectin-like domain superfamily. Infect. Genet. Evol. 2020, 83, 104321. [Google Scholar] [CrossRef] [PubMed]
- Athukorala, A.; Forwood, J.K.; Phalen, D.N.; Sarker, S. Molecular Characterisation of a Novel and Highly Divergent Passerine Adenovirus 1. Viruses 2020, 12, 1036. [Google Scholar] [CrossRef]
- Benkö, M.; Harrach, B. A proposal for a new (third) genus within the family Adenoviridae. Arch. Virol. 1998, 143, 829–837. [Google Scholar] [CrossRef]
- Simón, D.; Cristina, J.; Musto, H. Nucleotide Composition and Codon Usage Across Viruses and Their Respective Hosts. Front. Microbiol. 2021, 12, 646300. [Google Scholar] [CrossRef]
- Maeda, K.; Hondo, E.; Terakawa, J.; Kiso, Y.; Nakaichi, N.; Endoh, D.; Sakai, K.; Morikawa, S.; Mizutani, T. Isolation of Novel Adenovirus from Fruit Bat (Pteropus dasymallus yayeyamae). Emerg. Infect. Dis. 2008, 14, 347–349. [Google Scholar] [CrossRef]
- Prado-Irwin, S.R.; van de Schoot, M.; Geneva, A.J. Detection and phylogenetic analysis of adenoviruses occurring in a single anole species. PeerJ 2018, 6, e5521. [Google Scholar] [CrossRef]
- Wellehan, J.F.X.; Greenacre, C.B.; Fleming, G.J.; Stetter, M.D.; Childress, A.L.; Terrell, S.P. Siadenovirus infection in two psittacine bird species. Avian Pathol. 2009, 38, 413–417. [Google Scholar] [CrossRef]
- Conrardy, C.; Tao, Y.; Kuzmin, I.V.; Niezgoda, M.; Agwanda, B.; Breiman, R.F.; Anderson, L.J.; Rupprecht, C.E.; Tong, S. Molecular Detection of Adenoviruses, Rhabdoviruses, and Paramyxoviruses in Bats from Kenya. Am. J. Trop. Med. Hyg. 2014, 91, 258–266. [Google Scholar] [CrossRef]
- Geisbert, T.W.; Bailey, M.; Hensley, L.; Asiedu, C.; Geisbert, J.; Stanley, D.; Honko, A.; Johnson, J.; Mulangu, S.; Pau, M.G.; et al. Recombinant Adenovirus Serotype 26 (Ad26) and Ad35 Vaccine Vectors Bypass Immunity to Ad5 and Protect Nonhuman Primates against Ebolavirus Challenge. J. Virol. 2011, 85, 4222–4233. [Google Scholar] [CrossRef]
- Adenoviridae. In Virus Taxonomy; Elsevier: Amsterdam, The Netherlands, 2012; pp. 125–141. ISBN 978-0-12-384684-6.
- Capesius, I.; Bopp, M. New classification of liverworts based on molecular and morphological data. Plant Syst. Evol. 1997, 207, 87–97. [Google Scholar] [CrossRef]
- Nei, M.; Kumar, S.; Takahashi, K. The optimization principle in phylogenetic analysis tends to give incorrect topologies when the number of nucleotides or amino acids used is small. Proc. Natl. Acad. Sci. USA 1998, 95, 12390–12397. [Google Scholar] [CrossRef]
- Poe, S.; Swofford, D.L. Taxon sampling revisited. Nature 1999, 398, 299–300. [Google Scholar] [CrossRef]
- Nickrent, D.L.; Parkinson, C.L.; Palmer, J.D.; Duff, R.J. Multigene Phylogeny of Land Plants with Special Reference to Bryophytes and the Earliest Land Plants. Mol. Biol. Evol. 2000, 17, 1885–1895. [Google Scholar] [CrossRef]
- Hervé, P. Opinion: Long Branch Attraction and Protist Phylogeny. Protist 2000, 151, 307–316. [Google Scholar] [CrossRef]
- Hoef-Emden, K.; Marin, B.; Melkonian, M. Nuclear and Nucleomorph SSU rDNA Phylogeny in the Cryptophyta and the Evolution of Cryptophyte Diversity. J. Mol. Evol. 2002, 55, 161–179. [Google Scholar] [CrossRef]
- Singh, G.; Robinson, C.M.; Dehghan, S.; Schmidt, T.; Seto, D.; Jones, M.S.; Dyer, D.W.; Chodosh, J. Overreliance on the Hexon Gene, Leading to Misclassification of Human Adenoviruses: Fig 1. J. Virol. 2012, 86, 4693–4695. [Google Scholar] [CrossRef]
- Hillis, D.M. Inferring complex phytogenies. Nature 1996, 383, 130–131. [Google Scholar] [CrossRef]
- Gontcharov, A.A. Are Combined Analyses Better Than Single Gene Phylogenies? A Case Study Using SSU rDNA and rbcL Sequence Comparisons in the Zygnematophyceae (Streptophyta). Mol. Biol. Evol. 2003, 21, 612–624. [Google Scholar] [CrossRef]
- Young, A.D.; Gillung, J.P. Phylogenomics—Principles, opportunities and pitfalls of big-data phylogenetics. Syst. Entomol. 2020, 45, 225–247. [Google Scholar] [CrossRef]
- Teng, J.L.L.; Tang, Y.; Huang, Y.; Guo, F.-B.; Wei, W.; Chen, J.H.K.; Wong, S.S.Y.; Lau, S.K.P.; Woo, P.C.Y. Phylogenomic Analyses and Reclassification of Species within the Genus Tsukamurella: Insights to Species Definition in the Post-genomic Era. Front. Microbiol. 2016, 7, 1137. [Google Scholar] [CrossRef]
- Mahadevan, P. An Analysis of Adenovirus Genomes Using Whole Genome Software Tools. Bioinformation 2016, 12, 301–310. [Google Scholar] [CrossRef][Green Version]
- Konstantinidis, K.T.; Tiedje, J.M. Genomic insights that advance the species definition for prokaryotes. Proc. Natl. Acad. Sci. USA 2005, 102, 2567–2572. [Google Scholar] [CrossRef]
- Arrigoni, R.; Berumen, M.L.; Chen, C.A.; Terraneo, T.I.; Baird, A.H.; Payri, C.; Benzoni, F. Species delimitation in the reef coral genera Echinophyllia and Oxypora (Scleractinia, Lobophylliidae) with a description of two new species. Mol. Phylogenet. Evol. 2016, 105, 146–159. [Google Scholar] [CrossRef]
- Renner, M.A.M.; Heslewood, M.M.; Patzak, S.D.F.; Schäfer-Verwimp, A.; Heinrichs, J. By how much do we underestimate species diversity of liverworts using morphological evidence? An example from Australasian Plagiochila (Plagiochilaceae: Jungermanniopsida). Mol. Phylogenet. Evol. 2017, 107, 576–593. [Google Scholar] [CrossRef] [PubMed]
- Nieto-Montes de Oca, A.; Barley, A.J.; Meza-Lázaro, R.N.; García-Vázquez, U.O.; Zamora-Abrego, J.G.; Thomson, R.C.; Leaché, A.D. Phylogenomics and species delimitation in the knob-scaled lizards of the genus Xenosaurus (Squamata: Xenosauridae) using ddRADseq data reveal a substantial underestimation of diversity. Mol. Phylogenet. Evol. 2017, 106, 241–253. [Google Scholar] [CrossRef] [PubMed]
- Tang, C.Q.; Humphreys, A.M.; Fontaneto, D.; Barraclough, T.G. Effects of phylogenetic reconstruction method on the robustness of species delimitation using single-locus data. Methods Ecol. Evol. 2014, 5, 1086–1094. [Google Scholar] [CrossRef] [PubMed]
- Ducasse, J.; Ung, V.; Lecointre, G.; Miralles, A. LIMES: A tool for comparing species partition. Bioinformatics 2020, 36, 2282–2283. [Google Scholar] [CrossRef] [PubMed]
- International Committee on Taxonomy of Viruses Virus Taxonomy: The ICTV Report on Virus Classification and Taxon Nomenclature. Available online: https://talk.ictvonline.org/ictv-reports/ictv_online_report/ (accessed on 30 November 2021).
- Spielman, S.J.; Weaver, S.; Shank, S.D.; Magalis, B.R.; Li, M.; Kosakovsky Pond, S.L. Evolution of Viral Genomes: Interplay Between Selection, Recombination, and Other Forces. In Evolutionary Genomics; Anisimova, M., Ed.; Springer: New York, NY, USA, 2019; Volume 1910, pp. 427–468. ISBN 978-1-4939-9073-3. [Google Scholar]
- Van Valen, L. Molecular evolution as predicted by natural selection. J. Mol. Evol. 1974, 3, 89–101. [Google Scholar] [CrossRef]
- Stenseth, N.; Smith, J.M. Coevolution in Ecosystems: Red Queen Evolution or Stasis? Evolution 1984, 38, 870. [Google Scholar] [CrossRef]
- Hedrick, P.W. Evolutionary Genetics of the Major Histocompatibility Complex. Am. Nat. 1994, 143, 945–964. [Google Scholar] [CrossRef]
- Obbard, D.J.; Jiggins, F.M.; Halligan, D.L.; Little, T.J. Natural Selection Drives Extremely Rapid Evolution in Antiviral RNAi Genes. Curr. Biol. 2006, 16, 580–585. [Google Scholar] [CrossRef]
- Drosophila 12 Genomes Consortium Evolution of genes and genomes on the Drosophila phylogeny. Nature 2007, 450, 203–218. [CrossRef]
- Blanc, G.; Ngwamidiba, M.; Ogata, H.; Fournier, P.-E.; Claverie, J.-M.; Raoult, D. Molecular Evolution of Rickettsia Surface Antigens: Evidence of Positive Selection. Mol. Biol. Evol. 2005, 22, 2073–2083. [Google Scholar] [CrossRef]
- Mu, J.; Awadalla, P.; Duan, J.; McGee, K.M.; Keebler, J.; Seydel, K.; McVean, G.A.T.; Su, X. Genome-wide variation and identification of vaccine targets in the Plasmodium falciparum genome. Nat. Genet. 2007, 39, 126–130. [Google Scholar] [CrossRef]
- Barrett, L.G.; Thrall, P.H.; Dodds, P.N.; van der Merwe, M.; Linde, C.C.; Lawrence, G.J.; Burdon, J.J. Diversity and Evolution of Effector Loci in Natural Populations of the Plant Pathogen Melampsora lini. Mol. Biol. Evol. 2009, 26, 2499–2513. [Google Scholar] [CrossRef]
- Streicker, D.G.; Altizer, S.M.; Velasco-Villa, A.; Rupprecht, C.E. Variable evolutionary routes to host establishment across repeated rabies virus host shifts among bats. Proc. Natl. Acad. Sci. USA 2012, 109, 19715–19720. [Google Scholar] [CrossRef]
- Brockhurst, M.A.; Chapman, T.; King, K.C.; Mank, J.E.; Paterson, S.; Hurst, G.D.D. Running with the Red Queen: The role of biotic conflicts in evolution. Proc. R. Soc. B Biol. Sci. 2014, 281, 20141382. [Google Scholar] [CrossRef]
- Frank, H.K.; Enard, D.; Boyd, S.D. Exceptional diversity and selection pressure on SARS-CoV and SARS-CoV-2 host receptor in bats compared to other mammals. bioRxiv 2020. [Google Scholar] [CrossRef]
- Shih, A.C.-C.; Hsiao, T.-C.; Ho, M.-S.; Li, W.-H. Simultaneous amino acid substitutions at antigenic sites drive influenza A hemagglutinin evolution. Proc. Natl. Acad. Sci. USA 2007, 104, 6283–6288. [Google Scholar] [CrossRef]
- Bhatt, S.; Holmes, E.C.; Pybus, O.G. The Genomic Rate of Molecular Adaptation of the Human Influenza A Virus. Mol. Biol. Evol. 2011, 28, 2443–2451. [Google Scholar] [CrossRef]
- Strelkowa, N.; Lässig, M. Clonal Interference in the Evolution of Influenza. Genetics 2012, 192, 671–682. [Google Scholar] [CrossRef]
- Illingworth, C.J.R.; Mustonen, V. Components of Selection in the Evolution of the Influenza Virus: Linkage Effects Beat Inherent Selection. PLoS Pathog. 2012, 8, e1003091. [Google Scholar] [CrossRef]
- Meyer, A.G.; Dawson, E.T.; Wilke, C.O. Cross-species comparison of site-specific evolutionary-rate variation in influenza haemagglutinin. Philos. Trans. R. Soc. B Biol. Sci. 2013, 368, 20120334. [Google Scholar] [CrossRef]
- Nozawa, M.; Suzuki, Y.; Nei, M. Reliabilities of identifying positive selection by the branch-site and the site-prediction methods. Proc. Natl. Acad. Sci. USA 2009, 106, 6700–6705. [Google Scholar] [CrossRef]
- Longdon, B.; Day, J.P.; Alves, J.M.; Smith, S.C.L.; Houslay, T.M.; McGonigle, J.E.; Tagliaferri, L.; Jiggins, F.M. Host shifts result in parallel genetic changes when viruses evolve in closely related species. PLoS Pathog. 2018, 14, e1006951. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| TeAdV-1 | Gene | Strand | Size (aa) | DAdV-1 | PsAdV-3 | |
|---|---|---|---|---|---|---|
| p32 K | 236 | 1174 | − | 312 | p32 K | p32 K |
| LH2 | 1209 | 1625 | + | 138 | LH2 | E1B protein, small T-antigen |
| LH1 | 1656 | 2804 | + | 382 | E1B 55 K | |
| IVa2 protein | 2875 | 3696 | − | 296 | IVa2 protein | IVa2 protein |
| 4614 | 4682 | |||||
| DNA polymerase | 3945 | 7181 | − | 1078 | DNA polymerase | DNA polymerase |
| pTP | 7157 | 8950 | − | 602 | pTP | pTP |
| 11680 | 11694 | |||||
| 52 K protein | 8985 | 9971 | + | 328 | 52 K protein | 52 K protein |
| pIIIa protein | 9955 | 11664 | + | 569 | pIIIa protein | pIIIa protein |
| penton base protein | 11704 | 13062 | + | 452 | penton base protein | penton base protein |
| pVII protein | 13104 | 13559 | + | 151 | pVII | pVII |
| pX protein | 13568 | 13765 | + | 65 | pX | pX |
| pVI protein | 13800 | 14435 | + | 211 | pVI | pVI |
| hexon protein | 14456 | 17188 | + | 910 | hexon protein | hexon protein |
| protease | 17185 | 17790 | + | 201 | protease | protease |
| DNA-binding protein | 17809 | 18951 | − | 380 | DNA-binding protein | DNA-binding protein |
| 100 K protein | 19005 | 20924 | + | 639 | 100 K protein | 100 K protein |
| 22 K protein | 20758 | 20982 | + | 75 | ||
| 33 K protein | 20758 | 20973 | + | 150 | 33 K protein | 33 K protein |
| 21059 | 21292 | |||||
| pVIII protein | 21323 | 22129 | + | 268 | pVIII protein | pVIII protein |
| U-exon | 22142 | 22306 | − | 54 | U-exon | U-exon |
| fiber protein | 22324 | 24369 | + | 681 | fiber protein | fiber 2 protein |
| E4.3 protein | 24383 | 25273 | − | 296 | 34 K-2 | E4.3 protein |
| E4.2 protein | 25221 | 26027 | − | 268 | 34 K-1 | E4.2 protein |
| E4.1 protein | 25948 | 26385 | − | 145 | E4.1 protein | |
| ORF8 | 26628 | 26870 | − | 80 | ||
| ORF7 | 26888 | 27451 | − | 187 | ||
| ORF1 | 27508 | 27966 | + | 152 | ||
| ORF2 | 28072 | 28359 | + | 95 | ||
| ORF3 | 28366 | 28698 | + | 110 | ||
| ORF6 | 28872 | 29558 | − | 228 | ||
| ORF5 | 29658 | 30296 | − | 212 | ||
| ORF4 | 30424 | 31146 | + | 240 | ||
| Gene | Annotation |
|---|---|
| 100 K protein | participation in the transport of hexon monomers to the nucleus and trimerization [61] |
| 23 K protein (endopeptidase, protease) | participation in the cleavage of some AdV precursor proteins [62,63] |
| 52 K protein | participation in the packaging of the viral DNA into the capsid [62,63] |
| DBP (DNA-binding protein) | participation in the elongation phase of AdV DNA replication by unwinding the template [64] |
| hexon | major capsid protein [62,63] |
| III (penton base) | major capsid protein [62,63] |
| pIIIa | minor capsid protein [62,63,65] |
| IVa2 | participation in the packaging of the viral DNA into the capsid [62,63] |
| Pol (DNA polymerase) | participation in the elongation phase of AdV DNA replication [64] |
| pTP (preterminal protein) | the protein primer for AdV DNA replication [64] |
| pVI | minor capsid protein [62,63,65] |
| pVIII | minor capsid protein [62,63,65] |
| ASAP | PTP | GMYC (Single-Threshold) | GMYC (Multiple-Threshold) |
|---|---|---|---|
| TAdV-1 | TAdV-1 | TAdV-1 | TAdV-1 |
| LC606503.1 BoAdV-F LC597488.1 BoAdV-F MN901942.2 BoAdV-F | LC606503.1 BoAdV-F LC597488.1 BoAdV-F MN901942.2 BoAdV-F | LC606503.1 BoAdV-F LC597488.1 BoAdV-F MN901942.2 BoAdV-F | LC606503.1 BoAdV-F LC597488.1 BoAdV-F MN901942.2 BoAdV-F |
| U40839.3 OvAdV-D | U40839.3 OvAdV-D | U40839.3 OvAdV-D | U40839.3 OvAdV-D |
| MK537328.1 OdAdV-A KY748210.1 OdAdV-A KY468403.1 OdAdV-A KY468402.1 OdAdV-A MK343439.1 OdAdV-A KY468406.1 OdAdV-A KY468407.1 OdAdV-A KY468404.1 OdAdV-A KY468405.1 OdAdV-A | MK537328.1 OdAdV-A KY748210.1 OdAdV-A KY468403.1 OdAdV-A KY468402.1 OdAdV-A MK343439.1 OdAdV-A KY468406.1 OdAdV-A KY468407.1 OdAdV-A KY468404.1 OdAdV-A KY468405.1 OdAdV-A | MK537328.1 OdAdV-A KY748210.1 OdAdV-A KY468403.1 OdAdV-A KY468402.1 OdAdV-A MK343439.1 OdAdV-A KY468406.1 OdAdV-A KY468407.1 OdAdV-A KY468404.1 OdAdV-A KY468405.1 OdAdV-A | MK537328.1 OdAdV-A KY748210.1 OdAdV-A KY468403.1 OdAdV-A KY468402.1 OdAdV-A MK343439.1 OdAdV-A KY468406.1 OdAdV-A KY468407.1 OdAdV-A KY468404.1 OdAdV-A KY468405.1 OdAdV-A |
| AF036092.3 BoAdV-D JQ345700.1 BoAdV-E | AF036092.3 BoAdV-D | AF036092.3 BoAdV-D JQ345700.1 BoAdV-E | AF036092.3 BoAdV-D |
| JQ345700.1 BoAdV-E | JQ345700.1 BoAdV-E | ||
| MT050041.1 LiAdV-B | MT050041.1 LiAdV-B | MT050041.1 LiAdV-B | MT050041.1 LiAdV-B |
| KJ156523.1 LiAdV-A | KJ156523.1 LiAdV-A | KJ156523.1 LiAdV-A | KJ156523.1 LiAdV-A |
| KJ675568.1 PsAdV-A MN025529.1 PsAdV-A | KJ675568.1 PsAdV-A | KJ675568.1 PsAdV-A MN025529.1 PsAdV-A | KJ675568.1 PsAdV-A |
| MN025529.1 PsAdV-A | MN025529.1 PsAdV-A | ||
| KJ452170.1 DAdV-A KJ452171.1 DAdV-A | KJ452170.1 DAdV-A KJ452171.1 DAdV-A | KJ452170.1 DAdV-A KJ452171.1 DAdV-A | KJ452170.1 DAdV-A KJ452171.1 DAdV-A |
| KF286430.1 DAdV-A KJ452172.1 DAdV-A MT646045.1 DAdV-A MN310513.1 DAdV-A | KF286430.1 DAdV-A KJ452172.1 DAdV-A MT646045.1 DAdV-A MN310513.1 DAdV-A | KF286430.1 DAdV-A KJ452172.1 DAdV-A MT646045.1 DAdV-A MN310513.1 DAdV-A | KF286430.1 DAdV-A KJ452172.1 DAdV-A |
| MT646045.1 DAdV-A | |||
| MN310513.1 DAdV-A | |||
| 9 | 11 | 8 | 14 |
| Protein | Amino Acid Coordinate | PP (CODEML) | PP (HyPhy) |
|---|---|---|---|
| 100 K protein | 118 | 0.886 | 0.962 |
| 230 | 0.807 | 0.951 | |
| 450 | 0.905 | 0.972 | |
| 96 | 0.87 | 0.972 | |
| 13 | 0.903 | 0.955 | |
| 162 | 0.904 | 0.950 | |
| 180 | 0.936 | 0.980 | |
| 192 | 0.974 | 0.990 | |
| 35 | 0.953 | 0.946 | |
| pIVa2 | 123 | 0.937 | 0.983 |
| 137 | 0.894 | 0.982 | |
| 152 | 0.946 | 0.971 | |
| 94 | 0.935 | 0.964 | |
| DNA polymerase | 1044 | 0.862 | 0.951 |
| 366 | 0.934 | 0.973 | |
| 367 | 0.927 | 0.962 | |
| 773 | 0.941 | 0.961 | |
| pTP | 145 | 0.85 | 0.961 |
| 187 | 0.946 | 0.966 | |
| 297 | 0.859 | 0.975 | |
| 405 | 0.926 | 0.952 | |
| 44 | 0.946 | 0.972 | |
| 445 | 0.939 | 0.966 | |
| 565 | 0.795 | 0.950 | |
| 89 | 0.93 | 0.966 | |
| 96 | 0.939 | 0.988 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Matsvay, A.; Dyachkova, M.; Mikhaylov, I.; Kiselev, D.; Say, A.; Burskaia, V.; Artyushin, I.; Khafizov, K.; Shipulin, G. Complete Genome Sequence, Molecular Characterization and Phylogenetic Relationships of a Novel Tern Atadenovirus. Microorganisms 2022, 10, 31. https://doi.org/10.3390/microorganisms10010031
Matsvay A, Dyachkova M, Mikhaylov I, Kiselev D, Say A, Burskaia V, Artyushin I, Khafizov K, Shipulin G. Complete Genome Sequence, Molecular Characterization and Phylogenetic Relationships of a Novel Tern Atadenovirus. Microorganisms. 2022; 10(1):31. https://doi.org/10.3390/microorganisms10010031
Chicago/Turabian StyleMatsvay, Alina, Marina Dyachkova, Ivan Mikhaylov, Daniil Kiselev, Anna Say, Valentina Burskaia, Ilya Artyushin, Kamil Khafizov, and German Shipulin. 2022. "Complete Genome Sequence, Molecular Characterization and Phylogenetic Relationships of a Novel Tern Atadenovirus" Microorganisms 10, no. 1: 31. https://doi.org/10.3390/microorganisms10010031
APA StyleMatsvay, A., Dyachkova, M., Mikhaylov, I., Kiselev, D., Say, A., Burskaia, V., Artyushin, I., Khafizov, K., & Shipulin, G. (2022). Complete Genome Sequence, Molecular Characterization and Phylogenetic Relationships of a Novel Tern Atadenovirus. Microorganisms, 10(1), 31. https://doi.org/10.3390/microorganisms10010031