Emerging Roles of Heparanase in Viral Pathogenesis



Abstract
1. Introduction
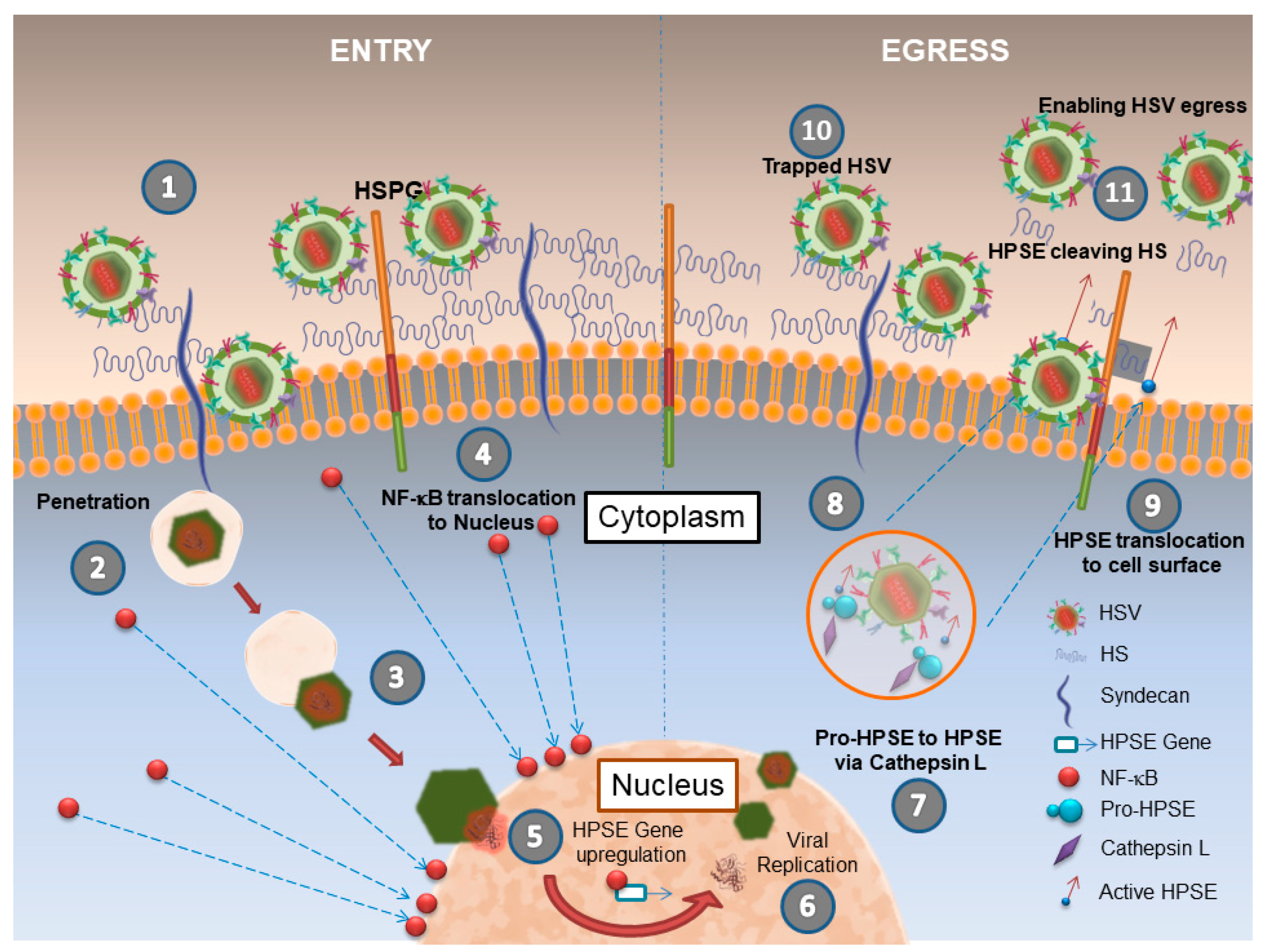
2. Herpes Simplex Virus (HSV-1) and HPSE
3. Dengue Virus (DENV) and HPSE
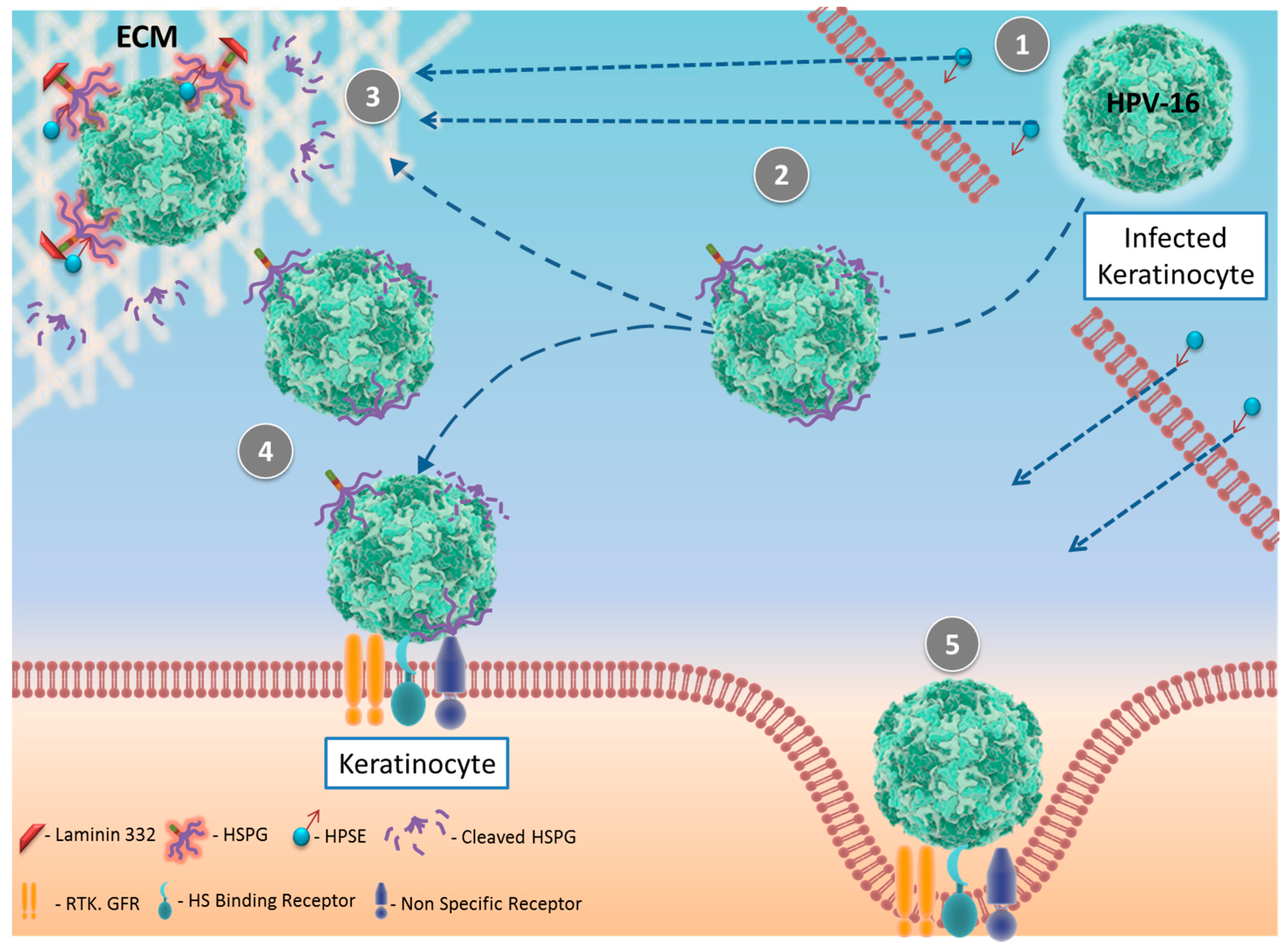
4. Human Papilloma Virus (HPV) and HPSE
5. Respiratory Syncytial Virus (RSV) and HPSE
6. Adenovirus (ADNV) and HPSE
7. Hepatitis C Virus (HCV) and HPSE
8. Porcine Respiratory and Reproductive Syncytial Virus (PRRSV) and HPSE
9. Future Therapeutic Potential of Targeting HPSE in Viral Diseases
10. Future Prospects and Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Sarrazin, S.; Lamanna, W.C.; Esko, J.D. Heparan sulfate proteoglycans. Cold Spring Harb. Perspect. Biol. 2011, 3, 1–33. [Google Scholar] [CrossRef] [PubMed]
- Rivara, S.; Milazzo, F.M.; Giannini, G. Heparanase: A rainbow pharmacological target associated to multiple pathologies including rare diseases. Future Med. Chem. 2016, 8, 647–680. [Google Scholar] [CrossRef] [PubMed]
- Agelidis, A.M.; Shukla, D. Cell entry mechanisms of HSV: What we have learned in recent years. Future Virol. 2015, 10, 1145–1154. [Google Scholar] [CrossRef] [PubMed]
- Kreuger, J.; Kjellén, L. Heparan Sulfate Biosynthesis: Regulation and Variability. J. Histochem. Cytochem. 2012, 60, 898–907. [Google Scholar] [CrossRef] [PubMed]
- Shukla, D.; Spear, P.G. Herpesviruses and heparan sulfate: An intimate relationship in aid of viral entry. J. Clin. Investig. 2001, 108, 503–510. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.; Li, J.; Lian, G. How does cellular heparan sulfate function in viral pathogenicity? Biomed. Environ. Sci. 2011, 24, 81–87. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Thorp, S.C. Cell surface heparan sulfate and its roles in assisting viral infections. Med. Res. Rev. 2002, 22, 1–25. [Google Scholar] [CrossRef] [PubMed]
- Bose, S.; Banerjee, A.K. Role of heparan sulfate in human parainfluenza virus type 3 infection. Virology 2002, 298, 73–83. [Google Scholar] [CrossRef] [PubMed]
- Connell, B.J.; Lortat-Jacob, H. Human immunodeficiency virus and heparan sulfate: From attachment to entry inhibition. Front. Immunol. 2013, 4, 385. [Google Scholar] [CrossRef] [PubMed]
- Plochmann, K.; Horn, A.; Gschmack, E.; Armbruster, N.; Krieg, J.; Wiktorowicz, T.; Weber, C.; Stirnnagel, K.; Lindemann, D.; Rethwilm, A.; et al. Heparan sulfate is an attachment factor for foamy virus entry. J. Virol. 2012, 86, 10028–10035. [Google Scholar] [CrossRef] [PubMed]
- De Boer, S.M.; Kortekaas, J.; de Haan, C.A.; Rottier, P.J.; Moormann, R.J.; Bosch, B.J. Heparan sulfate facilitates Rift Valley fever virus entry into the cell. J. Virol. 2012, 86, 13767–13771. [Google Scholar] [CrossRef] [PubMed]
- Byrnes, A.P.; Griffin, D.E. Binding of Sindbis virus to cell surface heparan sulfate. J. Virol. 1998, 72, 7349–7356. [Google Scholar] [PubMed]
- Jin, H.; Zhou, S. The functions of heparanase in human diseases. Mini Rev. Med. Chem. 2017, 17, 541–548. [Google Scholar] [CrossRef] [PubMed]
- Vlodavsky, I.; Friedmann, Y.; Elkin, M.; Aingorn, H.; Atzmon, R.; Ishai-Michaeli, R.; Bitan, M.; Pappo, O.; Peretz, T.; Michal, I.; et al. Mammalian heparanase: Gene cloning, expression and function in tumor progression and metastasis. Nat. Med. 1999, 5, 793–802. [Google Scholar] [CrossRef] [PubMed]
- Hulett, M.D.; Freeman, C.; Hamdorf, B.J.; Baker, R.T.; Harris, M.J.; Parish, C.R. Cloning of mammalian heparanase, an important enzyme in tumor invasion and metastasis. Nat. Med. 1999, 5, 803–809. [Google Scholar] [CrossRef] [PubMed]
- Kussie, P.H.; Hulmes, J.D.; Ludwig, D.L.; Patel, S.; Navarro, E.C.; Seddon, A.P.; Giorgio, N.A.; Bohlen, P. Cloning and functional expression of a human heparanase gene. Biochem. Biophys. Res. Commun. 1999, 261, 183–187. [Google Scholar] [CrossRef] [PubMed]
- Fairbanks, M.B.; Mildner, A.M.; Leone, J.W.; Cavey, G.S.; Mathews, W.R.; Drong, R.F.; Slightom, J.L.; Bienkowski, M.J.; Smith, C.W.; Bannow, C.A.; et al. Processing of the human heparanase precursor and evidence that the active enzyme is a heterodimer. J. Biol. Chem. 1999, 274, 29587–29590. [Google Scholar] [CrossRef] [PubMed]
- Toyoshima, M.; Nakajima, M. Human heparanase. Purification, characterization, cloning, and expression. J. Biol. Chem. 1999, 274, 24153–24160. [Google Scholar] [CrossRef] [PubMed]
- Jiang, P.; Kumar, A.; Parrillo, J.E.; Dempsey, L.A.; Platt, J.L.; Prinz, R.A.; Xu, X. Cloning and characterization of the human heparanase-1 (HPR1) gene promoter. Role of GA-binding protein and Sp1 in regulating HPR1 basal promoter activity. J. Biol. Chem. 2002, 277, 8989–8998. [Google Scholar] [CrossRef] [PubMed]
- Agelidis, A.M.; Hadigal, S.R.; Jaishankar, D.; Shukla, D. Viral Activation of Heparanase Drives Pathogenesis of Herpes Simplex Virus-1. Cell Rep. 2017, 20, 439–450. [Google Scholar] [CrossRef] [PubMed]
- Hadigal, S.R.; Agelidis, A.M.; Karasneh, G.A.; Antoine, T.E.; Yakoub, A.M.; Ramani, V.C.; Djalilian, A.R.; Sanderson, R.D.; Shukla, D. Heparanase is a host enzyme required for herpes simplex virus-1 release from cells. Nat. Commun. 2015, 6, 6985. [Google Scholar] [CrossRef] [PubMed]
- Abboud-Jarrous, G.; Atzmon, R.; Peretz, T.; Palermo, C.; Gadea, B.B.; Joyce, J.A.; Vlodavsky, I. Cathepsin L. is responsible for processing and activation of proheparanase through multiple cleavages of a linker segment. J. Biol. Chem. 2008, 283, 18167–18176. [Google Scholar] [CrossRef] [PubMed]
- Mao, Y.; Huang, Y.; Buczek-Thomas, J.A.; Ethen, C.M.; Nugent, M.A.; Wu, Z.L.; Zaia, J. A liquid chromatography-mass spectrometry-based approach to characterize the substrate specificity of mammalian heparanase. J. Biol. Chem. 2014, 289, 34141–34151. [Google Scholar] [CrossRef] [PubMed]
- McKenzie, E.; Tyson, K.; Stamps, A.; Smith, P.; Turner, P.; Barry, R.; Hircock, M.; Patel, S.; Barry, E.; Stubberfield, C.; et al. Cloning and expression profiling of Hpa2, a novel mammalian heparanase family member. Biochem. Biophys. Res. Commun. 2000, 276, 1170–1177. [Google Scholar] [CrossRef] [PubMed]
- Sanderson, R.D.; Elkin, M.; Rapraeger, A.C.; Ilan, N.; Vlodavsky, I. Heparanase regulation of cancer, autophagy and inflammation: New mechanisms and targets for therapy. FEBS J. 2017, 284, 42–55. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Viola, C.M.; Brzozowski, A.M.; Davies, G.J. Structural characterization of human heparanase reveals insights into substrate recognition. Nat. Struct. Mol. Biol. 2015, 22, 1016–1022. [Google Scholar] [CrossRef] [PubMed]
- Gandhi, N.S.; Freeman, C.; Parish, C.R.; Mancera, R.L. Computational analyses of the catalytic and heparin-binding sites and their interactions with glycosaminoglycans in glycoside hydrolase family 79 endo-β-d-glucuronidase (heparanase). Glycobiology 2012, 22, 35–55. [Google Scholar] [CrossRef] [PubMed]
- Meirovitz, A.; Goldberg, R.; Binder, A.; Rubinstein, A.M.; Hermano, E.; Elkin, M. Heparanase in inflammation and inflammation-associated cancer. FEBS J. 2013, 280, 2307–2319. [Google Scholar] [CrossRef] [PubMed]
- Xu, F.; Schillinger, J.A.; Sternberg, M.R.; Johnson, R.E.; Lee, F.K.; Nahmias, A.J.; Markowitz, L.E. Seroprevalence and coinfection with herpes simplex virus type 1 and type 2 in the United States, 1988–1994. J. Infect. Dis. 2002, 185, 1019–1024. [Google Scholar] [CrossRef] [PubMed]
- Thakkar, N.; Jaishankar, D.; Agelidis, A.; Yadavalli, T.; Mangano, K.; Patel, S.; Tekin, S.Z.; Shukla, D. Cultured corneas show dendritic spread and restrict herpes simplex virus infection that is not observed with cultured corneal cells. Sci. Rep. 2017, 7. [Google Scholar] [CrossRef] [PubMed]
- Knipe, D.M.; Howley, P.M. Fields of Virology; Wolters Kluwer Health/Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2007. [Google Scholar]
- Akhtar, J.; Shukla, D. Viral entry mechanisms: Cellular and viral mediators of herpes simplex virus entry. FEBS J. 2009, 276, 7228–7236. [Google Scholar] [CrossRef] [PubMed]
- Bhatt, S.; Gething, P.W.; Brady, O.J.; Messina, J.P.; Farlow, A.W.; Moyes, C.L.; Drake, J.M.; Brownstein, J.S.; Hoen, A.G.; Sankoh, O.; et al. The global distribution and burden of dengue. Nature 2013, 496, 504–507. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, E.R.A.; Mohana-Borges, R.; de Alencastro, R.B.; Horta, B.A.C. The flavivirus capsid protein: Structure, function and perspectives towards drug design. Virus Res. 2017, 227, 115–123. [Google Scholar] [CrossRef] [PubMed]
- Perera, R.; Kuhn, R.J. Structural proteomics of dengue virus. Curr. Opin. Microbiol. 2008, 11, 369–377. [Google Scholar] [CrossRef] [PubMed]
- Cruz-Oliveira, C.; Freire, J.M.; Conceição, T.M.; Higa, L.M.; Castanho, M.A.R.B.; Da Poian, A.T. Receptors and routes of dengue virus entry into the host cells. FEMS Microbiol. Rev. 2015, 39, 155–170. [Google Scholar] [CrossRef] [PubMed]
- Puerta-Guardo, H.; Glasner, D.R.; Harris, E. Dengue Virus NS1 Disrupts the Endothelial Glycocalyx, Leading to Hyperpermeability. PLoS Pathog. 2016, 12. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, H.P.; Ramírez-Fort, M.K.; Rady, P.L. The biology of human papillomaviruses. Curr. Probl. Dermatol. 2014, 45, 19–32. [Google Scholar] [CrossRef] [PubMed]
- Surviladze, Z.; Sterkand, R.T.; Ozbun, M.A. Interaction of human papillomavirus type 16 particles with heparan sulfate and syndecan-1 molecules in the keratinocyte extracellular matrix plays an active role in infection. J. Gen. Virol. 2015, 96, 2232–2241. [Google Scholar] [CrossRef] [PubMed]
- Hirshoren, N.; Bulvik, R.; Neuman, T.; Rubinstein, A.M.; Meirovitz, A.; Elkin, M. Induction of heparanase by HPV E6 oncogene in head and neck squamous cell carcinoma. J. Cell. Mol. Med. 2014, 18, 181–186. [Google Scholar] [CrossRef] [PubMed]
- Förster, A.; Maertens, G.N.; Farrell, P.J.; Bajorek, M. Dimerization of matrix protein is required for budding of respiratory syncytial virus. J. Virol. 2015, 89, 4624–4635. [Google Scholar] [CrossRef] [PubMed]
- Kiss, G.; Holl, J.M.; Williams, G.M.; Alonas, E.; Vanover, D.; Lifland, A.W.; Gudheti, M.; Guerrero-Ferreira, R.C.; Nair, V.; Yi, H.; et al. Structural analysis of respiratory syncytial virus reveals the position of M2-1 between the matrix protein and the ribonucleoprotein complex. J. Virol. 2014, 88, 7602–7617. [Google Scholar] [CrossRef] [PubMed]
- Tao, Y.; Wang, Z.; Zhou, Y. Expression of heparanase in kidney of rats with respiratory syncytial virus nephropathy and its relationship with proteinurina. J. Sichuan Univ. Med. Sci. Ed. 2014, 45, 212–215. [Google Scholar]
- Dong, L.; Wang, X.; Guo, Y.; Wu, J.; Li, S.; Yu, P.; Wang, Z. HS N-sulfation and iduronic acids play an important role in the infection of Respiratory Syncytial Virus in vitro. Eur. Rev. Med. Pharmacol. Sci. 2013, 17, 1864–1868. [Google Scholar] [PubMed]
- Mangel, W.F.; San Martin, C. Structure, function and dynamics in adenovirus maturation. Viruses 2014, 6, 4536–4570. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Rhee, E.G.; Masek-Hammerman, K.; Teigler, J.E.; Abbink, P.; Barouch, D.H. Adenovirus serotype 26 utilizes CD46 as a primary cellular receptor and only transiently activates T lymphocytes following vaccination of rhesus monkeys. J. Virol. 2012, 86, 10862–10865. [Google Scholar] [CrossRef] [PubMed]
- Goncalves, M.A.; de Vries, A.A. Adenovirus: From foe to friend. Rev. Med. Virol. 2006, 16, 167–186. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, M.; Curiel, D.T. Current issues and future directions of oncolytic adenoviruses. Mol. Ther. 2010, 18, 243–250. [Google Scholar] [CrossRef] [PubMed]
- Lasaro, M.O.; Ertl, H.C. New insights on adenovirus as vaccine vectors. Mol. Ther. 2009, 17, 1333–1339. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, Y.; Kojima, T.; Kagawa, S.; Uno, F.; Hashimoto, Y.; Kyo, S.; Mizuguchi, H.; Tanaka, N.; Kawamura, H.; Ichimaru, D.; et al. A novel translational approach for human malignant pleural mesothelioma: Heparanase-assisted dual virotherapy. Oncogene 2010, 29, 1145–1154. [Google Scholar] [CrossRef] [PubMed]
- Lindenbach, B.D.; Rice, C.M. The ins and outs of hepatitis C virus entry and assembly. Nat. Rev. Microbiol. 2013, 11, 688–700. [Google Scholar] [CrossRef] [PubMed]
- Appel, N.; Zayas, M.; Miller, S.; Krijnse-Locker, J.; Schaller, T.; Friebe, P.; Kallis, S.; Engel, U.; Bartenschlager, R. Essential role of domain III of nonstructural protein 5A for hepatitis C virus infectious particle assembly. PLoS Pathog. 2008, 4, e1000035. [Google Scholar] [CrossRef] [PubMed]
- Dong, S.; Wu, X. Heparanase and hepatocellular carcinoma: Promoter or inhibitor? World J. Gastroenterol. 2010, 16, 306–311. [Google Scholar] [CrossRef] [PubMed]
- Barth, H.; Schnober, E.K.; Zhang, F.; Linhardt, R.J.; Depla, E.; Boson, B.; Cosset, F.-L.; Patel, A.H.; Blum, H.E.; Baumert, T.F. Viral and cellular determinants of the hepatitis C virus envelope-heparan sulfate interaction. J. Virol. 2006, 80, 10579–10590. [Google Scholar] [CrossRef] [PubMed]
- El-Assal, O.N.; Yamanoi, A.; Ono, T.; Kohno, H.; Nagasue, N. The clinicopathological significance of heparanase and basic fibroblast growth factor expressions in hepatocellular carcinoma. Clin. Cancer Res. 2001, 7, 1299–1305. [Google Scholar] [PubMed]
- Kappes, M.A.; Faaberg, K.S. PRRSV structure, replication and recombination: Origin of phenotype and genotype diversity. Virology 2015, 479–480, 475–486. [Google Scholar] [CrossRef] [PubMed]
- Lunney, J.K.; Fang, Y.; Ladinig, A.; Chen, N.; Li, Y.; Rowland, B.; Renukaradhya, G.J. Porcine Reproductive and Respiratory Syndrome Virus (PRRSV): Pathogenesis and Interaction with the Immune System. Annu. Rev. Anim. Biosci. 2016, 4, 129–154. [Google Scholar] [CrossRef] [PubMed]
- Guo, C.; Zhu, Z.; Guo, Y.; Wang, X.; Yu, P.; Xiao, S.; Chen, Y.; Cao, Y.; Liu, X. Heparanase upregulation contributes to porcine reproductive and respiratory syndrome virus release. J. Virol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Guo, C.; Zhu, Z.; Wang, X.; Chen, Y.; Liu, X. Pyrithione inhibits porcine reproductive and respiratory syndrome virus replication through interfering with NF-κB and heparanase. Vet. Microbiol. 2017, 201, 231–239. [Google Scholar] [CrossRef] [PubMed]
- Cassinelli, G.; Zaffaroni, N.; Lanzi, C. The heparanase/heparan sulfate proteoglycan axis: A potential new therapeutic target in sarcomas. Cancer Lett. 2016, 382, 245–254. [Google Scholar] [CrossRef] [PubMed]
- Clercq, E.D. Antiviral agents active against influenza A viruses. Nat. Rev. Drug Discov. 2006, 5, 1015–1025. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Virus | Role of Viral Protein in Pathogenesis (Direct/Indirect) | Role of HPSE in Pathogenesis | References |
|---|---|---|---|
| HSV-1 | Direct | Increased virus release, ECM damage leading to disease pathologies | [20,21] |
| DENV | Indirect | Increased in severity of disease symptoms | [37] |
| HPV | Direct | Increased virus release | [39] |
| RSV | Direct | Increased rate of infection | [43,44] |
| ADNV | Indirect | Increased rate of infection, virotherapy of cancers | [47–49] |
| HCV | Not determined | Not determined | [54] |
| PRRSV | Direct | Increased virus release | [58,59] |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Thakkar, N.; Yadavalli, T.; Jaishankar, D.; Shukla, D. Emerging Roles of Heparanase in Viral Pathogenesis. Pathogens 2017, 6, 43. https://doi.org/10.3390/pathogens6030043
Thakkar N, Yadavalli T, Jaishankar D, Shukla D. Emerging Roles of Heparanase in Viral Pathogenesis. Pathogens. 2017; 6(3):43. https://doi.org/10.3390/pathogens6030043
Chicago/Turabian StyleThakkar, Neel, Tejabhiram Yadavalli, Dinesh Jaishankar, and Deepak Shukla. 2017. "Emerging Roles of Heparanase in Viral Pathogenesis" Pathogens 6, no. 3: 43. https://doi.org/10.3390/pathogens6030043
APA StyleThakkar, N., Yadavalli, T., Jaishankar, D., & Shukla, D. (2017). Emerging Roles of Heparanase in Viral Pathogenesis. Pathogens, 6(3), 43. https://doi.org/10.3390/pathogens6030043