Comparative Genome Analysis of 33 Chlamydia Strains Reveals Characteristic Features of Chlamydia Psittaci and Closely Related Species

 ,
,  , ,
, , 
Abstract
1. Introduction
2. Results
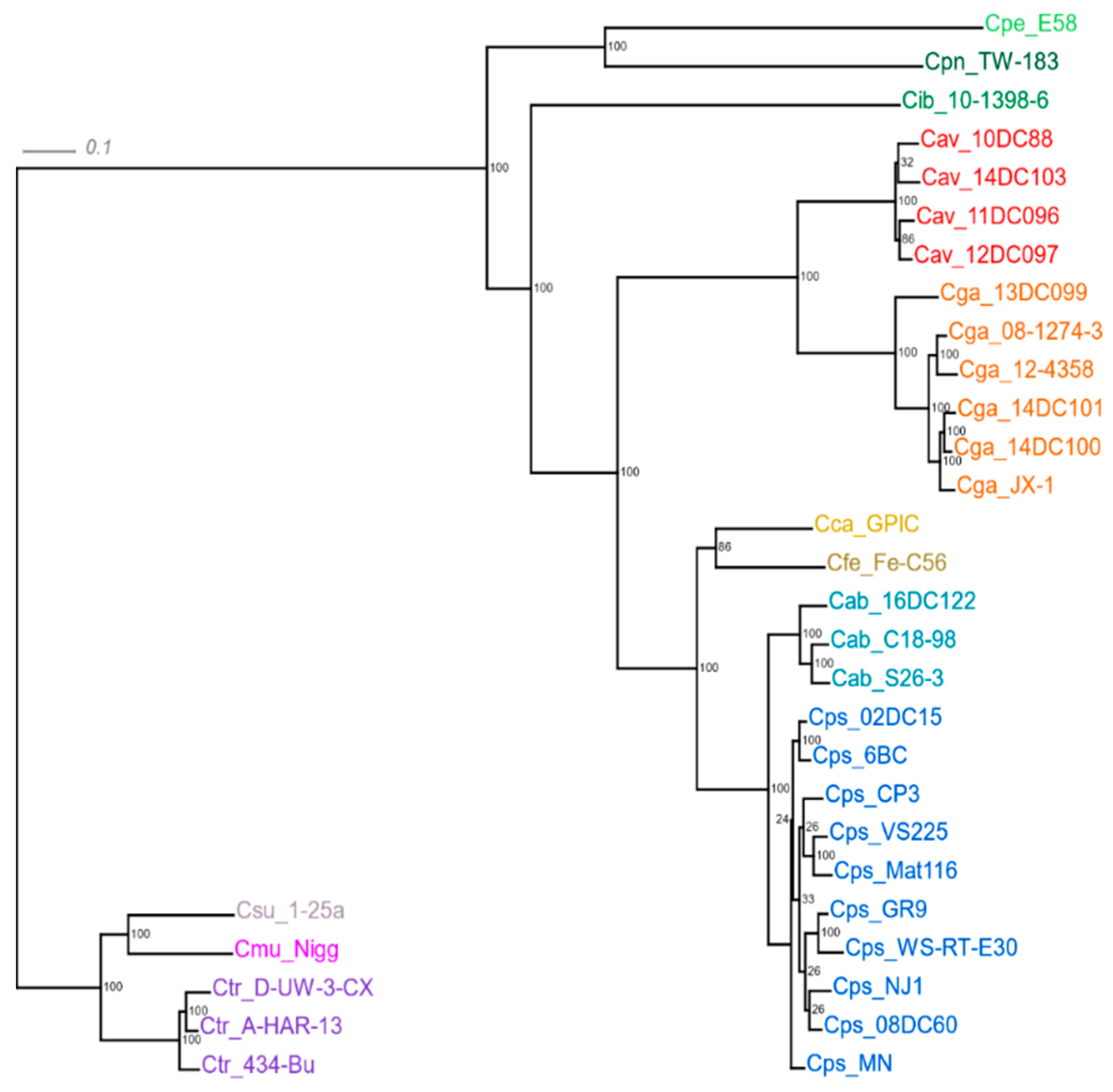
2.1. General Characteristics of the Genome Sequences
2.2. Common and Unique Elements in the Genomes of Chlamydia spp.
2.3. The Plasticity Zone (PZ)
2.4. Genes Encoding Polymorphic Membrane Proteins (pmps)
2.5. Inc Proteins
2.6. The Secreted Inner Nuclear Membrane-Associated Chlamydia Protein (SINC)
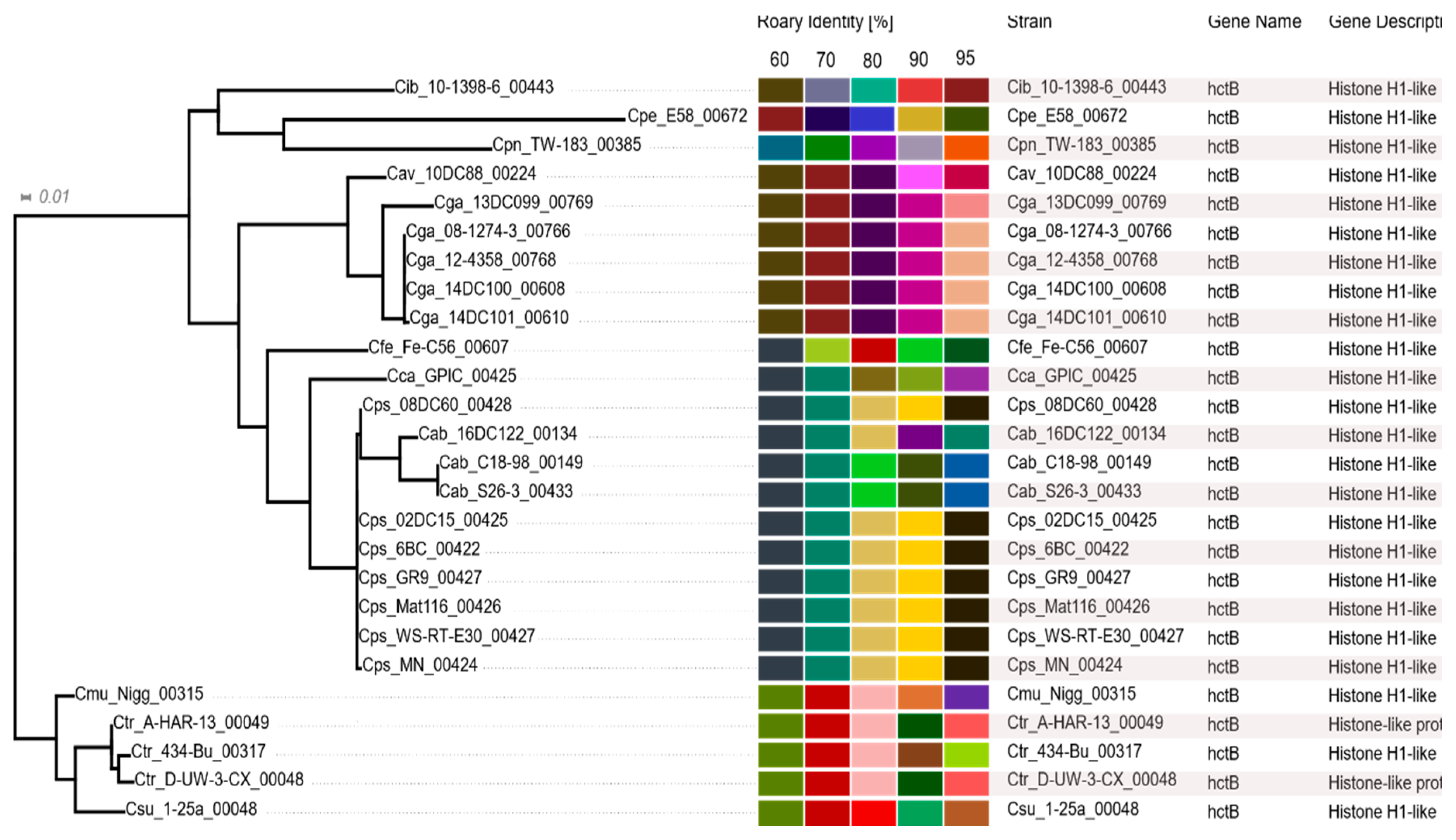
2.7. Histone-Like Proteins HctA and HctB
2.8. Pseudogenes
3. Discussion
3.1. Bioinformatics Tools: New and Unique Features of the RIBAP
3.2. Core Genome vs. Dispensable Genome
3.3. The Plasticity Zone
3.4. The Family of Polymorphic Membrane Proteins (Pmps)
3.5. Inclusion Membrane Proteins
3.6. SINC Protein
3.7. Histone-Like Proteins HctA and HctB
3.8. Pseudogenes
4. Materials and Methods
4.1. Chlamydial Strains
4.2. Genome Sequencing and Genome Assembly
4.3. Pan-Genome and Core Genome Calculation Using RIBAP
4.4. Annotation
4.5. Pan- Genome Scaffold
4.6. Integer Linear Programming and GLPK
4.7. Creating a RIBAP Group
4.8. The RIBAP Output
4.9. Phylogenetic Tree Based on Core Genomes
4.10. UpSet Diagrams
4.11. Multiple Blast to Identify Homologs of Pmp, Inc and SinC Genes
4.12. Calculation of Sequence Identities
4.13. Normalization of Genomes
5. Conclusions
6. Availability of Data and Materials
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Sigalova, O.M.; Chaplin, A.V.; Bochkareva, O.O.; Shelyakin, P.V.; Filaretov, V.A.; Akkuratov, E.E.; Burskaia, V.; Gelfand, M.S. Chlamydia pan-genomic analysis reveals balance between host adaptation and selective pressure to genome reduction. BMC Genom. 2019, 20, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Kuo, C.C.; Grayston, J.T. Amino acid requirements for growth of Chlamydia pneumoniae in cell cultures: Growth enhancement by lysine or methionine depletion. J. Clin. Microbiol. 1990, 28, 1098–1100. [Google Scholar] [CrossRef] [PubMed]
- Moulder, J.W. Interaction of chlamydiae and host cells in vitro. Microbiol. Rev. 1991, 55, 143–190. [Google Scholar] [CrossRef] [PubMed]
- Ouellette, S.P.; Dorsey, F.C.; Moshiach, S.; Cleveland, J.L.; Carabeo, R.A. Chlamydia Species-Dependent Differences in the Growth Requirement for Lysosomes. PLoS ONE 2011, 6, e16783. [Google Scholar] [CrossRef] [PubMed]
- Saka, H.A.; Valdivia, R.H. Acquisition of nutrients by Chlamydiae: Unique challenges of living in an intracellular compartment. Curr. Opin. Microbiol. 2010, 13, 4–10. [Google Scholar] [CrossRef] [PubMed]
- Moulder, J.W. The Biochemistry of Intracellular Parasitism; University of Chicago Press: Chicago, IL, USA, 1962. [Google Scholar]
- Stephens, R.S.; Kalman, S.; Lammel, C.; Fan, J.; Marathe, R.; Aravind, L.; Mitchell, W.; Olinger, L.; Tatusov, R.L.; Zhao, Q.; et al. Genome Sequence of an Obligate Intracellular Pathogen of Humans: Chlamydia trachomatis. Science 1998, 282, 754–759. [Google Scholar] [CrossRef]
- Iliffe-Lee, E.R.; McClarty, G. Glucose metabolism in Chlamydia trachomatis: The ‘energy parasite’ hypothesis revisited. Mol. Microbiol. 1999, 33, 177–187. [Google Scholar] [CrossRef]
- Omsland, A.; Sixt, B.S.; Horn, M.; Hackstadt, T. Chlamydial metabolism revisited: Interspecies metabolic variability and developmental stage-specific physiologic activities. FEMS Microbiol. Rev. 2014, 38, 779–801. [Google Scholar] [CrossRef]
- O’Connell, C.M.; Ferone, M.E. Chlamydia trachomatis Genital Infections. Microb. Cell 2016, 3, 390–403. [Google Scholar] [CrossRef]
- Taylor, H.R.; Burton, M.J.; Haddad, D.; West, S.; Wright, H. Trachoma. Lancet 2014, 384, 2142–2152. [Google Scholar] [CrossRef]
- Hahn, D.L.; Azenabor, A.A.; Beatty, W.L.; Byrne, G.I. Chlamydia pneumoniae as a respiratory pathogen. Front. Biosci. 2002, 7, e66–e76. [Google Scholar] [CrossRef]
- Knittler, M.R.; Sachse, K. Chlamydia psittaci: Update on an underestimated zoonotic agent (Minireview). FEMS Pathog. Dis. 2014, 73. [Google Scholar] [CrossRef] [PubMed]
- Sachse, K.; Laroucau, K.; Riege, K.; Wehner, S.; Dilcher, M.; Creasy, H.H.; Weidmann, M.; Myers, G.S.A.; Vorimore, F.; Vicari, N.; et al. Evidence for the existence of two new members of the family Chlamydiaceae and proposal of Chlamydia avium sp. nov. and Chlamydia gallinacea sp. nov. Syst. Appl. Microbiol. 2014, 37, 79–88. [Google Scholar] [CrossRef] [PubMed]
- Guo, W.; Jelocnik, M.; Li, J.; Sachse, K.; Polkinghorne, A.; Pannekoek, Y.; Kaltenboeck, B.; Gong, J.; You, J.; Wang, C. From genomes to genotypes: Molecular epidemiological analysis of Chlamydia gallinacea reveals a high level of genetic diversity for this newly emerging chlamydial pathogen. BMC Genom. 2017, 18, 949. [Google Scholar] [CrossRef] [PubMed]
- Floriano, A.M.; Rigamonti, S.; Comandatore, F.; Scaltriti, E.; Longbottom, D.; Livingstone, M.; Laroucau, K.; Gaffuri, A.; Pongolini, S.; Magnino, S.; et al. Complete Genome Sequence of Chlamydia avium PV 4360/2, Isolated from a Feral Pigeon in Italy. Microbiol. Resour. Announc. 2020, 9. [Google Scholar] [CrossRef] [PubMed]
- Hölzer, M.; Laroucau, K.; Creasy, H.H.; Ott, S.; Vorimore, F.; Bavoil, P.M.; Marz, M.; Sachse, K. Whole-Genome Sequence of Chlamydia gallinacea Type Strain 08-1274/3. Genome Announc. 2016, 4, e00708-16. [Google Scholar] [CrossRef]
- Vorimore, F.; Hsia, R.-C.; Huot-Creasy, H.; Bastian, S.; DeRuyter, L.; Passet, A.; Sachse, K.; Bavoil, P.; Myers, G.; Laroucau, K. Isolation of a New Chlamydia species from the Feral Sacred Ibis (Threskiornis aethiopicus): Chlamydia ibidis. PLoS ONE 2013, 8, e74823. [Google Scholar] [CrossRef]
- Laroucau, K.; Vorimore, F.; Aaziz, R.; Solmonson, L.; Hsia, R.C.; Bavoil, P.M.; Fach, P.; Holzer, M.; Wuenschmann, A.; Sachse, K. Chlamydia buteonis, a new Chlamydia species isolated from a red-shouldered hawk. Syst. Appl. Microbiol. 2019, 42, 125997. [Google Scholar] [CrossRef]
- Longbottom, D.; Coulter, L.J. Animal chlamydioses and zoonotic implications. J. Comp. Pathol. 2003, 128, 217–244. [Google Scholar] [CrossRef]
- Szymańska-Czerwińska, M.; Mitura, A.; Niemczuk, K.; Zaręba, K.; Jodełko, A.; Pluta, A.; Scharf, S.; Vitek, B.; Aaziz, R.; Vorimore, F.; et al. Dissemination and genetic diversity of chlamydial agents in Polish wildfowl: Isolation and molecular characterisation of avian Chlamydia abortus strains. PLoS ONE 2017, 12, e0174599. [Google Scholar] [CrossRef]
- Sachse, K.; Vretou, E.; Livingstone, M.; Borel, N.; Pospischil, A.; Longbottom, D. Recent developments in the laboratory diagnosis of chlamydial infections. Vet. Microbiol. 2009, 135, 2–21. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Kahane, S.; Cutcliffe, L.T.; Skilton, R.J.; Lambden, P.R.; Clarke, I.N. Development of a transformation system for Chlamydia trachomatis: Restoration of glycogen biosynthesis by acquisition of a plasmid shuttle vector. PLoS Pathog. 2011, 7, e1002258. [Google Scholar] [CrossRef] [PubMed]
- Hadfield, J.; Harris, S.R.; Seth-Smith, H.M.B.; Parmar, S.; Andersson, P.; Giffard, P.M.; Schachter, J.; Moncada, J.; Ellison, L.; Vaulet, M.L.G.; et al. Comprehensive global genome dynamics of Chlamydia trachomatis show ancient diversification followed by contemporary mixing and recent lineage expansion. Genome Res. 2017, 27, 1220–1229. [Google Scholar] [CrossRef] [PubMed]
- Kalman, S.; Mitchell, W.; Marathe, R.; Lammel, C.; Fan, J.; Hyman, R.W.; Olinger, L.; Grimwood, J.; Davis, R.W.; Stephens, R.S. Comparative genomes of Chlamydia pneumoniae and C. trachomatis. Nat. Genet. 1999, 21, 385–389. [Google Scholar] [CrossRef]
- Roulis, E.; Bachmann, N.L.; Myers, G.S.; Huston, W.; Summersgill, J.; Hudson, A.; Dreses-Werringloer, U.; Polkinghorne, A.; Timms, P. Comparative genomic analysis of human Chlamydia pneumoniae isolates from respiratory, brain and cardiac tissues. Genomics 2015, 106, 373–383. [Google Scholar] [CrossRef] [PubMed]
- Schofl, G.; Voigt, A.; Litsche, K.; Sachse, K.; Saluz, H.P. Complete genome sequences of four mammalian isolates of Chlamydophila psittaci. J. Bacteriol. 2011, 193, 4258. [Google Scholar] [CrossRef]
- Van Lent, S.; Piet, J.R.; Beeckman, D.; van der Ende, A.; Van Nieuwerburgh, F.; Bavoil, P.; Myers, G.; Vanrompay, D.; Pannekoek, Y. Full genome sequences of all nine Chlamydia psittaci genotype reference strains. J. Bacteriol. 2012, 194, 6930–6931. [Google Scholar] [CrossRef]
- Read, T.D.; Joseph, S.J.; Didelot, X.; Liang, B.; Patel, L.; Dean, D. Comparative analysis of Chlamydia psittaci genomes reveals the recent emergence of a pathogenic lineage with a broad host range. mBio 2013, 4. [Google Scholar] [CrossRef]
- Seth-Smith, H.M.B.; Buso, L.S.; Livingstone, M.; Sait, M.; Harris, S.R.; Aitchison, K.D.; Vretou, E.; Siarkou, V.I.; Laroucau, K.; Sachse, K.; et al. European Chlamydia abortus livestock isolate genomes reveal unusual stability and limited diversity, reflected in geographical signatures. BMC Genom. 2017, 18, 344. [Google Scholar] [CrossRef]
- Taylor-Brown, A.; Spang, L.; Borel, N.; Polkinghorne, A. Culture-independent metagenomics supports discovery of uncultivable bacteria within the genus Chlamydia. Sci. Rep. 2017, 7, 10661. [Google Scholar] [CrossRef]
- Joseph, S.J.; Marti, H.; Didelot, X.; Castillo-Ramirez, S.; Read, T.D.; Dean, D. Chlamydiaceae genomics reveals interspecies admixture and the recent evolution of Chlamydia abortus infecting lower mammalian species and humans. Genome Biol. Evol. 2015, 7, 3070–3084. [Google Scholar] [CrossRef]
- Collingro, A.; Tischler, P.; Weinmaier, T.; Penz, T.; Heinz, E.; Brunham, R.C.; Read, T.D.; Bavoil, P.M.; Sachse, K.; Kahane, S.; et al. Unity in variety–the pan-genome of the Chlamydiae. Mol. Biol. Evol. 2011, 28, 3253–3270. [Google Scholar] [CrossRef] [PubMed]
- Fehlner-Gardiner, C.; Roshick, C.; Carlson, J.H.; Hughes, S.; Belland, R.J.; Caldwell, H.D.; McClarty, G. Molecular basis defining human Chlamydia trachomatis tissue tropism. A possible role for tryptophan synthase. J. Biol. Chem. 2002, 277, 26893–26903. [Google Scholar] [CrossRef] [PubMed]
- Voigt, A.; Schofl, G.; Saluz, H.P. The Chlamydia psittaci genome: A comparative analysis of intracellular pathogens. PLoS ONE 2012, 7, e35097. [Google Scholar] [CrossRef] [PubMed]
- Sachse, K.; Rahman, K.S.; Schnee, C.; Müller, E.; Peisker, M.; Schumacher, T.; Schubert, E.; Ruettger, A.; Kaltenboeck, B.; Ehricht, R. A novel synthetic peptide microarray assay detects Chlamydia species-specific antibodies in animal and human sera. Sci. Rep. 2018, 8, 4701. [Google Scholar] [CrossRef] [PubMed]
- Mital, J.; Miller, N.J.; Dorward, D.W.; Dooley, C.A.; Hackstadt, T. Role for chlamydial inclusion membrane proteins in inclusion membrane structure and biogenesis. PLoS ONE 2013, 8, e63426. [Google Scholar] [CrossRef] [PubMed]
- Vasilevsky, S.; Stojanov, M.; Greub, G.; Baud, D. Chlamydial polymorphic membrane proteins: Regulation, function and potential vaccine candidates. Virulence 2016, 7, 11–22. [Google Scholar] [CrossRef]
- Molleken, K.; Schmidt, E.; Hegemann, J.H. Members of the Pmp protein family of Chlamydia pneumoniae mediate adhesion to human cells via short repetitive peptide motifs. Mol. Microbiol. 2010, 78, 1004–1017. [Google Scholar] [CrossRef]
- Hackstadt, T.; Baehr, W.; Ying, Y. Chlamydia trachomatis developmentally regulated protein is homologous to eukaryotic histone H1. Proc. Natl. Acad. Sci. USA 1991, 88, 3937–3941. [Google Scholar] [CrossRef]
- Medini, D.; Donati, C.; Tettelin, H.; Masignani, V.; Rappuoli, R. The microbial pan-genome. Curr. Opin. Genet. Dev. 2005, 15, 589–594. [Google Scholar] [CrossRef]
- Thomson, N.R.; Holden, M.T.; Carder, C.; Lennard, N.; Lockey, S.J.; Marsh, P.; Skipp, P.; O’Connor, C.D.; Goodhead, I.; Norbertzcak, H.; et al. Chlamydia trachomatis: Genome sequence analysis of lymphogranuloma venereum isolates. Genome Res. 2008, 18, 161–171. [Google Scholar] [CrossRef] [PubMed]
- Schmid, M.; Frei, D.; Patrignani, A.; Schlapbach, R.; Frey, J.E.; Remus-Emsermann, M.N.P.; Ahrens, C.H. Pushing the limits of de novo genome assembly for complex prokaryotic genomes harboring very long, near identical repeats. Nucleic Acids Res. 2018, 46, 8953–8965. [Google Scholar] [CrossRef] [PubMed]
- Ricker, N.; Qian, H.; Fulthorpe, R.R. The limitations of draft assemblies for understanding prokaryotic adaptation and evolution. Genomics 2012, 100, 167–175. [Google Scholar] [CrossRef] [PubMed]
- Conway, J.R.; Lex, A.; Gehlenborg, N. UpSetR: An R package for the visualization of intersecting sets and their properties. Bioinformatics 2017, 33, 2938–2940. [Google Scholar] [CrossRef]
- Collingro, A.; Kostlbacher, S.; Horn, M. Chlamydiae in the Environment. Trends Microbiol. 2020. [Google Scholar] [CrossRef]
- Pillonel, T.; Bertelli, C.; Salamin, N.; Greub, G. Taxogenomics of the order Chlamydiales. Int. J. Syst. Evol. Microbiol. 2015, 65, 1381–1393. [Google Scholar] [CrossRef]
- Aggelen, H.v.; Kolde, R.; Chamarthi, H.; Loving, J.; Fan, Y.; Fallon, J.T.; Huang, W.; Wa, G.; Fortunato-Habib, M.M.; Carmona, J.J.; et al. A novel core genome approach to enable prospective and dynamic monitoring of infectious outbreaks. bioRxiv 2018. [Google Scholar] [CrossRef]
- Segerman, B. The genetic integrity of bacterial species: The core genome and the accessory genome, two different stories. Front. Cell. Infect. Microbiol. 2012, 2, 116. [Google Scholar] [CrossRef]
- Rokas, A.; Williams, B.L.; King, N.; Carroll, S.B. Genome-scale approaches to resolving incongruence in molecular phylogenies. Nature 2003, 425, 798–804. [Google Scholar] [CrossRef]
- Sachse, K.; Bavoil, P.M.; Kaltenboeck, B.; Stephens, R.S.; Kuo, C.C.; Rossello-Mora, R.; Horn, M. Emendation of the family Chlamydiaceae: Proposal of a single genus, Chlamydia, to include all currently recognized species. Syst. Appl. Microbiol. 2015, 38, 99–103. [Google Scholar] [CrossRef]
- Xie, G.; Bonner, C.A.; Jensen, R.A. Dynamic diversity of the tryptophan pathway in chlamydiae: Reductive evolution and a novel operon for tryptophan recapture. Genome Biol. 2002, 3, research0051. [Google Scholar] [CrossRef]
- Klapproth, J.M. The role of lymphostatin/EHEC factor for adherence-1 in the pathogenesis of gram negative infection. Toxins 2010, 2, 954–962. [Google Scholar] [CrossRef] [PubMed]
- Reinert, D.J.; Jank, T.; Aktories, K.; Schulz, G.E. Structural basis for the function of Clostridium difficile toxin B. J. Mol. Biol. 2005, 351, 973–981. [Google Scholar] [CrossRef] [PubMed]
- Busch, C.; Hofmann, F.; Selzer, J.; Munro, S.; Jeckel, D.; Aktories, K. A common motif of eukaryotic glycosyltransferases is essential for the enzyme activity of large clostridial cytotoxins. J. Biol. Chem. 1998, 273, 19566–19572. [Google Scholar] [CrossRef] [PubMed]
- Read, T.D.; Myers, G.S.; Brunham, R.C.; Nelson, W.C.; Paulsen, I.T.; Heidelberg, J.; Holtzapple, E.; Khouri, H.; Federova, N.B.; Carty, H.A.; et al. Genome sequence of Chlamydophila caviae (Chlamydia psittaci GPIC): Examining the role of niche-specific genes in the evolution of the Chlamydiaceae. Nucleic Acids Res. 2003, 31, 2134–2147. [Google Scholar] [CrossRef] [PubMed]
- Carabeo, R. Bacterial subversion of host actin dynamics at the plasma membrane. Cell. Microbiol. 2011, 13, 1460–1469. [Google Scholar] [CrossRef]
- Taylor, L.D.; Nelson, D.E.; Dorward, D.W.; Whitmire, W.M.; Caldwell, H.D. Biological characterization of Chlamydia trachomatis plasticity zone MACPF domain family protein CT153. Infect. Immun. 2010, 78, 2691–2699. [Google Scholar] [CrossRef]
- Rajaram, K.; Giebel, A.M.; Toh, E.; Hu, S.; Newman, J.H.; Morrison, S.G.; Kari, L.; Morrison, R.P.; Nelson, D.E. Mutational analysis of the Chlamydia muridarum plasticity zone. Infect. Immun. 2015, 83, 2870–2881. [Google Scholar] [CrossRef]
- Nunes, A.; Gomes, J.P. Evolution, phylogeny, and molecular epidemiology of Chlamydia. Infect. Genet. Evol. 2014, 23, 49–64. [Google Scholar] [CrossRef]
- Becker, E.; Hegemann, J.H. All subtypes of the Pmp adhesin family are implicated in chlamydial virulence and show species-specific function. Microbiologyopen 2014, 3, 544–556. [Google Scholar] [CrossRef]
- Longbottom, D.; Russell, M.; Dunbar, S.M.; Jones, G.E.; Herring, A.J. Molecular cloning and characterization of the genes coding for the highly immunogenic cluster of 90-kilodalton envelope proteins from the Chlamydia psittaci subtype that causes abortion in sheep. Infect. Immun. 1998, 66, 1317–1324. [Google Scholar] [CrossRef]
- Tan, C.; Spitznagel, J.K.; Shou, H.; Hsia, R.C.; Bavoil, P.M. The polymorphic membrane protein gene family of the Chlamydiaceae. In Chlamydia: Genomics and Pathogenesis; Bavoil, P.M., Wyrick, P.B., Eds.; Horizon Bioscience: Norfolk, UK, 2006; pp. 195–218. [Google Scholar]
- Wolff, B.J.; Morrison, S.S.; Pesti, D.; Ganakammal, S.R.; Srinivasamoorthy, G.; Changayil, S.; Weil, M.R.; MacCannell, D.; Rowe, L.; Frace, M.; et al. Chlamydia psittaci comparative genomics reveals intraspecies variations in the putative outer membrane and type III secretion system genes. Microbiology 2015, 161, 1378–1391. [Google Scholar] [CrossRef] [PubMed]
- Tan, C.; Hsia, R.C.; Shou, H.; Haggerty, C.L.; Ness, R.B.; Gaydos, C.A.; Dean, D.; Scurlock, A.M.; Wilson, D.P.; Bavoil, P.M. Chlamydia trachomatis-infected patients display variable antibody profiles against the nine-member polymorphic membrane protein family. Infect. Immun. 2009, 77, 3218–3226. [Google Scholar] [CrossRef]
- Crane, D.D.; Carlson, J.H.; Fischer, E.R.; Bavoil, P.; Hsia, R.C.; Tan, C.; Kuo, C.C.; Caldwell, H.D. Chlamydia trachomatis polymorphic membrane protein D is a species-common pan-neutralizing antigen. Proc. Natl. Acad. Sci. USA 2006, 103, 1894–1899. [Google Scholar] [CrossRef] [PubMed]
- Kari, L.; Southern, T.R.; Downey, C.J.; Watkins, H.S.; Randall, L.B.; Taylor, L.D.; Sturdevant, G.L.; Whitmire, W.M.; Caldwell, H.D. Chlamydia trachomatis polymorphic membrane protein D is a virulence factor involved in early host-cell interactions. Infect. Immun. 2014, 82, 2756–2762. [Google Scholar] [CrossRef] [PubMed]
- Abdelsamed, H.; Peters, J.; Byrne, G.I. Genetic variation in Chlamydia trachomatis and their hosts: Impact on disease severity and tissue tropism. Future Microbiol. 2013, 8, 1129–1146. [Google Scholar] [CrossRef]
- Stanhope, R.; Flora, E.; Bayne, C.; Derre, I. IncV, a FFAT motif-containing Chlamydia protein, tethers the endoplasmic reticulum to the pathogen-containing vacuole. Proc. Natl. Acad. Sci. USA 2017, 114, 12039–12044. [Google Scholar] [CrossRef]
- Rahman, K.S.; Chowdhury, E.U.; Poudel, A.; Ruettger, A.; Sachse, K.; Kaltenboeck, B. Defining species-specific immunodominant B cell epitopes for molecular serology of Chlamydia species. Clin. Vaccine Immunol. 2015, 22, 539–552. [Google Scholar] [CrossRef]
- Mojica, S.A.; Hovis, K.M.; Frieman, M.B.; Tran, B.; Hsia, R.C.; Ravel, J.; Jenkins-Houk, C.; Wilson, K.L.; Bavoil, P.M. SINC, a type III secreted protein of Chlamydia psittaci, targets the inner nuclear membrane of infected cells and uninfected neighbors. Mol. Biol. Cell. 2015, 26, 1918–1934. [Google Scholar] [CrossRef]
- Tattersall, J.; Rao, G.V.; Runac, J.; Hackstadt, T.; Grieshaber, S.S.; Grieshaber, N.A. Translation inhibition of the developmental cycle protein HctA by the small RNA IhtA is conserved across Chlamydia. PLoS ONE 2012, 7, e47439. [Google Scholar] [CrossRef]
- Grieshaber, N.A.; Sager, J.B.; Dooley, C.A.; Hayes, S.F.; Hackstadt, T. Regulation of the Chlamydia trachomatis histone H1-like protein Hc2 is IspE dependent and IhtA independent. J. Bacteriol. 2006, 188, 5289–5292. [Google Scholar] [CrossRef]
- Gurevich, A.; Saveliev, V.; Vyahhi, N.; Tesler, G. QUAST: Quality assessment tool for genome assemblies. Bioinformatics 2013, 29, 1072–1075. [Google Scholar] [CrossRef] [PubMed]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [PubMed]
- Di Tommaso, P.; Chatzou, M.; Floden, E.W.; Barja, P.P.; Palumbo, E.; Notredame, C. Nextflow enables reproducible computational workflows. Nat. Biotechnol. 2017, 35, 316–319. [Google Scholar] [CrossRef] [PubMed]
- Seemann, T. Prokka: Rapid prokaryotic genome annotation. Bioinformatics 2014, 14, 2068–2069. [Google Scholar] [CrossRef]
- Page, A.J.; Cummins, C.A.; Hunt, M.; Wong, V.K.; Reuter, S.; Holden, M.T.; Fookes, M.; Falush, D.; Keane, J.A.; Parkhill, J. Roary: Rapid large-scale prokaryote pan genome analysis. Bioinformatics 2015, 31, 3691–3693. [Google Scholar] [CrossRef]
- Martinez, F.V.; Feijao, P.; Braga, M.D.; Stoye, J. On the family-free DCJ distance and similarity. Algorithms Mol. Biol. 2015, 10, 13. [Google Scholar] [CrossRef]
- Steinegger, M.; Soding, J. MMseqs2 enables sensitive protein sequence searching for the analysis of massive data sets. Nat. Biotechnol. 2017, 35, 1026–1028. [Google Scholar] [CrossRef]
- Dept. Appl. Informatics, Moscow Aviation Institute, Moscow, Russia. GNU Linear Programming Kit, v.4.65. Available online: http://www.gnu.org/software/glpk (accessed on 27 October 2020).
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Species_Strain | Genome Size [bp] | No. of Contigs a | No. of CDS (Prokka) | Hypothetical Proteins [%] | Coding Density | Max. Protein Length [aa] | Min. Protein Length [aa] | Signal Peptides | G+C [%] | rRNA Operons | tRNA | tmRNA | misc_RNA |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Cab_16DC122 | 1,131,589 | 1 | 992 | 44.15 | 0.89 | 3104 | 35 | 54 | 39.64 | 1 | 39 | 1 | 5 |
| Cab_C18-98 | 1,161,363 | 4 | 1017 | 44.14 | 0.89 | 1807 | 36 | 66 | 39.90 | 1 | 43 | 1 | 6 |
| Cab_S26-3 | 1,144,377 | 1 | 1002 | 43.51 | 0.90 | 1807 | 40 | 67 | 39.87 | 1 | 39 | 1 | 5 |
| Cav_10DC88 | 1,041,170 | 1 | 945 | 42.53 | 0.91 | 1780 | 32 | 43 | 36.92 | 1 | 39 | 1 | 6 |
| Cav_11DC096 | 1,040,930 | 1 | 902 | 41.35 | 0.90 | 1807 | 34 | 45 | 36.89 | 1 | 39 | 1 | 6 |
| Cav_12DC097 | 1,041,858 | 1 | 904 | 41.59 | 0.89 | 1807 | 34 | 45 | 36.89 | 1 | 39 | 1 | 6 |
| Cav_14DC103 | 1,042,060 | 1 | 907 | 41.45 | 0.89 | 1807 | 32 | 45 | 36.91 | 1 | 39 | 1 | 6 |
| Cca_GPIC | 1,173,390 | 1 | 989 | 42.87 | 0.91 | 3347 | 44 | 68 | 39.22 | 1 | 38 | 1 | 6 |
| Cfe_Fe-C56 | 1,166,239 | 1 | 981 | 41.99 | 0.91 | 3299 | 36 | 62 | 39.38 | 1 | 38 | 1 | 6 |
| Cga_08-1274-3 | 1,059,583 | 1 | 904 | 41.15 | 0.91 | 3121 | 35 | 43 | 37.94 | 1 | 39 | 1 | 7 |
| Cga_12-4358 | 1,058,551 | 1 | 905 | 41.21 | 0.90 | 3251 | 35 | 44 | 37.94 | 1 | 39 | 1 | 7 |
| Cga_13DC099 | 1,051,382 | 4 | 902 | 41.10 | 0.89 | 3257 | 46 | 38 | 37.83 | 1 | 39 | 1 | 7 |
| Cga_14DC100 | 1,051,382 | 3 | 902 | 41.35 | 0.89 | 3251 | 35 | 41 | 37.92 | 1 | 39 | 1 | 7 |
| Cga_14DC101 | 1,056,703 | 3 | 903 | 41.41 | 0.89 | 3251 | 35 | 40 | 37.92 | 1 | 39 | 1 | 7 |
| Cga_JX-1 | 1,059,522 | 1 | 918 | 41.17 | 0.91 | 2648 | 35 | 44 | 37.93 | 1 | 39 | 1 | 7 |
| Cib_10-1398-6 | 1,146,066 | 4 | 961 | 42.35 | 0.91 | 3126 | 31 | 60 | 38.32 | 1 | 38 | 1 | 5 |
| Cmu_Nigg | 1,072,950 | 1 | 887 | 39.12 | 0.90 | 3336 | 36 | 49 | 40.34 | 2 | 37 | 1 | 6 |
| Cpe_E58 | 1,106,197 | 1 | 938 | 40.19 | 0.93 | 3439 | 40 | 53 | 41.08 | 1 | 39 | 1 | 5 |
| Cpn_TW-183 | 1,225,935 | 1 | 1050 | 46.76 | 0.90 | 1827 | 32 | 60 | 40.58 | 1 | 38 | 1 | 5 |
| Cps_02DC15 | 1,172,182 | 1 | 991 | 42.78 | 0.91 | 3078 | 43 | 66 | 39.06 | 1 | 39 | 1 | 5 |
| Cps_08DC60 | 1,171,660 | 1 | 998 | 42.68 | 0.90 | 3255 | 43 | 67 | 39.05 | 1 | 39 | 1 | 5 |
| Cps_6BC | 1,172,032 | 1 | 984 | 42.78 | 0.91 | 3358 | 43 | 66 | 39.06 | 1 | 39 | 1 | 5 |
| Cps_CP3 | 1,168,150 | 1 | 1062 | 44.53 | 0.90 | 3131 | 35 | 60 | 39.06 | 1 | 39 | 1 | 5 |
| Cps_GR9 | 1,147,152 | 1 | 994 | 43.36 | 0.90 | 3104 | 39 | 60 | 39.08 | 1 | 39 | 1 | 5 |
| Cps_Mat116 | 1,163,362 | 1 | 1003 | 43.96 | 0.89 | 3165 | 35 | 57 | 39.06 | 1 | 39 | 0 | 5 |
| Cps_MN | 1,168,490 | 1 | 1001 | 43.05 | 0.90 | 3131 | 43 | 63 | 39.06 | 1 | 39 | 1 | 5 |
| Cps_NJ1 | 1,161,434 | 1 | 991 | 43.49 | 0.90 | 3253 | 43 | 60 | 38.96 | 1 | 39 | 1 | 5 |
| Cps_VS225 | 1,157,385 | 1 | 1054 | 44.11 | 0.90 | 2074 | 32 | 59 | 39.02 | 1 | 39 | 1 | 5 |
| Cps_WS-RT-E30 | 1,140,789 | 1 | 998 | 43.58 | 0.90 | 3104 | 39 | 59 | 39.03 | 1 | 39 | 1 | 5 |
| Csu_1-25a | 1,088,751 | 3 | 902 | 39.80 | 0.88 | 3363 | 31 | 47 | 42.07 | 2 | 37 | 1 | 6 |
| Ctr_434-Bu | 1,038,842 | 1 | 891 | 39.39 | 0.90 | 1787 | 43 | 55 | 41.33 | 2 | 37 | 1 | 6 |
| Ctr_A-HAR-13 | 1,044,459 | 1 | 903 | 39.86 | 0.90 | 1787 | 46 | 56 | 41.30 | 2 | 37 | 1 | 5 |
| Ctr_D-UW-3-CX | 1,042,519 | 1 | 892 | 39.68 | 0.90 | 1787 | 46 | 55 | 41.31 | 2 | 37 | 1 | 5 |
| Cab_16DC122 | Cab_S26-3 | Cav_10DC88 | Cga_08-1274-3 | Cca_GPIC | Cfe_Fe-C56 | Cib_10-1398-6 | Cps_6BC | Cpe_E58 | Cpn_TW-183 | Cmu_Nigg | Csu_1-25a | Ctr_D-UW-3-CX | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Cab_16DC122 | 64.71 | 47.94 | 30.15 | 35.61 | 35.78 | 32.00 | 65.12 | 28.32 | 35.41 | 29.39 | 29.56 | 29.68 | |
| Cab_S26-3 | 64.71 | 47.01 | 24.48 | 46.87 | 44.55 | 36.17 | 81.55 | 33.44 | 40.42 | 34.56 | 34.99 | 35.33 | |
| Cav_10DC88 | 47.94 | 47.01 | 0.00 | 46.38 | 45.67 | 43.29 | 48.04 | 42.16 | 44.86 | 43.78 | 44.15 | 43.71 | |
| Cga_08-1274-3 | 30.15 | 24.48 | 0.00 | 31.10 | 45.30 | 52.16 | 29.73 | 27.58 | 0.00 | 28.40 | 28.07 | 28.58 | |
| Cca_GPIC | 35.61 | 46.87 | 46.38 | 31.10 | 58.50 | 35.12 | 35.57 | 30.01 | 39.36 | 28.12 | 28.46 | 28.45 | |
| Cfe_Fe-C56 | 35.78 | 44.55 | 45.67 | 45.30 | 58.50 | 43.39 | 36.64 | 34.13 | 39.95 | 31.80 | 31.97 | 32.11 | |
| Cib_10-1398-6 | 32.00 | 36.17 | 43.29 | 52.16 | 35.12 | 43.39 | 32.88 | 29.61 | 36.88 | 31.33 | 31.22 | 31.64 | |
| Cps_6BC | 65.12 | 81.55 | 48.04 | 29.73 | 35.57 | 36.64 | 32.88 | 30.07 | 40.63 | 31.15 | 31.30 | 31.51 | |
| Cpe_E58 | 28.32 | 33.44 | 42.16 | 27.58 | 30.01 | 34.13 | 29.61 | 30.07 | 39.82 | 30.16 | 30.33 | 30.23 | |
| Cpn_TW-183 | 35.41 | 40.42 | 44.86 | 0.00 | 39.36 | 39.95 | 36.88 | 40.63 | 39.82 | 39.15 | 38.83 | 38.55 | |
| Cmu_Nigg | 29.39 | 34.56 | 43.78 | 28.40 | 28.12 | 31.80 | 31.33 | 31.15 | 30.16 | 39.15 | 66.24 | 62.96 | |
| Csu_1-25a | 29.56 | 34.99 | 44.15 | 28.07 | 28.46 | 31.97 | 31.22 | 31.30 | 30.33 | 38.83 | 66.24 | 62.41 | |
| Ctr_D-UW-3-CX | 29.68 | 35.33 | 43.71 | 28.58 | 28.45 | 32.11 | 31.64 | 31.51 | 30.23 | 38.55 | 62.96 | 62.41 |
| Species_Strain | PZ Total Size [nt] | # CDS in PZ | Biotin Modi-Fication | toxB [nt] | MAC/ Perforin a [nt] | Trp Operon | Purine Synthesis and Recycling |
|---|---|---|---|---|---|---|---|
| Cab_16DC122 | 22,240 | 13 | accB, accC | 9312 | - | - | - |
| Cab_S26-3 | 11,776 | 14 | accB, accC | - | (681) | - | guaB_1/2b |
| Cav_10DC88 | 5694 | 6 | accB, accC | - | - | - | - |
| Cca_GPIC | 34,753 | 21 | accB, accC | 10,041 | (504) b | trpA, trpB_1/2, trpD, trpF, trpR, kynU | guaA, guaB, ADA |
| Cfe_Fe-C56 | 39,924 | 28 | accB, accC | 9897 | 2442; (501) | trpA, trpB, trpD, trpF, trpR, kynU | guaA, guaB, ADA |
| Cga_08-1274-3 | 15,845 | 8 | accB, accC | 9363 | - | - | - |
| Cib_10-1398-6 | 31,344 | 20 | accB, accC | 9378 | 2505; 2484 | - | - |
| Cmu_Nigg | 82,115 | 45 | accB, accC | 9657; 10,008; 9768 | 2430 | - | guaA, guaB, ADA |
| Cpe_E58 | 42,163 | 18 | accB, accC | 10,134; 10,317 | 2418 | -c | guaA, guaB, ADA |
| Cpn_TW-183 | 8759 | 11 | accB, accC | - | 1236 | - | guaB_1/2b |
| Cps_6BC | 29,145 | 16 | accB, accC | 10,074 | 2469; (627) | - | guaA, guaB, ADA |
| Csu_1-25a | 82,505 | 52 | accB, accC | 10,089; 9675 | 2433 | trpA, trpB, trpR | - |
| Ctr_D-UW-3-CX | 55,445 | 49 | accB, accC | - | 2433 | trpA, trpB, trpR | - |
| Strain | # pmp | Individual Pmps in Subtypes | |||||
|---|---|---|---|---|---|---|---|
| A | B | D | E | G | H | ||
| Cab_S26-3 | 18 | 2 | 1 | 18 | 3,4,5 | 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17 | 6 |
| Cav_10DC88 | 7 | 19 | B | 21 | 15 | G, 13 or G-I | 14 |
| Cca_GPIC | 18 | A | B | D | E/F [5] | G [9] | H |
| Cfe_Fe-C56 | 20 | 19 | 20 | 1 | 14, 15, 16, 17, 18 | 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12 | 13 |
| Cga_08-1274-3 | 10 | A | B | D | E, F | G/I [4] | H |
| Cib_10-1398-6 | 22 | 2 | 1 | 22 | 3, 4, 5 | 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21 | 6, 7 |
| Cpe_E58 | 15 | A | B | D | E [2] | G [9] | H |
| Cpn_TW-183 | 21 | 19 | 20 | 21 | 15, 16, 17, 18 | 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13 | 14 |
| Cps_6BC | 21 | 2 | 1 | 22 | 3, 4, 5 | 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 19, 20, 21 | 6 |
| Cmu_Nigg | 9 | A | B, C | D | E, F | G, I | H |
| Csu_1-25a | 9 | A | B, C | D | E, F | G, I | H |
| Ctr_D-UW-3-CX | 9 | A | B, C | D | E, F | G, I | H |
| Strain | PZ Size [nt] | ToxB [aa] | Trp Operon * | # Pmps | Inc Family Subtypes | SINC [aa] | HctA/HctB [aa] | Main Host |
|---|---|---|---|---|---|---|---|---|
| Cab_S26-3 | 11,776 | 18 | A, B, C, V, X, Y | 361 | 123/154 | ruminant | ||
| Cps_6BC | 29,145 | 3358 | 21 | A, B, C, V, X, Y | 502 | 116-123/197 | avian | |
| Cav_10DC88 | 5694 | 7 | A, B, C, V | 234 | -/86 ** | avian | ||
| Cga_08-1274-3 | 15,845 | 3121 | 10 | A, B, C, V | 237 | -/187 | avian | |
| Cib_10-1398-6 | 31,344 | 3126 | 22 | A, B, C, V | 242 | 118/199 | avian | |
| Cca_GPIC | 34,753 | 3347 | 7 | 18 | A, B, C, V | 238 | 125/152 | rodent |
| Cfe_Fe-C56 | 39,924 | 3299 | 6 | 20 | A, B, C, V | 240 | 126/168 | feline |
| Cpe_E58 | 42,163 | 3378; 3439 | *** | 21 | A, B, C | 118/190 | ruminant | |
| Cpn_TW-183 | 8759 | 21 | B, C | 123/172 | human | |||
| Cmu_Nigg | 82,115 | 3219; 3336; 3256 | 15 | A, B, X | 125/207 | rodent | ||
| Csu_1-25a | 82,505 | 3363; 3225 | 3 | 9 | A, B, C, D, E, F, G | 126/203 | porcine | |
| Ctr_D-UW-3-CX | 55,445 | 3 | 9 | A, B, C, D, E, F, G, V | 125/201 | human |
| Species | Strain 1 | Source | NCBI acc. no. | ENA | de novo seq. (Source) |
|---|---|---|---|---|---|
| Chlamydia abortus | Cab_16DC122 | Muscovy duck | FLI 2 | ||
| Cab_C18-98 (B577T) | Sheep | SAMEA1094359 | |||
| Cab_S26-3 | Sheep | NC_004552.2 | |||
| Chlamydia avium | Cav_10DC88T | Pigeon | NZ_CP006571.1 | GCA_000583875.1 ASM58387v1 | |
| Cav_11DC096 | Pigeon | FLI 2 | |||
| Cav_12DC097 | Pigeon | FLI 2 | |||
| Cav_14DC103 | Pigeon | FLI 2 | |||
| Chlamydia caviae | Cca_GPICT | Guinea pig | NC_003361.3 | ||
| Chlamydia felis | Cfe_Fe-C56T | Cat | NC_007899.1 | ||
| Chlamydia gallinacea | Cga_08-1274-3T | Chicken | NZ_CP015840.1 | ||
| Cga_12-4358 | Chicken | ANSES 3 | |||
| 4 contigs | Cga_13DC099 | Turkey | FLI 2 | ||
| 3 contigs | Cga_14DC100 | Chicken | FLI 2 | ||
| 3 contigs | Cga_14DC101 | Chicken | FLI 2 | ||
| Cga_JX-1 | Chicken | NZ_CP019792.1. CP019792.1 | GCA_002007725.1. ASM200772v1 | ||
| Cand. Chlamydia ibidis | Cib_10-1398-6 | Ibis | NZ_APJW01000001.1 | ||
| Chlamydia muridarum | Cmu_NiggT | Mouse | NC_002620.2 | ||
| Chlamydia pecorum | Cpe_E58T | Cattle | NC_015408.1 | ||
| Chlamydia pneumoniae | Cpn_TW-183T | Human | NC_005043.1 | ||
| Chlamydia psittaci | Cps_02DC15 | Cattle | NC_017292.1 | GCA_000415545.1 | |
| Cps_08DC60 | Human | NC_017290.1 | GCA_000270445.1 | ||
| Cps_6BCT | Parakeet | NC_015470.1 | GCA_000191925.1 | ||
| Cps_CP3 | Pigeon | NC_018625.1 | GCA_000298535.2 | ||
| Cps_GR9 | Duck | NC_018620.1 | GCA_000298415.1 | ||
| Cps_Mat116 | Psittacine | CP002744.1 | |||
| Cps_MN | Human | NC_018627.1 | GCA_000298435.2 | ||
| Cps_NJ1 | Turkey | CP003798.1 | |||
| Cps_VS225 | Psittacine | CP003793.1 | |||
| Cps_WS-RT-E30 | Duck | NC_018622.1 | GCA_000298475.2 | ||
| Chlamydia suis | Csu_1-25a | Swine | FTQL01000001 | ||
| Chlamydia trachomatis | Ctr_434-Bu | Human | NC_010287.1 | ||
| Ctr_A-HAR-13T | Human | NC_007429.1 | |||
| Ctr_D-UW-3-CX | Human | NC_000117.1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hölzer, M.; Barf, L.-M.; Lamkiewicz, K.; Vorimore, F.; Lataretu, M.; Favaroni, A.; Schnee, C.; Laroucau, K.; Marz, M.; Sachse, K. Comparative Genome Analysis of 33 Chlamydia Strains Reveals Characteristic Features of Chlamydia Psittaci and Closely Related Species. Pathogens 2020, 9, 899. https://doi.org/10.3390/pathogens9110899
Hölzer M, Barf L-M, Lamkiewicz K, Vorimore F, Lataretu M, Favaroni A, Schnee C, Laroucau K, Marz M, Sachse K. Comparative Genome Analysis of 33 Chlamydia Strains Reveals Characteristic Features of Chlamydia Psittaci and Closely Related Species. Pathogens. 2020; 9(11):899. https://doi.org/10.3390/pathogens9110899
Chicago/Turabian StyleHölzer, Martin, Lisa-Marie Barf, Kevin Lamkiewicz, Fabien Vorimore, Marie Lataretu, Alison Favaroni, Christiane Schnee, Karine Laroucau, Manja Marz, and Konrad Sachse. 2020. "Comparative Genome Analysis of 33 Chlamydia Strains Reveals Characteristic Features of Chlamydia Psittaci and Closely Related Species" Pathogens 9, no. 11: 899. https://doi.org/10.3390/pathogens9110899
APA StyleHölzer, M., Barf, L.-M., Lamkiewicz, K., Vorimore, F., Lataretu, M., Favaroni, A., Schnee, C., Laroucau, K., Marz, M., & Sachse, K. (2020). Comparative Genome Analysis of 33 Chlamydia Strains Reveals Characteristic Features of Chlamydia Psittaci and Closely Related Species. Pathogens, 9(11), 899. https://doi.org/10.3390/pathogens9110899