
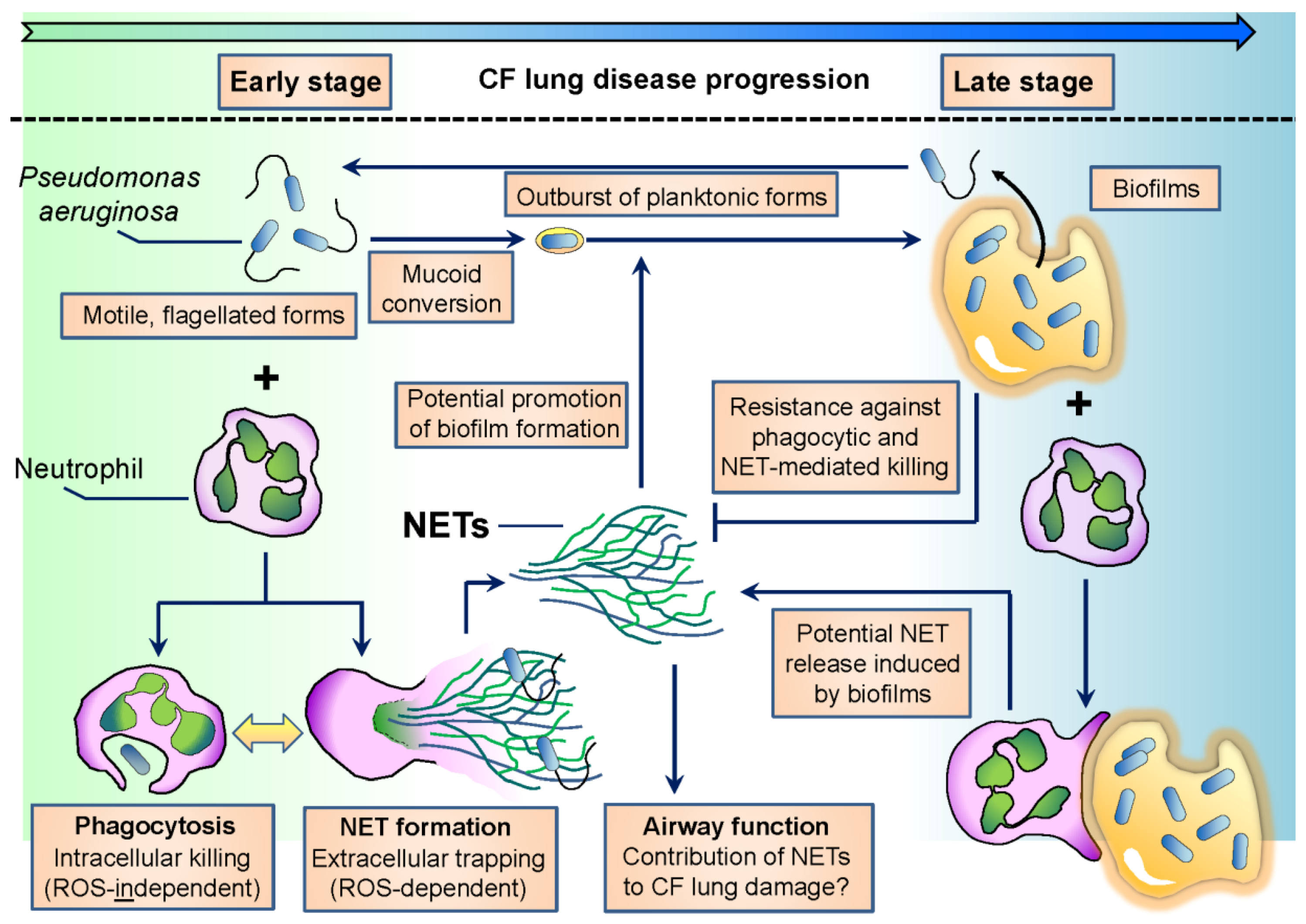
Interactions between Neutrophils and Pseudomonas aeruginosa in Cystic Fibrosis

{kind=link}
Abstract
:1. Antimicrobial Mechanisms of Neutrophils against Pseudomonas aeruginosa
1.1. P. aeruginosa, an Opportunistic Pathogen
1.2. Cystic Fibrosis
1.3. Neutrophils Are Key to Eliminate P. aeruginosa
1.4. PMN Recruitment to the Airways
1.5. Intracellular Killing Following Phagocytosis
1.6. Neutrophil Extracellular Traps
1.7. PMN Microvesicles
1.8. Oxidative and Non-Oxidative PMN Mechanisms against P. aeruginosa
2. PMN Dysfunction in CF
2.1. PMN Components Correlate with CF Lung Disease Severity
2.2. CFTR Deficiency in PMNs
2.3. CF Airway PMNs
2.4. NETs in CF
2.5. Anti-Inflammatory Strategies in CF
3. Adaptation of P. aeruginosa to Neutrophil-Mediated Attacks in CF
3.1. Loss of Flagellar Motility
3.2. Characteristic Image of Chronic CF Airways: Suspension Biofilms of P. aeruginosa Surrounded by PMNs
3.3. P. aeruginosa Resists NET-Mediated Attacks
3.4. Neutrophil Components Promote Biofilm Formation of P. aeruginosa
4. Conclusions
Conflicts of Interest
References
- Vasil, M.L. Pseudomonas aeruginosa: Biology, mechanisms of virulence, epidemiology. J. Pediatr. 1986, 108, 800–805. [Google Scholar] [CrossRef]
- Pollack, M. The virulence of Pseudomonas aeruginosa. Rev. Infect. Dis. 1984, 6 (Suppl. 3), S617–S626. [Google Scholar] [CrossRef] [PubMed]
- Silby, M.W.; Winstanley, C.; Godfrey, S.A.; Levy, S.B.; Jackson, R.W. Pseudomonas genomes: Diverse and adaptable. FEMS Microbiol. Rev. 2011, 35, 652–680. [Google Scholar] [CrossRef] [PubMed]
- Rahme, L.G.; Stevens, E.J.; Wolfort, S.F.; Shao, J.; Tompkins, R.G.; Ausubel, F.M. Common virulence factors for bacterial pathogenicity in plants and animals. Science 1995, 268, 1899–1902. [Google Scholar] [CrossRef] [PubMed]
- Mulcahy, L.R.; Isabella, V.M.; Lewis, K. Pseudomonas aeruginosa biofilms in disease. Microb. Ecol. 2014, 68, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Huber, P.; Basso, P.; Reboud, E.; Attree, I. Pseudomonas aeruginosa renews its virulence factors. Environ. Microbiol. Rep. 2016, 8, 564–571. [Google Scholar] [CrossRef] [PubMed]
- Lyczak, J.B.; Cannon, C.L.; Pier, G.B. Establishment of Pseudomonas aeruginosa infection: Lessons from a versatile opportunist. Microbes Infect. 2000, 2, 1051–1060. [Google Scholar] [CrossRef]
- Rybtke, M.; Hultqvist, L.D.; Givskov, M.; Tolker-Nielsen, T. Pseudomonas aeruginosa biofilm infections: Community structure, antimicrobial tolerance and immune response. J. Mol. Biol. 2015, 427, 3628–3645. [Google Scholar] [CrossRef] [PubMed]
- Rasamiravaka, T.; Labtani, Q.; Duez, P.; El Jaziri, M. The formation of biofilms by Pseudomonas aeruginosa: A review of the natural and synthetic compounds interfering with control mechanisms. BioMed Res. Int. 2015, 2015, 759348. [Google Scholar] [CrossRef] [PubMed]
- Tolker-Nielsen, T. Pseudomonas aeruginosa biofilm infections: From molecular biofilm biology to new treatment possibilities. APMIS Suppl. 2014, 122, 1–51. [Google Scholar] [CrossRef] [PubMed]
- Spencer, R.C. Predominant pathogens found in the European prevalence of infection in intensive care study. Eur. J. Clin. Microbiol. Infect. Dis. 1996, 15, 281–285. [Google Scholar] [CrossRef] [PubMed]
- Koulenti, D.; Lisboa, T.; Brun-Buisson, C.; Krueger, W.; Macor, A.; Sole-Violan, J.; Diaz, E.; Topeli, A.; DeWaele, J.; Carneiro, A.; et al. Spectrum of practice in the diagnosis of nosocomial pneumonia in patients requiring mechanical ventilation in european intensive care units. Crit. Care Med. 2009, 37, 2360–2368. [Google Scholar] [CrossRef] [PubMed]
- Walker, J.; Moore, G. Pseudomonas aeruginosa in hospital water systems: Biofilms, guidelines, and practicalities. J. Hosp. Infect. 2015, 89, 324–327. [Google Scholar] [CrossRef] [PubMed]
- Fujitani, S.; Sun, H.Y.; Yu, V.L.; Weingarten, J.A. Pneumonia due to Pseudomonas aeruginosa: Part I: Epidemiology, clinical diagnosis, and source. Chest 2011, 139, 909–919. [Google Scholar] [CrossRef] [PubMed]
- Trautmann, M.; Lepper, P.M.; Haller, M. Ecology of Pseudomonas aeruginosa in the intensive care unit and the evolving role of water outlets as a reservoir of the organism. Am. J. Infect. Control 2005, 33, S41–S49. [Google Scholar] [CrossRef] [PubMed]
- Beer, D.; Vandermeer, B.; Brosnikoff, C.; Shokoples, S.; Rennie, R.; Forgie, S. Bacterial contamination of health care workers’ pagers and the efficacy of various disinfecting agents. Pediatr. Infect. Dis. J. 2006, 25, 1074–1075. [Google Scholar] [CrossRef] [PubMed]
- Adair, C.G.; Gorman, S.P.; Feron, B.M.; Byers, L.M.; Jones, D.S.; Goldsmith, C.E.; Moore, J.E.; Kerr, J.R.; Curran, M.D.; Hogg, G.; et al. Implications of endotracheal tube biofilm for ventilator-associated pneumonia. Intensive Care Med. 1999, 25, 1072–1076. [Google Scholar] [CrossRef] [PubMed]
- Richards, M.J.; Edwards, J.R.; Culver, D.H.; Gaynes, R.P. Nosocomial infections in combined medical-surgical intensive care units in the united states. Infect. Control Hosp. Epidemiol. 2000, 21, 510–515. [Google Scholar] [CrossRef] [PubMed]
- Rolston, K.V.; Bodey, G.P. Pseudomonas aeruginosa infection in cancer patients. Cancer Investig. 1992, 10, 43–59. [Google Scholar] [CrossRef]
- Carratala, J.; Roson, B.; Fernandez-Sevilla, A.; Alcaide, F.; Gudiol, F. Bacteremic pneumonia in neutropenic patients with cancer: Causes, empirical antibiotic therapy, and outcome. Arch. Intern. Med. 1998, 158, 868–872. [Google Scholar] [CrossRef] [PubMed]
- Chatzinikolaou, I.; Abi-Said, D.; Bodey, G.P.; Rolston, K.V.; Tarrand, J.J.; Samonis, G. Recent experience with Pseudomonas aeruginosa bacteremia in patients with cancer: Retrospective analysis of 245 episodes. Arch. Intern. Med. 2000, 160, 501–509. [Google Scholar] [CrossRef] [PubMed]
- Papi, A.; Bellettato, C.M.; Braccioni, F.; Romagnoli, M.; Casolari, P.; Caramori, G.; Fabbri, L.M.; Johnston, S.L. Infections and airway inflammation in chronic obstructive pulmonary disease severe exacerbations. Am. J. Respir. Crit. Care Med. 2006, 173, 1114–1121. [Google Scholar] [CrossRef] [PubMed]
- Patel, I.S.; Seemungal, T.A.; Wilks, M.; Lloyd-Owen, S.J.; Donaldson, G.C.; Wedzicha, J.A. Relationship between bacterial colonisation and the frequency, character, and severity of COPD exacerbations. Thorax 2002, 57, 759–764. [Google Scholar] [CrossRef] [PubMed]
- Williams, B.J.; Dehnbostel, J.; Blackwell, T.S. Pseudomonas aeruginosa: Host defence in lung diseases. Respirology 2010, 15, 1037–1056. [Google Scholar] [CrossRef] [PubMed]
- Bush, A.; Chodhari, R.; Collins, N.; Copeland, F.; Hall, P.; Harcourt, J.; Hariri, M.; Hogg, C.; Lucas, J.; Mitchison, H.M.; et al. Primary ciliary dyskinesia: Current state of the art. Arch. Dis. Child. 2007, 92, 1136–1140. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, B.Z.; Haaf, J.B.; Leal, T.; Noel, S. Cystic fibrosis transmembrane conductance regulator modulators in cystic fibrosis: Current perspectives. Clin. Pharmacol. 2016, 8, 127–140. [Google Scholar] [PubMed]
- Cohen, T.S.; Prince, A. Cystic fibrosis: A mucosal immunodeficiency syndrome. Nat. Med. 2012, 18, 509–519. [Google Scholar] [CrossRef] [PubMed]
- Hoegger, M.J.; Fischer, A.J.; McMenimen, J.D.; Ostedgaard, L.S.; Tucker, A.J.; Awadalla, M.A.; Moninger, T.O.; Michalski, A.S.; Hoffman, E.A.; Zabner, J.; et al. Impaired mucus detachment disrupts mucociliary transport in a piglet model of cystic fibrosis. Science 2014, 345, 818–822. [Google Scholar] [CrossRef] [PubMed]
- Miao, X.Y.; Ji, X.B.; Lu, H.W.; Yang, J.W.; Xu, J.F. Distribution of major pathogens from sputum and bronchoalveolar lavage fluid in patients with noncystic fibrosis bronchiectasis: A systematic review. Chin. Med. J. (Engl.) 2015, 128, 2792–2797. [Google Scholar] [PubMed]
- Jacques, I.; Derelle, J.; Weber, M.; Vidailhet, M. Pulmonary evolution of cystic fibrosis patients colonized by Pseudomonas aeruginosa and/or burkholderia cepacia. Eur. J. Pediatr. 1998, 157, 427–431. [Google Scholar] [CrossRef] [PubMed]
- Rowe, S.M.; Miller, S.; Sorscher, E.J. Cystic fibrosis. N. Engl. J. Med. 2005, 352, 1992–2001. [Google Scholar] [CrossRef] [PubMed]
- Talwalkar, J.S.; Murray, T.S. The approach to Pseudomonas aeruginosa in cystic fibrosis. Clin. Chest Med. 2016, 37, 69–81. [Google Scholar] [CrossRef] [PubMed]
- Burns, J.L.; Gibson, R.L.; McNamara, S.; Yim, D.; Emerson, J.; Rosenfeld, M.; Hiatt, P.; McCoy, K.; Castile, R.; Smith, A.L.; et al. Longitudinal assessment of Pseudomonas aeruginosa in young children with cystic fibrosis. J. Infect. Dis. 2001, 183, 444–452. [Google Scholar] [CrossRef] [PubMed]
- Costerton, J.W. Cystic fibrosis pathogenesis and the role of biofilms in persistent infection. Trends Microbiol. 2001, 9, 50–52. [Google Scholar] [CrossRef]
- Costerton, J.W.; Stewart, P.S.; Greenberg, E.P. Bacterial biofilms: A common cause of persistent infections. Science 1999, 284, 1318–1322. [Google Scholar] [CrossRef] [PubMed]
- Hoiby, N.; Ciofu, O.; Bjarnsholt, T. Pseudomonas aeruginosa biofilms in cystic fibrosis. Future Microbiol. 2010, 5, 1663–1674. [Google Scholar] [CrossRef] [PubMed]
- Folkesson, A.; Jelsbak, L.; Yang, L.; Johansen, H.K.; Ciofu, O.; Hoiby, N.; Molin, S. Adaptation of Pseudomonas aeruginosa to the cystic fibrosis airway: An evolutionary perspective. Nat. Rev. Microbiol. 2012, 10, 841–851. [Google Scholar] [CrossRef] [PubMed]
- Haiko, J.; Westerlund-Wikstrom, B. The role of the bacterial flagellum in adhesion and virulence. Biology 2013, 2, 1242–1267. [Google Scholar] [CrossRef] [PubMed]
- Balloy, V.; Verma, A.; Kuravi, S.; Si-Tahar, M.; Chignard, M.; Ramphal, R. The role of flagellin versus motility in acute lung disease caused by Pseudomonas aeruginosa. J. Infect. Dis. 2007, 196, 289–296. [Google Scholar] [CrossRef] [PubMed]
- Wolfgang, M.C.; Jyot, J.; Goodman, A.L.; Ramphal, R.; Lory, S. Pseudomonas aeruginosa regulates flagellin expression as part of a global response to airway fluid from cystic fibrosis patients. Proc. Natl. Acad. Sci. USA 2004, 101, 6664–6668. [Google Scholar] [CrossRef] [PubMed]
- Picioreanu, C.; Kreft, J.U.; Klausen, M.; Haagensen, J.A.; Tolker-Nielsen, T.; Molin, S. Microbial motility involvement in biofilm structure formation—A 3D modelling study. Water Sci. Technol. 2007, 55, 337–343. [Google Scholar] [CrossRef] [PubMed]
- Klausen, M.; Aaes-Jorgensen, A.; Molin, S.; Tolker-Nielsen, T. Involvement of bacterial migration in the development of complex multicellular structures in Pseudomonas aeruginosa biofilms. Mol. Microbiol. 2003, 50, 61–68. [Google Scholar] [CrossRef] [PubMed]
- Alhede, M.; Bjarnsholt, T.; Givskov, M.; Alhede, M. Pseudomonas aeruginosa biofilms: Mechanisms of immune evasion. Adv. Appl. Microbiol. 2014, 86, 1–40. [Google Scholar] [PubMed]
- Jensen, P.O.; Givskov, M.; Bjarnsholt, T.; Moser, C. The immune system vs. Pseudomonas aeruginosa biofilms. FEMS Immunol. Med. Microbiol. 2010, 59, 292–305. [Google Scholar] [CrossRef]
- Wagner, V.E.; Iglewski, B.H.P. Aeruginosa biofilms in cf infection. Clin. Rev. Allergy Immunol. 2008, 35, 124–134. [Google Scholar] [CrossRef] [PubMed]
- Sousa, A.M.; Pereira, M.O. Pseudomonas aeruginosa diversification during infection development in cystic fibrosis lungs—A review. Pathogens 2014, 3, 680–703. [Google Scholar] [CrossRef] [PubMed]
- Andrews, T.; Sullivan, K.E. Infections in patients with inherited defects in phagocytic function. Clin. Microbiol. Rev. 2003, 16, 597–621. [Google Scholar] [CrossRef] [PubMed]
- Koh, A.Y.; Priebe, G.P.; Ray, C.; Van Rooijen, N.; Pier, G.B. Inescapable need for neutrophils as mediators of cellular innate immunity to acute Pseudomonas aeruginosa pneumonia. Infect. Immun. 2009, 77, 5300–5310. [Google Scholar] [CrossRef] [PubMed]
- Tsai, W.C.; Strieter, R.M.; Mehrad, B.; Newstead, M.W.; Zeng, X.; Standiford, T.J. CXC chemokine receptor CXCR2 is essential for protective innate host response in murine Pseudomonas aeruginosa pneumonia. Infect. Immun. 2000, 68, 4289–4296. [Google Scholar] [CrossRef] [PubMed]
- Kurahashi, K.; Sawa, T.; Ota, M.; Kajikawa, O.; Hong, K.; Martin, T.R.; Wiener-Kronish, J.P. Depletion of phagocytes in the reticuloendothelial system causes increased inflammation and mortality in rabbits with Pseudomonas aeruginosa pneumonia. Am. J. Physiol Lung Cell. Mol. Physiol. 2009, 296, 198–209. [Google Scholar] [CrossRef] [PubMed]
- Mijares, L.A.; Wangdi, T.; Sokol, C.; Homer, R.; Medzhitov, R.; Kazmierczak, B.I. Airway epithelial MyD88 restores control of Pseudomonas aeruginosa murine infection via an IL-1-dependent pathway. J. Immunol. 2011, 186, 7080–7088. [Google Scholar] [CrossRef] [PubMed]
- Anas, A.A.; van Lieshout, M.H.; Claushuis, T.A.; de Vos, A.F.; Florquin, S.; de Boer, O.J.; Hou, B.; Van’t Veer, C.; van der Poll, T. Lung epithelial MyD88 drives early pulmonary clearance of Pseudomonas aeruginosa by a flagellin dependent mechanism. Am. J. Physiol Lung Cell. Mol. Physiol. 2016, 311, 219–228. [Google Scholar] [CrossRef] [PubMed]
- Beaumont, P.E.; McHugh, B.; Gwyer Findlay, E.; Mackellar, A.; Mackenzie, K.J.; Gallo, R.L.; Govan, J.R.; Simpson, A.J.; Davidson, D.J. Cathelicidin host defence peptide augments clearance of pulmonary Pseudomonas aeruginosa infection by its influence on neutrophil function in vivo. PLoS ONE 2014, 9, e99029. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.C.; Reins, R.Y.; Gallo, R.L.; McDermott, A.M. Cathelicidin-deficient (Cnlp−/−) mice show increased susceptibility to Pseudomonas aeruginosa keratitis. Investig. Ophthalmol. Vis. Sci. 2007, 48, 4498–4508. [Google Scholar] [CrossRef] [PubMed]
- Guan, X.; Hou, Y.; Sun, F.; Yang, Z.; Li, C. Dysregulated chemokine signaling in cystic fibrosis lung disease: A potential therapeutic target. Curr. Drug Targets 2016, 17, 1535–1544. [Google Scholar] [CrossRef] [PubMed]
- Colombo, C.; Costantini, D.; Rocchi, A.; Cariani, L.; Garlaschi, M.L.; Tirelli, S.; Calori, G.; Copreni, E.; Conese, M. Cytokine levels in sputum of cystic fibrosis patients before and after antibiotic therapy. Pediatr. Pulmonol. 2005, 40, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Mayer-Hamblett, N.; Aitken, M.L.; Accurso, F.J.; Kronmal, R.A.; Konstan, M.W.; Burns, J.L.; Sagel, S.D.; Ramsey, B.W. Association between pulmonary function and sputum biomarkers in cystic fibrosis. Am. J. Respir. Crit. Care Med. 2007, 175, 822–828. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.S.; Okamoto, K.; Rubin, B.K. Pulmonary function is negatively correlated with sputum inflammatory markers and cough clearability in subjects with cystic fibrosis but not those with chronic bronchitis. Chest 2006, 129, 1148–1154. [Google Scholar] [CrossRef] [PubMed]
- Osika, E.; Cavaillon, J.M.; Chadelat, K.; Boule, M.; Fitting, C.; Tournier, G.; Clement, A. Distinct sputum cytokine profiles in cystic fibrosis and other chronic inflammatory airway disease. Eur. Respir J. 1999, 14, 339–346. [Google Scholar] [CrossRef] [PubMed]
- Zoumot, Z.; Wilson, R. Respiratory infection in noncystic fibrosis bronchiectasis. Curr. Opin. Infect. Dis. 2010, 23, 165–170. [Google Scholar] [CrossRef] [PubMed]
- Bodini, A.; D’Orazio, C.; Peroni, D.; Corradi, M.; Folesani, G.; Baraldi, E.; Assael, B.M.; Boner, A.; Piacentini, G.L. Biomarkers of neutrophilic inflammation in exhaled air of cystic fibrosis children with bacterial airway infections. Pediatr. Pulmonol. 2005, 40, 494–499. [Google Scholar] [CrossRef] [PubMed]
- Conese, M.; Copreni, E.; Di Gioia, S.; De Rinaldis, P.; Fumarulo, R. Neutrophil recruitment and airway epithelial cell involvement in chronic cystic fibrosis lung disease. J. Cyst. Fibros 2003, 2, 129–135. [Google Scholar] [CrossRef]
- Rada, B.; Gardina, P.; Myers, T.G.; Leto, T.L. Reactive oxygen species mediate inflammatory cytokine release and EGFR-dependent mucin secretion in airway epithelial cells exposed to pseudomonas pyocyanin. Mucosal Immunol. 2011, 4, 158–171. [Google Scholar] [CrossRef] [PubMed]
- Rada, B.; Leto, T.L. Pyocyanin effects on respiratory epithelium: Relevance in Pseudomonas aeruginosa airway infections. Trends Microbiol. 2013, 21, 73–81. [Google Scholar] [CrossRef] [PubMed]
- DiMango, E.; Zar, H.J.; Bryan, R.; Prince, A. Diverse Pseudomonas aeruginosa gene products stimulate respiratory epithelial cells to produce interleukin-8. J. Clin. Investig. 1995, 96, 2204–2210. [Google Scholar] [CrossRef] [PubMed]
- Massion, P.P.; Inoue, H.; Richman-Eisenstat, J.; Grunberger, D.; Jorens, P.G.; Housset, B.; Pittet, J.F.; Wiener-Kronish, J.P.; Nadel, J.A. Novel pseudomonas product stimulates interleukin-8 production in airway epithelial cells in vitro. J. Clin. Investig. 1994, 93, 26–32. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, H.; Yoshimura, K.; McElvaney, N.G.; Crystal, R.G. Neutrophil elastase in respiratory epithelial lining fluid of individuals with cystic fibrosis induces interleukin-8 gene expression in a human bronchial epithelial cell line. J. Clin Investig. 1992, 89, 1478–1484. [Google Scholar] [CrossRef] [PubMed]
- Bonfield, T.L.; Konstan, M.W.; Berger, M. Altered respiratory epithelial cell cytokine production in cystic fibrosis. J. Allergy Clin. Immunol. 1999, 104, 72–78. [Google Scholar] [CrossRef]
- Tabary, O.; Zahm, J.M.; Hinnrasky, J.; Couetil, J.P.; Cornillet, P.; Guenounou, M.; Gaillard, D.; Puchelle, E.; Jacquot, J. Selective up-regulation of chemokine IL-8 expression in cystic fibrosis bronchial gland cells in vivo and in vitro. Am. J. Pathol. 1998, 153, 921–930. [Google Scholar] [CrossRef]
- Tabary, O.; Escotte, S.; Couetil, J.P.; Hubert, D.; Dusser, D.; Puchelle, E.; Jacquot, J. Genistein inhibits constitutive and inducible nfkappab activation and decreases IL-8 production by human cystic fibrosis bronchial gland cells. Am. J. Pathol. 1999, 155, 473–481. [Google Scholar] [CrossRef]
- Tang, A.C.; Saferali, A.; He, G.; Sandford, A.J.; Strug, L.J.; Turvey, S.E. Endoplasmic reticulum stress regulates chemokine production in cystic fibrosis airway cells through STAT3 modulation. J. Infect. Dis. 2017, 215, 293–302. [Google Scholar] [CrossRef] [PubMed]
- Stecenko, A.A.; King, G.; Torii, K.; Breyer, R.M.; Dworski, R.; Blackwell, T.S.; Christman, J.W.; Brigham, K.L. Dysregulated cytokine production in human cystic fibrosis bronchial epithelial cells. Inflammation 2001, 25, 145–155. [Google Scholar] [CrossRef] [PubMed]
- Venkatakrishnan, A.; Stecenko, A.A.; King, G.; Blackwell, T.R.; Brigham, K.L.; Christman, J.W.; Blackwell, T.S. Exaggerated activation of nuclear factor-kappab and altered ikappab-beta processing in cystic fibrosis bronchial epithelial cells. Am. J. Respir. Cell Mol. Biol. 2000, 23, 396–403. [Google Scholar] [CrossRef] [PubMed]
- Mackerness, K.J.; Jenkins, G.R.; Bush, A.; Jose, P.J. Characterisation of the range of neutrophil stimulating mediators in cystic fibrosis sputum. Thorax 2008, 63, 614–620. [Google Scholar] [CrossRef] [PubMed]
- Hopken, U.E.; Lu, B.; Gerard, N.P.; Gerard, C. The C5a chemoattractant receptor mediates mucosal defence to infection. Nature 1996, 383, 86–89. [Google Scholar] [CrossRef] [PubMed]
- Fick, R.B., Jr.; Robbins, R.A.; Squier, S.U.; Schoderbek, W.E.; Russ, W.D. Complement activation in cystic fibrosis respiratory fluids: In vivo and in vitro generation of C5a and chemotactic activity. Pediatr. Res. 1986, 20, 1258–1268. [Google Scholar] [CrossRef] [PubMed]
- Van den Berg, C.W.; Tambourgi, D.V.; Clark, H.W.; Hoong, S.J.; Spiller, O.B.; McGreal, E.P. Mechanism of neutrophil dysfunction: Neutrophil serine proteases cleave and inactivate the C5a receptor. J. Immunol. 2014, 192, 1787–1795. [Google Scholar] [CrossRef] [PubMed]
- Sagel, S.D.; Wagner, B.D.; Anthony, M.M.; Emmett, P.; Zemanick, E.T. Sputum biomarkers of inflammation and lung function decline in children with cystic fibrosis. Am. J. Respir. Crit. Care Med. 2012, 186, 857–865. [Google Scholar] [CrossRef] [PubMed]
- Tang, A.; Sharma, A.; Jen, R.; Hirschfeld, A.F.; Chilvers, M.A.; Lavoie, P.M.; Turvey, S.E. Inflammasome-mediated IL-1beta production in humans with cystic fibrosis. PLoS ONE 2012, 7, e37689. [Google Scholar]
- Bakele, M.; Joos, M.; Burdi, S.; Allgaier, N.; Poschel, S.; Fehrenbacher, B.; Schaller, M.; Marcos, V.; Kummerle-Deschner, J.; Rieber, N.; et al. Localization and functionality of the inflammasome in neutrophils. J. Biol. Chem. 2014, 289, 5320–5329. [Google Scholar] [CrossRef] [PubMed]
- Cromwell, O.; Walport, M.J.; Morris, H.R.; Taylor, G.W.; Hodson, M.E.; Batten, J.; Kay, A.B. Identification of leukotrienes d and b in sputum from cystic fibrosis patients. Lancet 1981, 2, 164–165. [Google Scholar] [CrossRef]
- Cromwell, O.; Walport, M.J.; Taylor, G.W.; Morris, H.R.; O’Driscoll, B.R.; Kay, A.B. Identification of leukotrienes in the sputum of patients with cystic fibrosis. Adv. Prostaglandin Thromboxane Leukot. Res. 1982, 9, 251–257. [Google Scholar] [CrossRef]
- Konstan, M.W.; Walenga, R.W.; Hilliard, K.A.; Hilliard, J.B. Leukotriene B4 markedly elevated in the epithelial lining fluid of patients with cystic fibrosis. Am. Rev. Respir. Dis. 1993, 148, 896–901. [Google Scholar] [CrossRef] [PubMed]
- Dayer Pastore, F.; Schlegel-Haueter, S.E.; Belli, D.C.; Rochat, T.; Dudez, T.S.; Suter, S. Chemotactic factors in bronchial secretions of cystic fibrosis patients. J. Infect. Dis. 1998, 177, 1413–1417. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, R.H.; Sorrelli, T.C. Decreased polymorphonuclear leucocyte chemotactic response to leukotriene B4 in cystic fibrosis. Clin. Exp. Immunol. 1992, 89, 321–324. [Google Scholar] [CrossRef] [PubMed]
- Bayes, H.K.; Ritchie, N.D.; Evans, T.J. IL-17 is required for control of chronic lung infection caused by Pseudomonas aeruginosa. Infect. Immun. 2016, 84, 3507–3516. [Google Scholar] [CrossRef] [PubMed]
- Decraene, A.; Willems-Widyastuti, A.; Kasran, A.; De Boeck, K.; Bullens, D.M.; Dupont, L.J. Elevated expression of both mRNA and protein levels of IL-17a in sputum of stable cystic fibrosis patients. Respir. Res. 2010, 11, 177. [Google Scholar] [CrossRef] [PubMed]
- Dubin, P.J.; Kolls, J.K. IL-23 mediates inflammatory responses to mucoid Pseudomonas aeruginosa lung infection in mice. Am. J. Physiol Lung Cell. Mol. Physiol. 2007, 292, L519–L528. [Google Scholar] [CrossRef] [PubMed]
- Brodlie, M.; McKean, M.C.; Johnson, G.E.; Anderson, A.E.; Hilkens, C.M.; Fisher, A.J.; Corris, P.A.; Lordan, J.L.; Ward, C. Raised interleukin-17 is immunolocalised to neutrophils in cystic fibrosis lung disease. Eur. Respir. J. 2011, 37, 1378–1385. [Google Scholar] [CrossRef] [PubMed]
- Taylor, P.R.; Bonfield, T.L.; Chmiel, J.F.; Pearlman, E. Neutrophils from F508del cystic fibrosis patients produce IL-17a and express IL-23—Dependent IL-17RC. Clin. Immunol. 2016, 170, 53–60. [Google Scholar] [CrossRef] [PubMed]
- Dubin, P.J.; Kolls, J.K. Il-17 in cystic fibrosis: More than just Th17 cells. Am. J. Respir. Crit. Care Med. 2011, 184, 155–157. [Google Scholar] [CrossRef] [PubMed]
- Dubin, P.J.; McAllister, F.; Kolls, J.K. Is cystic fibrosis a Th17 disease? Inflamm. Res. 2007, 56, 221–227. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.Y.; Lee, M.; Bae, Y.S. Formyl peptide receptors in cellular differentiation and inflammatory diseases. J. Cell Biochem. 2017, 9999, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Balloy, V.; Thevenot, G.; Bienvenu, T.; Morand, P.; Corvol, H.; Clement, A.; Ramphal, R.; Hubert, D.; Chignard, M. Flagellin concentrations in expectorations from cystic fibrosis patients. BMC Pulm. Med. 2014, 14, 100. [Google Scholar] [CrossRef] [PubMed]
- Sutterwala, F.S.; Mijares, L.A.; Li, L.; Ogura, Y.; Kazmierczak, B.I.; Flavell, R.A. Immune recognition of Pseudomonas aeruginosa mediated by the IPAF/NLRC4 inflammasome. J. Exp. Med. 2007, 204, 3235–3245. [Google Scholar] [CrossRef] [PubMed]
- Hansch, G.M.; Prior, B.; Brenner-Weiss, G.; Obst, U.; Overhage, J. The pseudomonas quinolone signal (PQS) stimulates chemotaxis of polymorphonuclear neutrophils. J. Appl. Biomater. Funct. Mater. 2014, 12, 21–26. [Google Scholar] [CrossRef] [PubMed]
- Rada, B.K.; Geiszt, M.; Kaldi, K.; Timar, C.; Ligeti, E. Dual role of phagocytic nadph oxidase in bacterial killing. Blood 2004, 104, 2947–2953. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Utomo, A.; Cullere, X.; Choi, M.M.; Milner, D.A., Jr.; Venkatesh, D.; Yun, S.H.; Mayadas, T.N. The beta-glucan receptor dectin-1 activates the integrin Mac-1 in neutrophils via Vav protein signaling to promote Candida albicans clearance. Cell Host Microbe 2011, 10, 603–615. [Google Scholar] [CrossRef] [PubMed]
- Ford, J.W.; McVicar, D.W. TREM and Trem-like receptors in inflammation and disease. Curr. Opin. Immunol. 2009, 21, 38–46. [Google Scholar] [CrossRef] [PubMed]
- Mayadas, T.N.; Cullere, X.; Lowell, C.A. The multifaceted functions of neutrophils. Annu. Rev. Pathol. 2014, 9, 181–218. [Google Scholar] [CrossRef] [PubMed]
- Travassos, L.H.; Carneiro, L.A.; Girardin, S.E.; Boneca, I.G.; Lemos, R.; Bozza, M.T.; Domingues, R.C.; Coyle, A.J.; Bertin, J.; Philpott, D.J.; et al. Nod1 participates in the innate immune response to Pseudomonas aeruginosa. J. Biol. Chem. 2005, 280, 36714–36718. [Google Scholar] [CrossRef]
- Pollard, A.J.; Heale, J.P.; Tsang, A.; Massing, B.; Speert, D.P. Nonopsonic phagocytosis of pseudomonas aeruginoas: Insights from an infant with leukocyte adhesion deficiency. Pediatr. Infect. Dis. J. 2001, 20, 452–454. [Google Scholar] [CrossRef] [PubMed]
- Lovewell, R.R.; Patankar, Y.R.; Berwin, B. Mechanisms of phagocytosis and host clearance of Pseudomonas aeruginosa. Am. J. Physiol Lung Cell. Mol. Physiol. 2014, 306, L591–L603. [Google Scholar] [CrossRef] [PubMed]
- Heale, J.P.; Pollard, A.J.; Stokes, R.W.; Simpson, D.; Tsang, A.; Massing, B.; Speert, D.P. Two distinct receptors mediate nonopsonic phagocytosis of different strains of Pseudomonas aeruginosa. J. Infect. Dis. 2001, 183, 1214–1220. [Google Scholar] [CrossRef] [PubMed]
- Amiel, E.; Lovewell, R.R.; O’Toole, G.A.; Hogan, D.A.; Berwin, B. Pseudomonas aeruginosa evasion of phagocytosis is mediated by loss of swimming motility and is independent of flagellum expression. Infect. Immun. 2010, 78, 2937–2945. [Google Scholar] [CrossRef] [PubMed]
- Cowland, J.B.; Borregaard, N. Granulopoiesis and granules of human neutrophils. Immunol. Rev. 2016, 273, 11–28. [Google Scholar] [CrossRef] [PubMed]
- Rada, B.; Leto, T.L. Oxidative innate immune defenses by Nox/Duox family nadph oxidases. Contrib. Microbiol. 2008, 15, 164–187. [Google Scholar] [PubMed]
- Rada, B.; Hably, C.; Meczner, A.; Timar, C.; Lakatos, G.; Enyedi, P.; Ligeti, E. Role of nox2 in elimination of microorganisms. Semin. Immunopathol. 2008, 30, 237–253. [Google Scholar] [CrossRef] [PubMed]
- Rada, B.K.; Geiszt, M.; Hably, C.; Ligeti, E. Consequences of the electrogenic function of the phagocytic nadph oxidase. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2005, 360, 2293–2300. [Google Scholar] [CrossRef] [PubMed]
- Reeves, E.P.; Lu, H.; Jacobs, H.L.; Messina, C.G.; Bolsover, S.; Gabella, G.; Potma, E.O.; Warley, A.; Roes, J.; Segal, A.W. Killing activity of neutrophils is mediated through activation of proteases by K+ flux. Nature 2002, 416, 291–297. [Google Scholar] [CrossRef] [PubMed]
- Schultz, H.; Weiss, J.P. The bactericidal/permeability-increasing protein (BPI) in infection and inflammatory disease. Clin. Chim. Acta 2007, 384, 12–23. [Google Scholar] [CrossRef] [PubMed]
- Wittmann, I.; Schonefeld, M.; Aichele, D.; Groer, G.; Gessner, A.; Schnare, M. Murine bactericidal/permeability-increasing protein inhibits the endotoxic activity of lipopolysaccharide and gram-negative bacteria. J. Immunol. 2008, 180, 7546–7552. [Google Scholar] [CrossRef] [PubMed]
- Pham, C.T. Neutrophil serine proteases: Specific regulators of inflammation. Nat. Rev. Immunol 2006, 6, 541–550. [Google Scholar] [CrossRef]
- Hirche, T.O.; Benabid, R.; Deslee, G.; Gangloff, S.; Achilefu, S.; Guenounou, M.; Lebargy, F.; Hancock, R.E.; Belaaouaj, A. Neutrophil elastase mediates innate host protection against Pseudomonas aeruginosa. J. Immunol. 2008, 181, 4945–4954. [Google Scholar] [CrossRef] [PubMed]
- Sedor, J.; Hogue, L.; Akers, K.; Boslaugh, S.; Schreiber, J.; Ferkol, T. Cathepsin-G interferes with clearance of Pseudomonas aeruginosa from mouse lungs. Pediatr. Res. 2007, 61, 26–31. [Google Scholar] [CrossRef] [PubMed]
- Tai, K.P.; Kamdar, K.; Yamaki, J.; Le, V.V.; Tran, D.; Tran, P.; Selsted, M.E.; Ouellette, A.J.; Wong-Beringer, A. Microbicidal effects of alpha- and theta-defensins against antibiotic-resistant Staphylococcus aureus and Pseudomonas aeruginosa. Innate Immun. 2015, 21, 17–29. [Google Scholar] [CrossRef] [PubMed]
- Bhavsar, T.; Liu, M.; Hardej, D.; Liu, X.; Cantor, J. Aerosolized recombinant human lysozyme ameliorates Pseudomonas aeruginosa-induced pneumonia in hamsters. Exp. Lung Res. 2010, 36, 94–100. [Google Scholar] [CrossRef] [PubMed]
- Cole, A.M.; Thapa, D.R.; Gabayan, V.; Liao, H.I.; Liu, L.; Ganz, T. Decreased clearance of Pseudomonas aeruginosa from airways of mice deficient in lysozyme M. J. Leuk. Biol. 2005, 78, 1081–1085. [Google Scholar] [CrossRef] [PubMed]
- Segal, A.W. How neutrophils kill microbes. Annu. Rev. Immunol. 2005, 23, 197–223. [Google Scholar] [CrossRef] [PubMed]
- Brinkmann, V.; Reichard, U.; Goosmann, C.; Fauler, B.; Uhlemann, Y.; Weiss, D.S.; Weinrauch, Y.; Zychlinsky, A. Neutrophil extracellular traps kill bacteria. Science 2004, 303, 1532–1535. [Google Scholar] [CrossRef] [PubMed]
- Winterbourn, C.C.; Kettle, A.J. Redox reactions and microbial killing in the neutrophil phagosome. Antioxid. Redox Signal. 2013, 18, 642–660. [Google Scholar] [CrossRef] [PubMed]
- Yoo, D.G.; Winn, M.; Pang, L.; Moskowitz, S.M.; Malech, H.L.; Leto, T.L.; Rada, B. Release of cystic fibrosis airway inflammatory markers from Pseudomonas aeruginosa-stimulated human neutrophils involves nadph oxidase-dependent extracellular DNA trap formation. J. Immunol. 2014, 192, 4728–4738. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, T.A.; Abed, U.; Goosmann, C.; Hurwitz, R.; Schulze, I.; Wahn, V.; Weinrauch, Y.; Brinkmann, V.; Zychlinsky, A. Novel cell death program leads to neutrophil extracellular traps. J. Cell Biol. 2007, 176, 231–241. [Google Scholar] [CrossRef] [PubMed]
- Remijsen, Q.; Kuijpers, T.W.; Wirawan, E.; Lippens, S.; Vandenabeele, P.; Vanden Berghe, T. Dying for a cause: Netosis, mechanisms behind an antimicrobial cell death modality. Cell Death Differ. 2011, 18, 581–588. [Google Scholar] [CrossRef] [PubMed]
- Yoo, D.G.; Floyd, M.; Winn, M.; Moskowitz, S.M.; Rada, B. Net formation induced by Pseudomonas aeruginosa cystic fibrosis isolates measured as release of myeloperoxidase-DNA and neutrophil elastase-DNA complexes. Immunol. Lett. 2014, 160, 186–194. [Google Scholar] [CrossRef] [PubMed]
- Kessenbrock, K.; Krumbholz, M.; Schonermarck, U.; Back, W.; Gross, W.L.; Werb, Z.; Grone, H.J.; Brinkmann, V.; Jenne, D.E. Netting neutrophils in autoimmune small-vessel vasculitis. Nat. Med. 2009, 15, 623–625. [Google Scholar] [CrossRef] [PubMed]
- Schimmel, M.; Nur, E.; Biemond, B.J.; van Mierlo, G.J.; Solati, S.; Brandjes, D.P.; Otten, H.M.; Schnog, J.J.; Zeerleder, S.; Curama Study, G. Nucleosomes and neutrophil activation in sickle cell disease painful crisis. Haematologica 2013, 98, 1797–1803. [Google Scholar] [CrossRef] [PubMed]
- Masuda, S.; Nakazawa, D.; Shida, H.; Miyoshi, A.; Kusunoki, Y.; Tomaru, U.; Ishizu, A. Netosis markers: Quest for specific, objective, and quantitative markers. Clin. Chim. Acta 2016, 459, 89–93. [Google Scholar] [CrossRef] [PubMed]
- Stoiber, W.; Obermayer, A.; Steinbacher, P.; Krautgartner, W.D. The role of reactive oxygen species (ROS) in the formation of extracellular traps (ETs) in humans. Biomolecules 2015, 5, 702–723. [Google Scholar] [CrossRef] [PubMed]
- Rada, B.; Jendrysik, M.A.; Pang, L.; Hayes, C.P.; Yoo, D.G.; Park, J.J.; Moskowitz, S.M.; Malech, H.L.; Leto, T.L. Pyocyanin-enhanced neutrophil extracellular trap formation requires the nadph oxidase. PLoS ONE 2013, 8, e54205. [Google Scholar] [CrossRef] [PubMed]
- Papayannopoulos, V.; Metzler, K.D.; Hakkim, A.; Zychlinsky, A. Neutrophil elastase and myeloperoxidase regulate the formation of neutrophil extracellular traps. J. Cell Biol. 2010, 191, 677–691. [Google Scholar] [CrossRef] [PubMed]
- Pang, L.; Hayes, C.P.; Buac, K.; Yoo, D.G.; Rada, B. Pseudogout-associated inflammatory calcium pyrophosphate dihydrate microcrystals induce formation of neutrophil extracellular traps. J. Immunol. 2013, 190, 6488–6500. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Li, M.; Stadler, S.; Correll, S.; Li, P.; Wang, D.; Hayama, R.; Leonelli, L.; Han, H.; Grigoryev, S.A.; et al. Histone hypercitrullination mediates chromatin decondensation and neutrophil extracellular trap formation. J. Cell Biol. 2009, 184, 205–213. [Google Scholar] [CrossRef] [PubMed]
- Leshner, M.; Wang, S.; Lewis, C.; Zheng, H.; Chen, X.A.; Santy, L.; Wang, Y. PAD4 mediated histone hypercitrullination induces heterochromatin decondensation and chromatin unfolding to form neutrophil extracellular trap-like structures. Front. Immunol. 2012, 3, 307. [Google Scholar] [CrossRef]
- Li, P.; Li, M.; Lindberg, M.R.; Kennett, M.J.; Xiong, N.; Wang, Y. PAD4 is essential for antibacterial innate immunity mediated by neutrophil extracellular traps. J. Exp. Med. 2010, 207, 1853–1862. [Google Scholar] [CrossRef] [PubMed]
- Nauseef, W.M.; Kubes, P. Pondering neutrophil extracellular traps with healthy skepticism. Cell. Microbiol. 2016, 18, 1349–1357. [Google Scholar] [CrossRef] [PubMed]
- Halverson, T.W.; Wilton, M.; Poon, K.K.; Petri, B.; Lewenza, S. DNA is an antimicrobial component of neutrophil extracellular traps. PLoS Pathog. 2015, 11, e1004593. [Google Scholar] [CrossRef] [PubMed]
- Mulcahy, H.; Charron-Mazenod, L.; Lewenza, S. Extracellular DNA chelates cations and induces antibiotic resistance in Pseudomonas aeruginosa biofilms. PLoS Pathog. 2008, 4, e1000213. [Google Scholar] [CrossRef] [PubMed]
- Yipp, B.G.; Kubes, P. Netosis: How vital is it? Blood 2013, 122, 2784–2794. [Google Scholar] [CrossRef] [PubMed]
- Parker, H.; Albrett, A.M.; Kettle, A.J.; Winterbourn, C.C. Myeloperoxidase associated with neutrophil extracellular traps is active and mediates bacterial killing in the presence of hydrogen peroxide. J. Leuk. Biol. 2012, 91, 369–376. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Kaplan, M.J. The role of neutrophils and netosis in autoimmune and renal diseases. Nat. Rev. Nephrol 2016, 12, 402–413. [Google Scholar] [CrossRef] [PubMed]
- Schauer, C.; Janko, C.; Munoz, L.E.; Zhao, Y.; Kienhofer, D.; Frey, B.; Lell, M.; Manger, B.; Rech, J.; Naschberger, E.; et al. Aggregated neutrophil extracellular traps limit inflammation by degrading cytokines and chemokines. Nat. Med. 2014, 20, 511–517. [Google Scholar] [PubMed]
- Wong, S.L.; Demers, M.; Martinod, K.; Gallant, M.; Wang, Y.; Goldfine, A.B.; Kahn, C.R.; Wagner, D.D. Diabetes primes neutrophils to undergo NETosis, which impairs wound healing. Nat. Med. 2015, 21, 815–819. [Google Scholar] [CrossRef] [PubMed]
- Schonrich, G.; Raftery, M.J. Neutrophil extracellular traps go viral. Front. Immunol. 2016, 7, 366. [Google Scholar] [CrossRef] [PubMed]
- Olsson, A.K.; Cedervall, J. Netosis in cancer—Platelet-neutrophil crosstalk promotes tumor-associated pathology. Front. Immunol. 2016, 7, 373. [Google Scholar] [CrossRef] [PubMed]
- Timar, C.I.; Lorincz, A.M.; Csepanyi-Komi, R.; Valyi-Nagy, A.; Nagy, G.; Buzas, E.I.; Ivanyi, Z.; Kittel, A.; Powell, D.W.; McLeish, K.R.; et al. Antibacterial effect of microvesicles released from human neutrophilic granulocytes. Blood 2013, 121, 510–518. [Google Scholar] [CrossRef] [PubMed]
- Cocucci, E.; Racchetti, G.; Meldolesi, J. Shedding microvesicles: Artefacts no more. Trends Cell Biol. 2009, 19, 43–51. [Google Scholar] [CrossRef] [PubMed]
- Johnson, B.L., III; Kuethe, J.W.; Caldwell, C.C. Neutrophil derived microvesicles: Emerging role of a key mediator to the immune response. Endocr. Metab. Immune Disord. Drug Targets 2014, 14, 210–217. [Google Scholar] [CrossRef] [PubMed]
- Ben-Ari, J.; Wolach, O.; Gavrieli, R.; Wolach, B. Infections associated with chronic granulomatous disease: Linking genetics to phenotypic expression. Expert Rev. Anti-Infect. Ther. 2012, 10, 881–894. [Google Scholar] [CrossRef] [PubMed]
- Dinauer, M.C. Disorders of neutrophil function: An overview. Methods Mol. Biol. 2014, 1124, 501–515. [Google Scholar] [PubMed]
- Nauseef, W.M. Myeloperoxidase deficiency. Hematol. Pathol. 1990, 4, 165–178. [Google Scholar] [PubMed]
- Speert, D.P.; Bond, M.; Woodman, R.C.; Curnutte, J.T. Infection with pseudomonas cepacia in chronic granulomatous disease: Role of nonoxidative killing by neutrophils in host defense. J. Infect. Dis. 1994, 170, 1524–1531. [Google Scholar] [CrossRef] [PubMed]
- Mathee, K.; Ciofu, O.; Sternberg, C.; Lindum, P.W.; Campbell, J.I.; Jensen, P.; Johnsen, A.H.; Givskov, M.; Ohman, D.E.; Molin, S.; et al. Mucoid conversion of Pseudomonas aeruginosa by hydrogen peroxide: A mechanism for virulence activation in the cystic fibrosis lung. Microbiology 1999, 145 Pt 6, 1349–1357. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, M.I.; Mukherjee, S. Treatment for chronic methicillin-sensitive staphylococcus aureus pulmonary infection in people with cystic fibrosis. Cochrane Database Syst. Rev. 2016, 3, CD011581. [Google Scholar] [PubMed]
- Buvelot, H.; Posfay-Barbe, K.M.; Linder, P.; Schrenzel, J.; Krause, K.H. Staphylococcus aureus, phagocyte NADPH oxidase and chronic granulomatous disease. FEMS Microbiol. Rev. 2016. [Google Scholar] [CrossRef] [PubMed]
- Regelmann, W.E.; Siefferman, C.M.; Herron, J.M.; Elliott, G.R.; Clawson, C.C.; Gray, B.H. Sputum peroxidase activity correlates with the severity of lung disease in cystic fibrosis. Pediatr. Pulmonol. 1995, 19, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Garner, H.P.; Phillips, J.R.; Herron, J.G.; Severson, S.J.; Milla, C.E.; Regelmann, W.E. Peroxidase activity within circulating neutrophils correlates with pulmonary phenotype in cystic fibrosis. J. Lab. Clin. Med. 2004, 144, 127–133. [Google Scholar] [CrossRef] [PubMed]
- Sly, P.D.; Gangell, C.L.; Chen, L.; Ware, R.S.; Ranganathan, S.; Mott, L.S.; Murray, C.P.; Stick, S.M.; Investigators, A.C. Risk factors for bronchiectasis in children with cystic fibrosis. N. Engl. J. Med. 2013, 368, 1963–1970. [Google Scholar] [CrossRef]
- Davis, S.D.; Ferkol, T. Identifying the origins of cystic fibrosis lung disease. N. Engl. J. Med. 2013, 368, 2026–2028. [Google Scholar] [CrossRef] [PubMed]
- Watt, A.P.; Courtney, J.; Moore, J.; Ennis, M.; Elborn, J.S. Neutrophil cell death, activation and bacterial infection in cystic fibrosis. Thorax 2005, 60, 659–664. [Google Scholar] [CrossRef] [PubMed]
- Waters, V.J.; Stanojevic, S.; Sonneveld, N.; Klingel, M.; Grasemann, H.; Yau, Y.C.; Tullis, E.; Wilcox, P.; Freitag, A.; Chilvers, M.; et al. Factors associated with response to treatment of pulmonary exacerbations in cystic fibrosis patients. J. Cyst. Fibros 2015, 14, 755–762. [Google Scholar] [CrossRef] [PubMed]
- Yoshimura, K.; Nakamura, H.; Trapnell, B.C.; Chu, C.S.; Dalemans, W.; Pavirani, A.; Lecocq, J.P.; Crystal, R.G. Expression of the cystic fibrosis transmembrane conductance regulator gene in cells of non-epithelial origin. Nucleic Acids Res. 1991, 19, 5417–5423. [Google Scholar] [CrossRef] [PubMed]
- Painter, R.G.; Bonvillain, R.W.; Valentine, V.G.; Lombard, G.A.; LaPlace, S.G.; Nauseef, W.M.; Wang, G. The role of chloride anion and CFTR in killing of Pseudomonas aeruginosa by normal and CF neutrophils. J. Leuk. Biol. 2008, 83, 1345–1353. [Google Scholar] [CrossRef] [PubMed]
- Ng, H.P.; Zhou, Y.; Song, K.; Hodges, C.A.; Drumm, M.L.; Wang, G. Neutrophil-mediated phagocytic host defense defect in myeloid CFTR-inactivated mice. PLoS ONE 2014, 9, e106813. [Google Scholar] [CrossRef] [PubMed]
- Pohl, K.; Hayes, E.; Keenan, J.; Henry, M.; Meleady, P.; Molloy, K.; Jundi, B.; Bergin, D.A.; McCarthy, C.; McElvaney, O.J.; et al. A neutrophil intrinsic impairment affecting Rab27a and degranulation in cystic fibrosis is corrected by cftr potentiator therapy. Blood 2014, 124, 999–1009. [Google Scholar] [CrossRef] [PubMed]
- Graff, I.; Schram-Doumont, A.; Szpirer, C. Defective protein kinase C-mediated actions in cystic fibrosis neutrophils. Cell. Signal. 1991, 3, 259–266. [Google Scholar] [CrossRef]
- Kemp, T.; Schram-Doumont, A.; van Geffel, R.; Kram, R.; Szpirer, C. Alteration of the N-formyl-methionyl-leucyl-phenylalanine-induced response in cystic fibrosis neutrophils. Pediatr. Res. 1986, 20, 520–526. [Google Scholar] [CrossRef] [PubMed]
- Cabrini, G.; De Togni, P. Increased cytosolic calcium in cystic fibrosis neutrophils effect on stimulus-secretion coupling. Life Sci. 1985, 36, 1561–1567. [Google Scholar] [CrossRef]
- Galant, S.P.; Norton, L.; Herbst, J.; Wood, C. Impaired beta adrenergic receptor binding and function in cystic fibrosis neutrophils. J. Clin. Investig. 1981, 68, 253–258. [Google Scholar] [CrossRef] [PubMed]
- Young, R.L.; Malcolm, K.C.; Kret, J.E.; Caceres, S.M.; Poch, K.R.; Nichols, D.P.; Taylor-Cousar, J.L.; Saavedra, M.T.; Randell, S.H.; Vasil, M.L.; et al. Neutrophil extracellular trap (net)-mediated killing of Pseudomonas aeruginosa: Evidence of acquired resistance within the CF airway, independent of cftr. PLoS ONE 2011, 6, e23637. [Google Scholar] [CrossRef] [PubMed]
- McKeon, D.J.; Cadwallader, K.A.; Idris, S.; Cowburn, A.S.; Pasteur, M.C.; Barker, H.; Haworth, C.S.; Bilton, D.; Chilvers, E.R.; Condliffe, A.M. Cystic fibrosis neutrophils have normal intrinsic reactive oxygen species generation. Eur. Respir. J. 2010, 35, 1264–1272. [Google Scholar] [CrossRef] [PubMed]
- Dinauer, M.C. Primary immune deficiencies with defects in neutrophil function. Hematol. Am. Soc. Hematol. Educ. Program 2016, 2016, 43–50. [Google Scholar] [CrossRef] [PubMed]
- Laval, J.; Ralhan, A.; Hartl, D. Neutrophils in cystic fibrosis. Biol. Chem. 2016, 397, 485–496. [Google Scholar] [CrossRef] [PubMed]
- Gifford, A.M.; Chalmers, J.D. The role of neutrophils in cystic fibrosis. Curr. Opin. Hematol. 2014, 21, 16–22. [Google Scholar] [CrossRef] [PubMed]
- Hayes, E.; Pohl, K.; McElvaney, N.G.; Reeves, E.P. The cystic fibrosis neutrophil: A specialized yet potentially defective cell. Arch. Immunol. Ther. Exp. 2011, 59, 97–112. [Google Scholar] [CrossRef] [PubMed]
- Laval, J.; Touhami, J.; Herzenberg, L.A.; Conrad, C.; Taylor, N.; Battini, J.L.; Sitbon, M.; Tirouvanziam, R. Metabolic adaptation of neutrophils in cystic fibrosis airways involves distinct shifts in nutrient transporter expression. J. Immunol. 2013, 190, 6043–6050. [Google Scholar] [CrossRef] [PubMed]
- Makam, M.; Diaz, D.; Laval, J.; Gernez, Y.; Conrad, C.K.; Dunn, C.E.; Davies, Z.A.; Moss, R.B.; Herzenberg, L.A.; Herzenberg, L.A.; et al. Activation of critical, host-induced, metabolic and stress pathways marks neutrophil entry into cystic fibrosis lungs. Proc. Natl. Acad. Sci. USA 2009, 106, 5779–5783. [Google Scholar] [CrossRef] [PubMed]
- Tirouvanziam, R.; Gernez, Y.; Conrad, C.K.; Moss, R.B.; Schrijver, I.; Dunn, C.E.; Davies, Z.A.; Herzenberg, L.A.; Herzenberg, L.A. Profound functional and signaling changes in viable inflammatory neutrophils homing to cystic fibrosis airways. Proc. Natl. Acad. Sci. USA 2008, 105, 4335–4339. [Google Scholar] [CrossRef] [PubMed]
- Petit-Bertron, A.F.; Tabary, O.; Corvol, H.; Jacquot, J.; Clement, A.; Cavaillon, J.M.; Adib-Conquy, M. Circulating and airway neutrophils in cystic fibrosis display different TLR expression and responsiveness to interleukin-10. Cytokine 2008, 41, 54–60. [Google Scholar] [CrossRef] [PubMed]
- Tabary, O.; Corvol, H.; Boncoeur, E.; Chadelat, K.; Fitting, C.; Cavaillon, J.M.; Clement, A.; Jacquot, J. Adherence of airway neutrophils and inflammatory response are increased in CF airway epithelial cell-neutrophil interactions. Am. J. Physiol Lung Cell. Mol. Physiol. 2006, 290, 588–596. [Google Scholar] [CrossRef]
- Conese, M.; Castellani, S.; Lepore, S.; Palumbo, O.; Manca, A.; Santostasi, T.; Polizzi, A.M.; Copetti, M.; Di Gioia, S.; Casavola, V.; et al. Evaluation of genome-wide expression profiles of blood and sputum neutrophils in cystic fibrosis patients before and after antibiotic therapy. PLoS ONE 2014, 9, e104080. [Google Scholar] [CrossRef] [PubMed]
- Houston, N.; Stewart, N.; Smith, D.S.; Bell, S.C.; Champion, A.C.; Reid, D.W. Sputum neutrophils in cystic fibrosis patients display a reduced respiratory burst. J. Cyst. Fibros 2013, 12, 352–362. [Google Scholar] [CrossRef] [PubMed]
- Koller, B.; Kappler, M.; Latzin, P.; Gaggar, A.; Schreiner, M.; Takyar, S.; Kormann, M.; Kabesch, M.; Roos, D.; Griese, M.; et al. TLR expression on neutrophils at the pulmonary site of infection: TLR1/TLR2-mediated up-regulation of TLR5 expression in cystic fibrosis lung disease. J. Immunol. 2008, 181, 2753–2763. [Google Scholar] [CrossRef] [PubMed]
- Shah, P.L.; Scott, S.F.; Knight, R.A.; Marriott, C.; Ranasinha, C.; Hodson, M.E. In vivo effects of recombinant human DNase I on sputum in patients with cystic fibrosis. Thorax 1996, 51, 119–125. [Google Scholar] [CrossRef] [PubMed]
- Shak, S.; Capon, D.J.; Hellmiss, R.; Marsters, S.A.; Baker, C.L. Recombinant human DNase I reduces the viscosity of cystic fibrosis sputum. Proc. Natl. Acad. Sci. USA 1990, 87, 9188–9192. [Google Scholar] [CrossRef] [PubMed]
- Henry, R.L.; Gibson, P.G.; Carty, K.; Cai, Y.; Francis, J.L. Airway inflammation after treatment with aerosolized deoxyribonuclease in cystic fibrosis. Pediatr. Pulmonol. 1998, 26, 97–100. [Google Scholar] [CrossRef]
- Bucki, R.; Cruz, K.; Pogoda, K.; Eggert, A.; Chin, L.; Ferrin, M.; Imbesi, G.; Hadjiliadis, D.; Janmey, P.A. Enhancement of pulmozyme activity in purulent sputum by combination with poly-aspartic acid or gelsolin. J. Cyst. Fibros 2015, 14, 587–593. [Google Scholar] [CrossRef] [PubMed]
- Shah, P.I.; Bush, A.; Canny, G.J.; Colin, A.A.; Fuchs, H.J.; Geddes, D.M.; Johnson, C.A.; Light, M.C.; Scott, S.F.; Tullis, D.E.; et al. Recombinant human DNase I in cystic fibrosis patients with severe pulmonary disease: A short-term, double-blind study followed by six months open-label treatment. Eur. Respir. J. 1995, 8, 954–958. [Google Scholar] [PubMed]
- Riethmueller, J.; Vonthein, R.; Borth-Bruhns, T.; Grassme, H.; Eyrich, M.; Schilbach, K.; Stern, M.; Gulbins, E. DNA quantification and fragmentation in sputum after inhalation of recombinant human deoxyribonuclease. Cell. Physiol. Biochem. 2008, 22, 347–352. [Google Scholar] [CrossRef] [PubMed]
- Lethem, M.I.; James, S.L.; Marriott, C.; Burke, J.F. The origin of DNA associated with mucus glycoproteins in cystic fibrosis sputum. Eur. Respir. J. 1990, 3, 19–23. [Google Scholar] [PubMed]
- Manzenreiter, R.; Kienberger, F.; Marcos, V.; Schilcher, K.; Krautgartner, W.D.; Obermayer, A.; Huml, M.; Stoiber, W.; Hector, A.; Griese, M.; et al. Ultrastructural characterization of cystic fibrosis sputum using atomic force and scanning electron microscopy. J. Cyst. Fibros 2012, 11, 84–92. [Google Scholar] [CrossRef] [PubMed]
- Papayannopoulos, V.; Staab, D.; Zychlinsky, A. Neutrophil elastase enhances sputum solubilization in cystic fibrosis patients receiving dnase therapy. PLoS ONE 2011, 6, e28526. [Google Scholar] [CrossRef] [PubMed]
- Carswell, F.; Robinson, D.W.; Ward, C.C.; Waterfield, M.R. Deoxyribonucleic acid output in the sputum from cystic fibrosis patients. Eur. J. Respir. Dis. 1984, 65, 53–57. [Google Scholar] [PubMed]
- Dubois, A.V.; Gauthier, A.; Brea, D.; Varaigne, F.; Diot, P.; Gauthier, F.; Attucci, S. Influence of DNA on the activities and inhibition of neutrophil serine proteases in cystic fibrosis sputum. Am. J. Respir. Cell Mol. Biol. 2012, 47, 80–86. [Google Scholar] [CrossRef] [PubMed]
- Torphy, T.J.; Allen, J.; Cantin, A.M.; Konstan, M.W.; Accurso, F.J.; Joseloff, E.; Ratjen, F.A.; Chmiel, J.F.; Antiinflammatory Therapy Working Group. Considerations for the conduct of clinical trials with antiinflammatory agents in cystic fibrosis. A cystic fibrosis foundation workshop report. Ann. Am. Thorac. Soc. 2015, 12, 1398–1406. [Google Scholar] [CrossRef] [PubMed]
- Dinwiddie, R. Anti-inflammatory therapy in cystic fibrosis. J. Cyst. Fibros 2005, 4 (Suppl. 2), 45–48. [Google Scholar] [CrossRef]
- Lands, L.C.; Stanojevic, S. Oral non-steroidal anti-inflammatory drug therapy for lung disease in cystic fibrosis. Cochrane Database Syst. Rev. 2016, 4, CD001505. [Google Scholar] [PubMed]
- Frerichs, C.; Smyth, A. Treatment strategies for cystic fibrosis: What’s in the pipeline? Expert Opin. Pharmacother. 2009, 10, 1191–1202. [Google Scholar] [CrossRef] [PubMed]
- Balfour-Lynn, I.M. The protease-antiprotease battle in the cystic fibrosis lung. J. R. Soc. Med. 1999, 92 (Suppl. 37), 23–30. [Google Scholar] [PubMed]
- Tsai, Y.F.; Hwang, T.L. Neutrophil elastase inhibitors: A patent review and potential applications for inflammatory lung diseases (2010–2014). Expert Opin. Ther. Pat. 2015, 25, 1145–1158. [Google Scholar] [CrossRef] [PubMed]
- Kelly, E.; Greene, C.M.; McElvaney, N.G. Targeting neutrophil elastase in cystic fibrosis. Expert Opin. Ther. Targets 2008, 12, 145–157. [Google Scholar] [CrossRef] [PubMed]
- Patel, D.D.; Lee, D.M.; Kolbinger, F.; Antoni, C. Effect of IL-17a blockade with secukinumab in autoimmune diseases. Ann. Rheum. Dis. 2013, 72 (Suppl. 2), ii116–ii123. [Google Scholar] [CrossRef] [PubMed]
- Leonardi, C.; Matheson, R.; Zachariae, C.; Cameron, G.; Li, L.; Edson-Heredia, E.; Braun, D.; Banerjee, S. Anti-interleukin-17 monoclonal antibody ixekizumab in chronic plaque psoriasis. N. Engl. J. Med. 2012, 366, 1190–1199. [Google Scholar] [CrossRef] [PubMed]
- Lovewell, R.R.; Hayes, S.M.; O’Toole, G.A.; Berwin, B. Pseudomonas aeruginosa flagellar motility activates the phagocyte PI3K/AKT pathway to induce phagocytic engulfment. Am. J. Physiol. Lung Cell. Mol. Physiol. 2014, 306, 698–707. [Google Scholar] [CrossRef] [PubMed]
- Floyd, M.; Winn, M.; Cullen, C.; Sil, P.; Chassaing, B.; Yoo, D.G.; Gewirtz, A.T.; Goldberg, J.B.; McCarter, L.L.; Rada, B. Swimming motility mediates the formation of neutrophil extracellular traps induced by flagellated Pseudomonas aeruginosa. PLoS Pathog. 2016, 12, e1005987. [Google Scholar] [CrossRef] [PubMed]
- Mahenthiralingam, E.; Campbell, M.E.; Speert, D.P. Nonmotility and phagocytic resistance of Pseudomonas aeruginosa isolates from chronically colonized patients with cystic fibrosis. Infect. Immun. 1994, 62, 596–605. [Google Scholar] [PubMed]
- Luzar, M.A.; Montie, T.C. Avirulence and altered physiological properties of cystic fibrosis strains of Pseudomonas aeruginosa. Infect. Immun. 1985, 50, 572–576. [Google Scholar] [PubMed]
- Luzar, M.A.; Thomassen, M.J.; Montie, T.C. Flagella and motility alterations in Pseudomonas aeruginosa strains from patients with cystic fibrosis: Relationship to patient clinical condition. Infect. Immun. 1985, 50, 577–582. [Google Scholar] [PubMed]
- Lopez-Boado, Y.S.; Espinola, M.; Bahr, S.; Belaaouaj, A. Neutrophil serine proteinases cleave bacterial flagellin, abrogating its host response-inducing activity. J. Immunol. 2004, 172, 509–515. [Google Scholar] [CrossRef] [PubMed]
- Sonawane, A.; Jyot, J.; During, R.; Ramphal, R. Neutrophil elastase, an innate immunity effector molecule, represses flagellin transcription in Pseudomonas aeruginosa. Infect. Immun. 2006, 74, 6682–6689. [Google Scholar] [CrossRef] [PubMed]
- Leid, J.G.; Kerr, M.; Selgado, C.; Johnson, C.; Moreno, G.; Smith, A.; Shirtliff, M.E.; O’Toole, G.A.; Cope, E.K. Flagellum-mediated biofilm defense mechanisms of Pseudomonas aeruginosa against host-derived lactoferrin. Infect. Immun. 2009, 77, 4559–4566. [Google Scholar] [CrossRef] [PubMed]
- Pritt, B.; O’Brien, L.; Winn, W. Mucoid pseudomonas in cystic fibrosis. Am. J. Clin. Pathol. 2007, 128, 32–34. [Google Scholar] [CrossRef] [PubMed]
- Kragh, K.N.; Alhede, M.; Jensen, P.O.; Moser, C.; Scheike, T.; Jacobsen, C.S.; Seier Poulsen, S.; Eickhardt-Sorensen, S.R.; Trostrup, H.; Christoffersen, L.; et al. Polymorphonuclear leukocytes restrict growth of Pseudomonas aeruginosa in the lungs of cystic fibrosis patients. Infect. Immun. 2014, 82, 4477–4486. [Google Scholar] [CrossRef] [PubMed]
- Caceres, S.M.; Malcolm, K.C.; Taylor-Cousar, J.L.; Nichols, D.P.; Saavedra, M.T.; Bratton, D.L.; Moskowitz, S.M.; Burns, J.L.; Nick, J.A. Enhanced in vitro formation and antibiotic resistance of nonattached Pseudomonas aeruginosa aggregates through incorporation of neutrophil products. Antimicrob. Agents Chemother. 2014, 58, 6851–6860. [Google Scholar] [CrossRef] [PubMed]
- Singh, P.K.; Schaefer, A.L.; Parsek, M.R.; Moninger, T.O.; Welsh, M.J.; Greenberg, E.P. Quorum-sensing signals indicate that cystic fibrosis lungs are infected with bacterial biofilms. Nature 2000, 407, 762–764. [Google Scholar] [CrossRef] [PubMed]
- Bjarnsholt, T.; Jensen, P.O.; Fiandaca, M.J.; Pedersen, J.; Hansen, C.R.; Andersen, C.B.; Pressler, T.; Givskov, M.; Hoiby, N. Pseudomonas aeruginosa biofilms in the respiratory tract of cystic fibrosis patients. Pediatr. Pulmonol. 2009, 44, 547–558. [Google Scholar] [CrossRef] [PubMed]
- Klebensberger, J.; Rui, O.; Fritz, E.; Schink, B.; Philipp, B. Cell aggregation of Pseudomonas aeruginosa strain pao1 as an energy-dependent stress response during growth with sodium dodecyl sulfate. Arch. Microbiol. 2006, 185, 417–427. [Google Scholar] [CrossRef] [PubMed]
- Schleheck, D.; Barraud, N.; Klebensberger, J.; Webb, J.S.; McDougald, D.; Rice, S.A.; Kjelleberg, S. Pseudomonas aeruginosa PAO1 preferentially grows as aggregates in liquid batch cultures and disperses upon starvation. PLoS ONE 2009, 4, e5513. [Google Scholar] [CrossRef] [PubMed]
- Fuxman Bass, J.I.; Russo, D.M.; Gabelloni, M.L.; Geffner, J.R.; Giordano, M.; Catalano, M.; Zorreguieta, A.; Trevani, A.S. Extracellular DNA: A major proinflammatory component of Pseudomonas aeruginosa biofilms. J. Immunol. 2010, 184, 6386–6395. [Google Scholar] [CrossRef] [PubMed]
- Alhede, M.; Kragh, K.N.; Qvortrup, K.; Allesen-Holm, M.; van Gennip, M.; Christensen, L.D.; Jensen, P.O.; Nielsen, A.K.; Parsek, M.; Wozniak, D.; et al. Phenotypes of non-attached Pseudomonas aeruginosa aggregates resemble surface attached biofilm. PLoS ONE 2011, 6, e27943. [Google Scholar] [CrossRef] [PubMed]
- Das, T.; Kutty, S.K.; Tavallaie, R.; Ibugo, A.I.; Panchompoo, J.; Sehar, S.; Aldous, L.; Yeung, A.W.; Thomas, S.R.; Kumar, N.; et al. Phenazine virulence factor binding to extracellular DNA is important for Pseudomonas aeruginosa biofilm formation. Sci. Rep. 2015, 5, 8398. [Google Scholar] [CrossRef] [PubMed]
- Das, T.; Manefield, M. Phenazine production enhances extracellular DNA release via hydrogen peroxide generation in Pseudomonas aeruginosa. Commun. Integr. Biol. 2013, 6, e23570. [Google Scholar] [CrossRef] [PubMed]
- Das, T.; Sehar, S.; Koop, L.; Wong, Y.K.; Ahmed, S.; Siddiqui, K.S.; Manefield, M. Influence of calcium in extracellular DNA mediated bacterial aggregation and biofilm formation. PLoS ONE 2014, 9, e91935. [Google Scholar] [CrossRef] [PubMed]
- Klebensberger, J.; Lautenschlager, K.; Bressler, D.; Wingender, J.; Philipp, B. Detergent-induced cell aggregation in subpopulations of Pseudomonas aeruginosa as a preadaptive survival strategy. Environ. Microbiol. 2007, 9, 2247–2259. [Google Scholar] [CrossRef] [PubMed]
- Kahle, N.A.; Brenner-Weiss, G.; Overhage, J.; Obst, U.; Hansch, G.M. Bacterial quorum sensing molecule induces chemotaxis of human neutrophils via induction of p38 and leukocyte specific protein 1 (LSP1). Immunobiology 2013, 218, 145–151. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, S.; Wagner, C.; Muller, W.; Brenner-Weiss, G.; Hug, F.; Prior, B.; Obst, U.; Hansch, G.M. Induction of neutrophil chemotaxis by the quorum-sensing molecule N-(3-oxododecanoyl)-l-homoserine lactone. Infect. Immun. 2006, 74, 5687–5692. [Google Scholar] [CrossRef] [PubMed]
- Pier, G.B.; Coleman, F.; Grout, M.; Franklin, M.; Ohman, D.E. Role of alginate o acetylation in resistance of mucoid Pseudomonas aeruginosa to opsonic phagocytosis. Infect. Immun. 2001, 69, 1895–1901. [Google Scholar] [CrossRef] [PubMed]
- Krieg, D.P.; Helmke, R.J.; German, V.F.; Mangos, J.A. Resistance of mucoid Pseudomonas aeruginosa to nonopsonic phagocytosis by alveolar macrophages in vitro. Infect. Immun. 1988, 56, 3173–3179. [Google Scholar] [PubMed]
- Dwyer, M.; Shan, Q.; D’Ortona, S.; Maurer, R.; Mitchell, R.; Olesen, H.; Thiel, S.; Huebner, J.; Gadjeva, M. Cystic fibrosis sputum DNA has NETosis characteristics and neutrophil extracellular trap release is regulated by macrophage migration-inhibitory factor. J. Innate Immun. 2014, 6, 765–779. [Google Scholar] [CrossRef] [PubMed]
- Jensen, E.T.; Kharazmi, A.; Garred, P.; Kronborg, G.; Fomsgaard, A.; Mollnes, T.E.; Hoiby, N. Complement activation by Pseudomonas aeruginosa biofilms. Microb. Pathog. 1993, 15, 377–388. [Google Scholar] [CrossRef] [PubMed]
- Jensen, E.T.; Kharazmi, A.; Hoiby, N.; Costerton, J.W. Some bacterial parameters influencing the neutrophil oxidative burst response to Pseudomonas aeruginosa biofilms. APMIS 1992, 100, 727–733. [Google Scholar] [CrossRef] [PubMed]
- Alhede, M.; Bjarnsholt, T.; Jensen, P.O.; Phipps, R.K.; Moser, C.; Christophersen, L.; Christensen, L.D.; van Gennip, M.; Parsek, M.; Hoiby, N.; et al. Pseudomonas aeruginosa recognizes and responds aggressively to the presence of polymorphonuclear leukocytes. Microbiology 2009, 155, 3500–3508. [Google Scholar] [CrossRef] [PubMed]
- Jensen, P.O.; Bjarnsholt, T.; Phipps, R.; Rasmussen, T.B.; Calum, H.; Christoffersen, L.; Moser, C.; Williams, P.; Pressler, T.; Givskov, M.; et al. Rapid necrotic killing of polymorphonuclear leukocytes is caused by quorum-sensing-controlled production of rhamnolipid by Pseudomonas aeruginosa. Microbiology 2007, 153, 1329–1338. [Google Scholar] [CrossRef] [PubMed]
- Kharami, A.; Bibi, Z.; Nielsen, H.; Hoiby, N.; Doring, G. Effect of Pseudomonas aeruginosa rhamnolipid on human neutrophil and monocyte function. APMIS 1989, 97, 1068–1072. [Google Scholar] [CrossRef] [PubMed]
- Van Gennip, M.; Christensen, L.D.; Alhede, M.; Phipps, R.; Jensen, P.O.; Christophersen, L.; Pamp, S.J.; Moser, C.; Mikkelsen, P.J.; Koh, A.Y.; et al. Inactivation of the rhla gene in Pseudomonas aeruginosa prevents rhamnolipid production, disabling the protection against polymorphonuclear leukocytes. APMIS 2009, 117, 537–546. [Google Scholar] [CrossRef] [PubMed]
- Jesaitis, A.J.; Franklin, M.J.; Berglund, D.; Sasaki, M.; Lord, C.I.; Bleazard, J.B.; Duffy, J.E.; Beyenal, H.; Lewandowski, Z. Compromised host defense on Pseudomonas aeruginosa biofilms: Characterization of neutrophil and biofilm interactions. J. Immunol. 2003, 171, 4329–4339. [Google Scholar] [CrossRef] [PubMed]
- Van Gennip, M.; Christensen, L.D.; Alhede, M.; Qvortrup, K.; Jensen, P.O.; Hoiby, N.; Givskov, M.; Bjarnsholt, T. Interactions between polymorphonuclear leukocytes and Pseudomonas aeruginosa biofilms on silicone implants in vivo. Infect. Immun. 2012, 80, 2601–2607. [Google Scholar] [CrossRef] [PubMed]
- Branzk, N.; Lubojemska, A.; Hardison, S.E.; Wang, Q.; Gutierrez, M.G.; Brown, G.D.; Papayannopoulos, V. Neutrophils sense microbe size and selectively release neutrophil extracellular traps in response to large pathogens. Nat. Immunol. 2014, 15, 1017–1025. [Google Scholar] [CrossRef] [PubMed]
- Sil, P.; Hayes, C.P.; Reaves, B.J.; Breen, P.; Quinn, S.; Sokolove, J.; Rada, B. P2Y6 receptor antagonist MRS2578 inhibits neutrophil activation and aggregated neutrophil extracellular trap formation induced by gout-associated monosodium urate crystals. J. Immunol. 2017, 198, 428–442. [Google Scholar] [CrossRef] [PubMed]
- Parks, Q.M.; Young, R.L.; Poch, K.R.; Malcolm, K.C.; Vasil, M.L.; Nick, J.A. Neutrophil enhancement of Pseudomonas aeruginosa biofilm development: Human f-actin and DNA as targets for therapy. J. Med. Microbiol. 2009, 58, 492–502. [Google Scholar] [CrossRef] [PubMed]
- Walker, T.S.; Tomlin, K.L.; Worthen, G.S.; Poch, K.R.; Lieber, J.G.; Saavedra, M.T.; Fessler, M.B.; Malcolm, K.C.; Vasil, M.L.; Nick, J.A. Enhanced Pseudomonas aeruginosa biofilm development mediated by human neutrophils. Infect. Immun. 2005, 73, 3693–3701. [Google Scholar] [CrossRef] [PubMed]
- Robertson, D.M.; Parks, Q.M.; Young, R.L.; Kret, J.; Poch, K.R.; Malcolm, K.C.; Nichols, D.P.; Nichols, M.; Zhu, M.; Cavanagh, H.D.; et al. Disruption of contact lens-associated Pseudomonas aeruginosa biofilms formed in the presence of neutrophils. Investig. Ophthalmol. Vis. Sci. 2011, 52, 2844–2850. [Google Scholar] [CrossRef] [PubMed]
© 2017 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rada, B. Interactions between Neutrophils and Pseudomonas aeruginosa in Cystic Fibrosis. Pathogens 2017, 6, 10. https://doi.org/10.3390/pathogens6010010
Rada B. Interactions between Neutrophils and Pseudomonas aeruginosa in Cystic Fibrosis. Pathogens. 2017; 6(1):10. https://doi.org/10.3390/pathogens6010010
Chicago/Turabian StyleRada, Balázs. 2017. "Interactions between Neutrophils and Pseudomonas aeruginosa in Cystic Fibrosis" Pathogens 6, no. 1: 10. https://doi.org/10.3390/pathogens6010010
APA StyleRada, B. (2017). Interactions between Neutrophils and Pseudomonas aeruginosa in Cystic Fibrosis. Pathogens, 6(1), 10. https://doi.org/10.3390/pathogens6010010