
Disruptions of Host Immunity and Inflammation by Giardia Duodenalis: Potential Consequences for Co-Infections in the Gastro-Intestinal Tract

{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Does Giardia duodenalis Induce Pro-Inflammatory Responses?
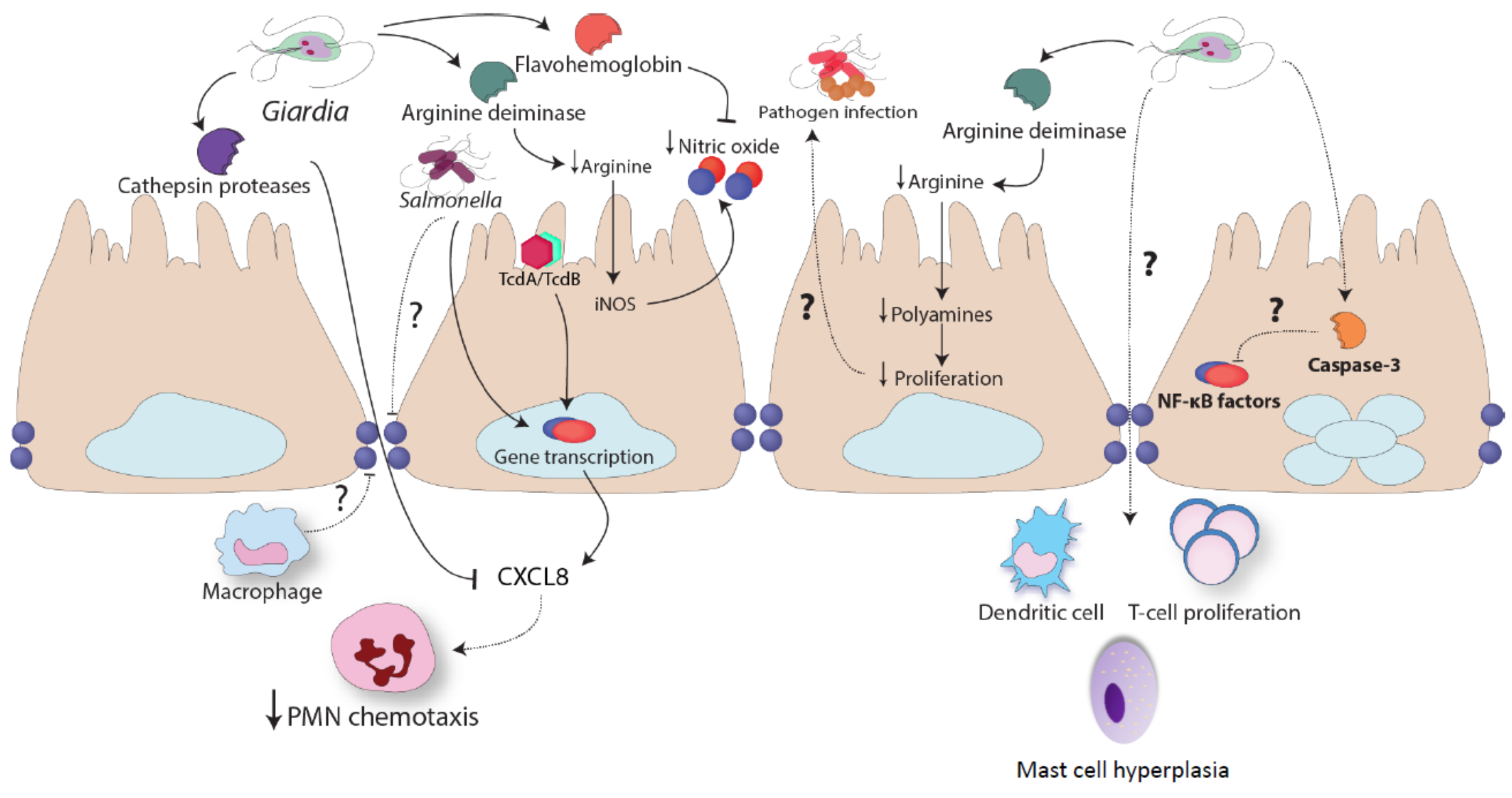
3. Polymicrobial GI Infections Involving Giardia
4. Giardia and Immunomodulation
4.1. Giardia and the Intestinal Mucus Layer
4.2. Giardia and Neutrophil Recruitment
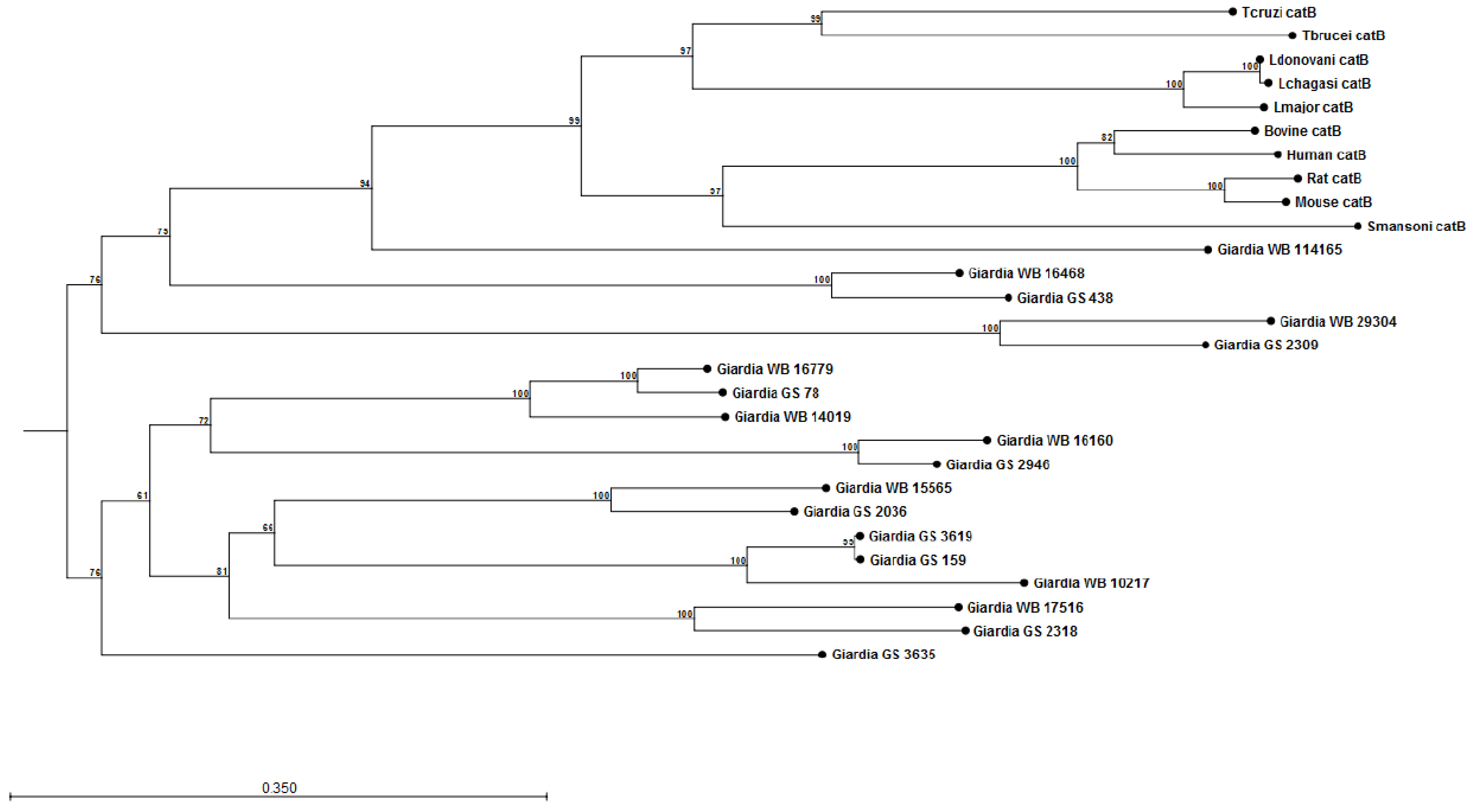
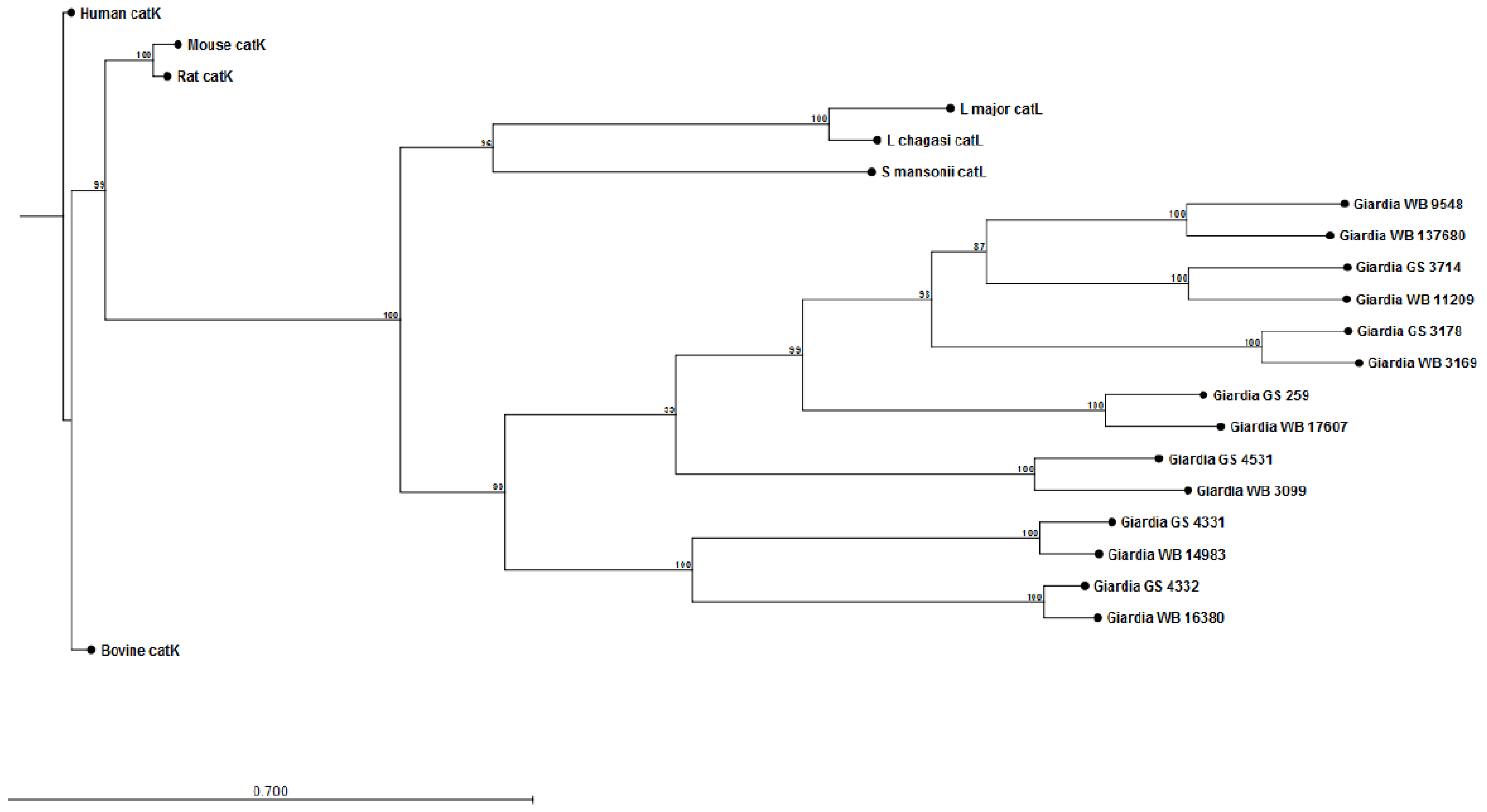
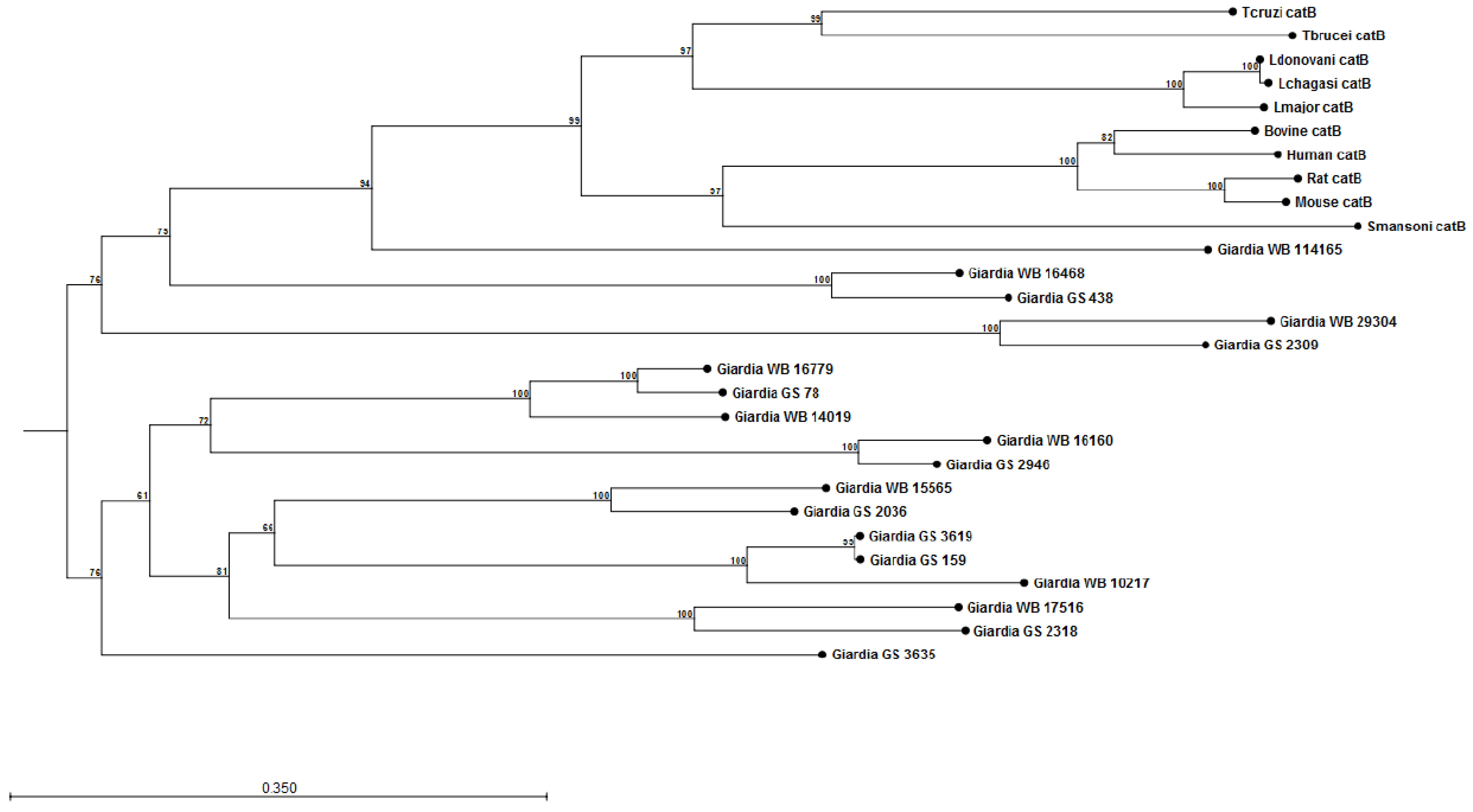
Modulation of Neutrophil Recruitment and Co-Infections
4.3. Giardia and L-Arginine
Parasite Arginine Consumption Inhibits NO Production
4.4. Intestinal Epithelial Cell Death
4.5. Dendritic Cells
4.6. Macrophages
5. Giardia and Distant Site Co-Infections
6. Summary
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Lane, S.; Lloyd, D. Current trends in research into the waterborne parasite Giardia. Crit. Rev. Microbiol. 2002, 28, 123–147. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Kabir, F.; Manneh, J.; Lertsethtakarn, P.; Begum, S.; Gratz, J.; Becker, S.M.; Operario, D.J.; Taniuchi, M.; Janaki, L.; et al. Development and assessment of molecular diagnostic tests for 15 enteropathogens causing childhood diarrhoea: A multicentre study. Lancet Infect. Dis. 2014, 14, 716–724. [Google Scholar] [CrossRef]
- Mejia, R.; Vicuna, Y.; Broncano, N.; Sandoval, C.; Vaca, M.; Chico, M.; Cooper, P.J.; Nutman, T.B. A novel, multi-parallel, real-time polymerase chain reaction approach for eight gastrointestinal parasites provides improved diagnostic capabilities to resource-limited at-risk populations. Am. J. Trop. Med. Hyg. 2013, 88, 1041–1047. [Google Scholar] [CrossRef] [PubMed]
- Nash, T.E.; Herrington, D.A.; Losonsky, G.A.; Levine, M.M. Experimental human infections with Giardia lamblia. J. Infect. Dis. 1987, 156, 974–984. [Google Scholar] [CrossRef] [PubMed]
- Savioli, L.; Smith, H.; Thompson, A. Giardia and Cryptosporidium join the “neglected diseases initiative”. Trends Parasitol. 2006, 22, 203–208. [Google Scholar] [CrossRef] [PubMed]
- Escobedo, A.A.; Arencibia, R.; Vega, R.L.; Rodriguez-Morales, A.J.; Almirall, P.; Alfonso, M. A bibliometric study of international scientific productivity in giardiasis covering the period 1971–2010. J. Infect. Dev. Ctries 2015, 9, 76–86. [Google Scholar] [CrossRef] [PubMed]
- Cotton, J.A.; Beatty, J.K.; Buret, A.G. Host parasite interactions and pathophysiology in Giardia infections. Int. J. Parasitol. 2011, 41, 925–933. [Google Scholar] [CrossRef] [PubMed]
- Halliez, M.C.; Buret, A.G. Extra-intestinal and long term consequences of Giardia duodenalis infections. World J. Gastroenterol. 2013, 19, 8974–8985. [Google Scholar] [CrossRef] [PubMed]
- Wensaas, K.A.; Langeland, N.; Hanevik, K.; Morch, K.; Eide, G.E.; Rortveit, G. Irritable bowel syndrome and chronic fatigue 3 years after acute giardiasis: Historic cohort study. Gut 2012, 61, 214–219. [Google Scholar] [CrossRef] [PubMed]
- Robertson, L.J.; Hanevik, K.; Escobedo, A.A.; Morch, K.; Langeland, N. Giardiasis—Why do the symptoms sometimes never stop? Trends Parasitol. 2010, 26, 75–82. [Google Scholar] [CrossRef] [PubMed]
- Istre, G.R.; Dunlop, T.S.; Gaspard, G.B.; Hopkins, R.S. Waterborne giardiasis at a mountain resort: Evidence for acquired immunity. Am. J. Public Health 1984, 74, 602–604. [Google Scholar] [CrossRef] [PubMed]
- Monis, P.T.; Caccio, S.M.; Thompson, R.C. Variation in Giardia: Towards a taxonomic revision of the genus. Trends Parasitol. 2009, 25, 93–100. [Google Scholar] [CrossRef] [PubMed]
- Lasek-Nesselquist, E.; Welch, D.M.; Sogin, M.L. The identification of a new Giardia duodenalis assemblage in marine vertebrates and a preliminary analysis of g. Duodenalis population biology in marine systems. Int. J. Parasitol. 2010, 40, 1063–1074. [Google Scholar] [CrossRef] [PubMed]
- Haque, R.; Roy, S.; Kabir, M.; Stroup, S.E.; Mondal, D.; Houpt, E.R. Giardia assemblage a infection and diarrhea in bangladesh. J. Infect. Dis. 2005, 192, 2171–2173. [Google Scholar] [CrossRef] [PubMed]
- Pestechian, N.; Rasekh, H.; Rostami-Nejad, M.; Yousofi, H.A.; Hosseini-Safa, A. Molecular identification of Giardia lamblia; is there any correlation between diarrhea and genotyping in iranian population? Gastroenterol. Hepatol. Bed Bench 2014, 7, 168–172. [Google Scholar] [PubMed]
- ElBakri, A.; Samie, A.; Bessong, P.; Potgieter, N.; Odeh, R.A. Detection and molecular characterisation of Giardia lamblia genotypes in sharjah, united arab emirates. Trans R. Soc. Trop. Med. Hyg. 2014, 108, 466–473. [Google Scholar] [CrossRef] [PubMed]
- Puebla, L.J.; Nunez, F.A.; Fernandez, Y.A.; Fraga, J.; Rivero, L.R.; Millan, I.A.; Valdes, L.A.; Silva, I.M. Correlation of Giardia duodenalis assemblages with clinical and epidemiological data in cuban children. Infect. Genet. Evol. 2014, 23, 7–12. [Google Scholar] [CrossRef] [PubMed]
- Minetti, C.; Lamden, K.; Durband, C.; Cheesbrough, J.; Platt, K.; Charlett, A.; O’Brien, S.J.; Fox, A.; Wastling, J.M. Case-control study of risk factors for sporadic giardiasis and parasite assemblages in north west england. J. Clin. Microbiol. 2015. [Google Scholar] [CrossRef] [PubMed]
- Solaymani-Mohammadi, S.; Singer, S.M. Host immunity and pathogen strain contribute to intestinal disaccharidase impairment following gut infection. J. Immunol. 2011, 187, 3769–3775. [Google Scholar] [CrossRef] [PubMed]
- Franzen, O.; Jerlstrom-Hultqvist, J.; Castro, E.; Sherwood, E.; Ankarklev, J.; Reiner, D.S.; Palm, D.; Andersson, J.O.; Andersson, B.; Svard, S.G. Draft genome sequencing of Giardia intestinalis assemblage B isolate GS: Is human giardiasis caused by two different species? PLoS Pathog. 2009. [Google Scholar] [CrossRef] [PubMed]
- Adam, R.D.; Dahlstrom, E.W.; Martens, C.A.; Bruno, D.P.; Barbian, K.D.; Ricklefs, S.M.; Hernandez, M.M.; Narla, N.P.; Patel, R.B.; Porcella, S.F.; et al. Genome sequencing of Giardia lamblia genotypes A2 and B isolates (DH and GS) and comparative analysis with the genomes of genotypes A1 and E (WB and Pig). Genome Biol. Evol. 2013, 5, 2498–2511. [Google Scholar] [CrossRef] [PubMed]
- Jerlstrom-Hultqvist, J.; Ankarklev, J.; Svard, S.G. Is human giardiasis caused by two different Giardia species? Gut Microbes 2010, 1, 379–382. [Google Scholar] [CrossRef] [PubMed]
- Bartelt, L.A.; Sartor, R.B. Advances in understanding Giardia: Determinants and mechanisms of chronic sequelae. F1000Prime Rep. 2015. [Google Scholar] [CrossRef] [PubMed]
- Klotz, C.; Aebischer, T. The immunological enigma of human giardiasis. Curr. Trop. Med. Rep. 2015, 2, 1–9. [Google Scholar] [CrossRef]
- Solaymani-Mohammadi, S.; Singer, S.M. Giardia duodenalis: The double-edged sword of immune responses in giardiasis. Exp. Parasitol. 2010, 126, 292–297. [Google Scholar] [CrossRef] [PubMed]
- Buret, A.; Amat, C.; Manko, A.; Beatty, J.; Halliez, M.M.; Bhargava, A.; Motta, J.-P.; Cotton, J. Giardia duodenalis: New research developments in pathophysiology, pathogenesis, and virulence factors. Curr. Trop. Med. Rep. 2015, 2, 110–118. [Google Scholar] [CrossRef]
- Maizels, R.M. Parasite immunomodulation and polymorphisms of the immune system. J. Biol. 2009, 8, 62. [Google Scholar] [CrossRef] [PubMed]
- McSorley, H.J.; Maizels, R.M. Helminth infections and host immune regulation. Clin. Microbiol. Rev. 2012, 25, 585–608. [Google Scholar] [CrossRef] [PubMed]
- Turner, J.R. Intestinal mucosal barrier function in health and disease. Nat. Rev. Immunol. 2009, 9, 799–809. [Google Scholar] [CrossRef] [PubMed]
- McGuckin, M.A.; Linden, S.K.; Sutton, P.; Florin, T.H. Mucin dynamics and enteric pathogens. Nat. Rev. Microbiol. 2011, 9, 265–278. [Google Scholar] [CrossRef] [PubMed]
- Hollander, D.; Vadheim, C.M.; Brettholz, E.; Petersen, G.M.; Delahunty, T.; Rotter, J.I. Increased intestinal permeability in patients with crohn’s disease and their relatives. A possible etiologic factor. Ann. Intern. Med. 1986, 105, 883–885. [Google Scholar] [CrossRef] [PubMed]
- Buhner, S.; Buning, C.; Genschel, J.; Kling, K.; Herrmann, D.; Dignass, A.; Kuechler, I.; Krueger, S.; Schmidt, H.H.; Lochs, H. Genetic basis for increased intestinal permeability in families with crohn’s disease: Role of card15 3020insc mutation? Gut 2006, 55, 342–347. [Google Scholar] [CrossRef] [PubMed]
- O’Hara, J.R.; Buret, A.G. Mechanisms of intestinal tight junctional disruption during infection. Front. Biosci. 2008, 13, 7008–7021. [Google Scholar] [PubMed]
- Berkes, J.; Viswanathan, V.K.; Savkovic, S.D.; Hecht, G. Intestinal epithelial responses to enteric pathogens: Effects on the tight junction barrier, ion transport, and inflammation. Gut 2003, 52, 439–451. [Google Scholar] [CrossRef] [PubMed]
- Chin, A.C.; Teoh, D.A.; Scott, K.G.; Meddings, J.B.; Macnaughton, W.K.; Buret, A.G. Strain-dependent induction of enterocyte apoptosis by Giardia lamblia disrupts epithelial barrier function in a caspase-3-dependent manner. Infect. Immun. 2002, 70, 3673–3680. [Google Scholar] [CrossRef] [PubMed]
- Scott, K.G.; Meddings, J.B.; Kirk, D.R.; Lees-Miller, S.P.; Buret, A.G. Intestinal infection with Giardia spp. Reduces epithelial barrier function in a myosin light chain kinase-dependent fashion. Gastroenterology 2002, 123, 1179–1190. [Google Scholar] [CrossRef] [PubMed]
- Troeger, H.; Epple, H.J.; Schneider, T.; Wahnschaffe, U.; Ullrich, R.; Burchard, G.D.; Jelinek, T.; Zeitz, M.; Fromm, M.; Schulzke, J.D. Effect of chronic Giardia lamblia infection on epithelial transport and barrier function in human duodenum. Gut 2007, 56, 328–335. [Google Scholar] [CrossRef] [PubMed]
- Teoh, D.A.; Kamieniecki, D.; Pang, G.; Buret, A.G. Giardia lamblia rearranges f-actin and alpha-actinin in human colonic and duodenal monolayers and reduces transepithelial electrical resistance. J. Parasitol. 2000, 86, 800–806. [Google Scholar] [PubMed]
- Panaro, M.A.; Cianciulli, A.; Mitolo, V.; Mitolo, C.I.; Acquafredda, A.; Brandonisio, O.; Cavallo, P. Caspase-dependent apoptosis of the hct-8 epithelial cell line induced by the parasite Giardia intestinalis. FEMS Immunol. Med. Microbiol. 2007, 51, 302–309. [Google Scholar] [CrossRef] [PubMed]
- Reynoso-Robles, R.; Ponce-Macotela, M.; Rosas-Lopez, L.E.; Ramos-Morales, A.; Martinez-Gordillo, M.N.; Gonzalez-Maciel, A. The invasive potential of Giardia intestinalis in an in vivo model. Sci. Rep. 2015. [Google Scholar] [CrossRef] [PubMed]
- Oberhuber, G.; Kastner, N.; Stolte, M. Giardiasis: A histologic analysis of 567 cases. Scand. J. Gastroenterol. 1997, 32, 48–51. [Google Scholar] [CrossRef] [PubMed]
- Campbell, D.I.; McPhail, G.; Lunn, P.G.; Elia, M.; Jeffries, D.J. Intestinal inflammation measured by fecal neopterin in gambian children with enteropathy: Association with growth failure, Giardia lamblia, and intestinal permeability. J. Pediatr. Gastroenterol. Nutr. 2004, 39, 153–157. [Google Scholar] [CrossRef] [PubMed]
- Craven, M.; Egan, C.E.; Dowd, S.E.; McDonough, S.P.; Dogan, B.; Denkers, E.Y.; Bowman, D.; Scherl, E.J.; Simpson, K.W. Inflammation drives dysbiosis and bacterial invasion in murine models of ileal crohn’s disease. PLoS ONE 2012. [Google Scholar] [CrossRef] [PubMed]
- Scott, K.G.; Yu, L.C.; Buret, A.G. Role of cd8+ and cd4+ t lymphocytes in jejunal mucosal injury during murine giardiasis. Infect. Immun. 2004, 72, 3536–3542. [Google Scholar] [CrossRef] [PubMed]
- Hardin, J.A.; Buret, A.G.; Olson, M.E.; Kimm, M.H.; Gall, D.G. Mast cell hyperplasia and increased macromolecular uptake in an animal model of giardiasis. J. Parasitol. 1997, 83, 908–912. [Google Scholar] [CrossRef] [PubMed]
- Dreesen, L.; Rinaldi, M.; Chiers, K.; Li, R.; Geurden, T.; van den Broeck, W.; Goddeeris, B.; Vercruysse, J.; Claerebout, E.; Geldhof, P. Microarray analysis of the intestinal host response in Giardia duodenalis assemblage E infected calves. PLoS ONE 2012. [Google Scholar] [CrossRef] [PubMed]
- Jung, H.C.; Eckmann, L.; Yang, S.K.; Panja, A.; Fierer, J.; Morzycka-Wroblewska, E.; Kagnoff, M.F. A distinct array of proinflammatory cytokines is expressed in human colon epithelial cells in response to bacterial invasion. J. Clin. Investig. 1995, 95, 55–65. [Google Scholar] [CrossRef] [PubMed]
- Eckmann, L.; Laurent, F.; Langford, T.D.; Hetsko, M.L.; Smith, J.R.; Kagnoff, M.F.; Gillin, F.D. Nitric oxide production by human intestinal epithelial cells and competition for arginine as potential determinants of host defense against the lumen-dwelling pathogen Giardia lamblia. J. Immunol. 2000, 164, 1478–1487. [Google Scholar] [CrossRef] [PubMed]
- Roxstrom-Lindquist, K.; Ringqvist, E.; Palm, D.; Svard, S. Giardia lamblia-induced changes in gene expression in differentiated caco-2 human intestinal epithelial cells. Infect. Immun. 2005, 73, 8204–8208. [Google Scholar] [CrossRef] [PubMed]
- Veenemans, J.; Mank, T.; Ottenhof, M.; Baidjoe, A.; Mbugi, E.V.; Demir, A.Y.; Wielders, J.P.; Savelkoul, H.F.; Verhoef, H. Protection against diarrhea associated with Giardia intestinalis is lost with multi-nutrient supplementation: A study in tanzanian children. PLoS Negl. Trop. Dis. 2011, 5. [Google Scholar] [CrossRef] [PubMed]
- Hanevik, K.; Hausken, T.; Morken, M.H.; Strand, E.A.; Morch, K.; Coll, P.; Helgeland, L.; Langeland, N. Persisting symptoms and duodenal inflammation related to Giardia duodenalis infection. J. Infect. 2007, 55, 524–530. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.L.; Chen, S.; Wu, H.W.; Lee, T.C.; Lu, Y.Z.; Wu, L.L.; Ni, Y.H.; Sun, C.H.; Yu, W.H.; Buret, A.G.; et al. Persistent gut barrier damage and commensal bacterial influx following eradication of Giardia infection in mice. Gut Pathog. 2013, 5, 26. [Google Scholar] [CrossRef] [PubMed]
- Benere, E.; van Assche, T.; van Ginneken, C.; Peulen, O.; Cos, P.; Maes, L. Intestinal growth and pathology of Giardia duodenalis assemblage subtype a(i), a(ii), b and e in the gerbil model. Parasitology 2012, 139, 424–433. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.Y.; Hyung, S.; Lee, N.Y.; Yong, T.S.; Han, S.H.; Park, S.J. Excretory-secretory products of Giardia lamblia induce interleukin-8 production in human colonic cells via activation of p38, erk1/2, nf-kappab and ap-1. Parasite Immunol. 2012, 34, 183–198. [Google Scholar] [CrossRef] [PubMed]
- Dos Santos, J.I.; Vituri Cde, L. Some hematimetric findings in human Giardia lamblia infection. Rev. Inst. Med. Trop. Sao Paulo 1996, 38, 91–95. [Google Scholar] [CrossRef] [PubMed]
- Koot, B.G.; ten Kate, F.J.; Juffrie, M.; Rosalina, I.; Taminiau, J.J.; Benninga, M.A. Does Giardia lamblia cause villous atrophy in children?: A retrospective cohort study of the histological abnormalities in giardiasis. J. Pediatr. Gastroenterol. Nutr. 2009, 49, 304–308. [Google Scholar] [CrossRef] [PubMed]
- Aloisio, F.; Filippini, G.; Antenucci, P.; Lepri, E.; Pezzotti, G.; Caccio, S.M.; Pozio, E. Severe weight loss in lambs infected with Giardia duodenalis assemblage b. Vet. Parasitol. 2006, 142, 154–158. [Google Scholar] [CrossRef] [PubMed]
- Bartelt, L.A.; Roche, J.; Kolling, G.; Bolick, D.; Noronha, F.; Naylor, C.; Hoffman, P.; Warren, C.; Singer, S.; Guerrant, R. Persistent g. Lamblia impairs growth in a murine malnutrition model. J. Clin. Investig. 2013, 123, 2672–2684. [Google Scholar] [CrossRef] [PubMed]
- Jimenez, J.C.; Fontaine, J.; Grzych, J.M.; Dei-Cas, E.; Capron, M. Systemic and mucosal responses to oral administration of excretory and secretory antigens from Giardia intestinalis. Clin. Diagn Lab. Immunol. 2004, 11, 152–160. [Google Scholar] [CrossRef] [PubMed]
- Ankarklev, J.; Jerlstrom-Hultqvist, J.; Ringqvist, E.; Troell, K.; Svard, S.G. Behind the smile: Cell biology and disease mechanisms of Giardia species. Nat. Rev. Microbiol. 2010, 8, 413–422. [Google Scholar] [CrossRef] [PubMed]
- Mahmoudi, M.R.; Kazemi, B.; Mohammadiha, A.; Mirzaei, A.; Karanis, P. Detection of Cryptosporidium and Giardia (oo)cysts by IFA, PCR and LAMP in surface water from Rasht, Iran. Trans. R. Soc. Trop. Med. Hyg. 2013, 107, 511–517. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, A.S.; Levine, M.; Camp, J.W., Jr.; Lund, E.; Yoder, J.S.; Glickman, L.T.; Moore, G.E. Temporal patterns of human and canine Giardia infection in the united states: 2003–2009. Prev. Vet. Med. 2014, 113, 249–256. [Google Scholar] [CrossRef] [PubMed]
- Muhsen, K.; Levine, M.M. A systematic review and meta-analysis of the association between Giardia lamblia and endemic pediatric diarrhea in developing countries. Clin. Infect. Dis. 2012, 55 (Suppl. S4), S271–S293. [Google Scholar] [CrossRef] [PubMed]
- Kotloff, K.L.; Nataro, J.P.; Blackwelder, W.C.; Nasrin, D.; Farag, T.H.; Panchalingam, S.; Wu, Y.; Sow, S.O.; Sur, D.; Breiman, R.F.; et al. Burden and aetiology of diarrhoeal disease in infants and young children in developing countries (the global enteric multicenter study, gems): A prospective, case-control study. Lancet 2013, 382, 209–222. [Google Scholar] [CrossRef]
- Ankarklev, J.; Hestvik, E.; Lebbad, M.; Lindh, J.; Kaddu-Mulindwa, D.H.; Andersson, J.O.; Tylleskar, T.; Tumwine, J.K.; Svard, S.G. Common coinfections of Giardia intestinalis and helicobacter pylori in non-symptomatic ugandan children. PLoS Negl. Trop. Dis. 2012. [Google Scholar] [CrossRef] [PubMed]
- Grazioli, B.; Matera, G.; Laratta, C.; Schipani, G.; Guarnieri, G.; Spiniello, E.; Imeneo, M.; Amorosi, A.; Foca, A.; Luzza, F. Giardia lamblia infection in patients with irritable bowel syndrome and dyspepsia: A prospective study. World J. Gastroenterol. 2006, 12, 1941–1944. [Google Scholar] [PubMed]
- Mukherjee, A.K.; Chowdhury, P.; Rajendran, K.; Nozaki, T.; Ganguly, S. Association between Giardia duodenalis and coinfection with other diarrhea-causing pathogens in india. Biomed. Res. Int. 2014. [Google Scholar] [CrossRef] [PubMed]
- Bilenko, N.; Levy, A.; Dagan, R.; Deckelbaum, R.J.; El-On, Y.; Fraser, D. Does co-infection with Giardia lamblia modulate the clinical characteristics of enteric infections in young children? Eur. J. Epidemiol. 2004, 19, 877–883. [Google Scholar] [CrossRef] [PubMed]
- Hagel, I.; Cabrera, M.; Puccio, F.; Santaella, C.; Buvat, E.; Infante, B.; Zabala, M.; Cordero, R.; Di Prisco, M.C. Co-infection with ascaris lumbricoides modulates protective immune responses against Giardia duodenalis in school venezuelan rural children. Acta Trop. 2011, 117, 189–195. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Xiao, L.; Duan, L.; Ye, J.; Guo, Y.; Guo, M.; Liu, L.; Feng, Y. Concurrent infections of Giardia duodenalis, enterocytozoon bieneusi, and clostridium difficile in children during a cryptosporidiosis outbreak in a pediatric hospital in china. PLoS Negl. Trop. Dis. 2013. [Google Scholar] [CrossRef] [PubMed]
- Oberhelman, R.A.; Flores-Abuxapqui, J.; Suarez-Hoil, G.; Puc-Franco, M.; Heredia-Navarrete, M.; Vivas-Rosel, M.; Mera, R.; Gutierrez-Cogco, L. Asymptomatic salmonellosis among children in day-care centers in merida, yucatan, mexico. Pediatr. Infect. Dis. J. 2001, 20, 792–797. [Google Scholar] [CrossRef] [PubMed]
- Muhsen, K.; Cohen, D.; Levine, M.M. Can Giardia lamblia infection lower the risk of acute diarrhea among preschool children? J. Trop. Pediatr. 2014, 60, 99–103. [Google Scholar] [CrossRef] [PubMed]
- Bhavnani, D.; Goldstick, J.E.; Cevallos, W.; Trueba, G.; Eisenberg, J.N. Synergistic effects between rotavirus and coinfecting pathogens on diarrheal disease: Evidence from a community-based study in northwestern ecuador. Am. J. Epidemiol. 2012, 176, 387–395. [Google Scholar] [CrossRef] [PubMed]
- Von Allmen, N.; Christen, S.; Forster, U.; Gottstein, B.; Welle, M.; Muller, N. Acute trichinellosis increases susceptibility to Giardia lamblia infection in the mouse model. Parasitology 2006, 133, 139–149. [Google Scholar] [CrossRef] [PubMed]
- Elliott, E.; Li, Z.; Bell, C.; Stiel, D.; Buret, A.; Wallace, J.; Brzuszczak, I.; O’Loughlin, E. Modulation of host response to escherichia coli o157:H7 infection by anti-cd18 antibody in rabbits. Gastroenterology 1994, 106, 1554–1561. [Google Scholar] [PubMed]
- Johansson, M.E.; Phillipson, M.; Petersson, J.; Velcich, A.; Holm, L.; Hansson, G.C. The inner of the two muc2 mucin-dependent mucus layers in colon is devoid of bacteria. Proc. Natl. Acad. Sci. USA 2008, 105, 15064–15069. [Google Scholar] [CrossRef] [PubMed]
- Johansson, M.E.; Larsson, J.M.; Hansson, G.C. The two mucus layers of colon are organized by the muc2 mucin, whereas the outer layer is a legislator of host-microbial interactions. Proc. Natl. Acad. Sci. USA 2011, 108, 4659–4665. [Google Scholar] [CrossRef] [PubMed]
- Gum, J.R., Jr.; Hicks, J.W.; Toribara, N.W.; Siddiki, B.; Kim, Y.S. Molecular cloning of human intestinal mucin (muc2) cdna. Identification of the amino terminus and overall sequence similarity to prepro-von willebrand factor. J. Biol. Chem. 1994, 269, 2440–2446. [Google Scholar] [PubMed]
- Tanaka, S.; Mizuno, M.; Maga, T.; Yoshinaga, F.; Tomoda, J.; Nasu, J.; Okada, H.; Yokota, K.; Oguma, K.; Shiratori, Y.; et al. H. Pylori decreases gastric mucin synthesis via inhibition of galactosyltransferase. Hepatogastroenterology 2003, 50, 1739–1742. [Google Scholar] [PubMed]
- Tse, S.K.; Chadee, K. Biochemical characterization of rat colonic mucins secreted in response to entamoeba histolytica. Infect. Immun. 1992, 60, 1603–1612. [Google Scholar] [PubMed]
- Lidell, M.E.; Moncada, D.M.; Chadee, K.; Hansson, G.C. Entamoeba histolytica cysteine proteases cleave the muc2 mucin in its c-terminal domain and dissolve the protective colonic mucus gel. Proc. Natl. Acad. Sci. USA 2006, 103, 9298–9303. [Google Scholar] [CrossRef] [PubMed]
- Hasnain, S.Z.; McGuckin, M.A.; Grencis, R.K.; Thornton, D.J. Serine protease(s) secreted by the nematode trichuris muris degrade the mucus barrier. PLoS Negl. Trop. Dis. 2012. [Google Scholar] [CrossRef] [PubMed]
- Song, K.S.; Lee, W.J.; Chung, K.C.; Koo, J.S.; Yang, E.J.; Choi, J.Y.; Yoon, J.H. Interleukin-1 beta and tumor necrosis factor-alpha induce muc5ac overexpression through a mechanism involving erk/p38 mitogen-activated protein kinases-msk1-creb activation in human airway epithelial cells. J. Biol. Chem. 2003, 278, 23243–23250. [Google Scholar] [CrossRef] [PubMed]
- Smirnova, M.G.; Guo, L.; Birchall, J.P.; Pearson, J.P. Lps up-regulates mucin and cytokine mrna expression and stimulates mucin and cytokine secretion in goblet cells. Cell. Immunol. 2003, 221, 42–49. [Google Scholar] [CrossRef]
- McKenzie, G.J.; Bancroft, A.; Grencis, R.K.; McKenzie, A.N. A distinct role for interleukin-13 in th2-cell-mediated immune responses. Curr. Biol. 1998, 8, 339–342. [Google Scholar] [CrossRef]
- Heazlewood, C.K.; Cook, M.C.; Eri, R.; Price, G.R.; Tauro, S.B.; Taupin, D.; Thornton, D.J.; Png, C.W.; Crockford, T.L.; Cornall, R.J.; et al. Aberrant mucin assembly in mice causes endoplasmic reticulum stress and spontaneous inflammation resembling ulcerative colitis. PLoS Med. 2008. [Google Scholar] [CrossRef] [PubMed]
- Bergstrom, K.S.; Kissoon-Singh, V.; Gibson, D.L.; Ma, C.; Montero, M.; Sham, H.P.; Ryz, N.; Huang, T.; Velcich, A.; Finlay, B.B.; et al. Muc2 protects against lethal infectious colitis by disassociating pathogenic and commensal bacteria from the colonic mucosa. PLoS Pathog. 2010. [Google Scholar] [CrossRef] [PubMed]
- Kissoon-Singh, V.; Moreau, F.; Trusevych, E.; Chadee, K. Entamoeba histolytica exacerbates epithelial tight junction permeability and proinflammatory responses in Muc2(−/−) mice. Am. J. Pathol. 2013, 182, 852–865. [Google Scholar] [CrossRef] [PubMed]
- Hasnain, S.Z.; Wang, H.; Ghia, J.E.; Haq, N.; Deng, Y.; Velcich, A.; Grencis, R.K.; Thornton, D.J.; Khan, W.I. Mucin gene deficiency in mice impairs host resistance to an enteric parasitic infection. Gastroenterology 2010, 138, 1763–1771. [Google Scholar] [CrossRef] [PubMed]
- Larsson, J.M.; Karlsson, H.; Crespo, J.G.; Johansson, M.E.; Eklund, L.; Sjovall, H.; Hansson, G.C. Altered o-glycosylation profile of muc2 mucin occurs in active ulcerative colitis and is associated with increased inflammation. Inflamm. Bowel Dis. 2011, 17, 2299–2307. [Google Scholar] [CrossRef] [PubMed]
- Tytgat, K.M.; van der Wal, J.W.; Einerhand, A.W.; Buller, H.A.; Dekker, J. Quantitative analysis of muc2 synthesis in ulcerative colitis. Biochem. Biophys. Res. Commun. 1996, 224, 397–405. [Google Scholar] [CrossRef] [PubMed]
- Van Klinken, B.J.; van der Wal, J.W.; Einerhand, A.W.; Buller, H.A.; Dekker, J. Sulphation and secretion of the predominant secretory human colonic mucin muc2 in ulcerative colitis. Gut 1999, 44, 387–393. [Google Scholar] [CrossRef] [PubMed]
- Amulic, B.; Cazalet, C.; Hayes, G.L.; Metzler, K.D.; Zychlinsky, A. Neutrophil function: From mechanisms to disease. Annu. Rev. Immunol. 2012, 30, 459–489. [Google Scholar] [CrossRef] [PubMed]
- Van Haastert, P.J.; Devreotes, P.N. Chemotaxis: Signalling the way forward. Nat. Rev. Mol. Cell. Biol. 2004, 5, 626–634. [Google Scholar] [CrossRef] [PubMed]
- Thelen, M. Dancing to the tune of chemokines. Nat. Immunol. 2001, 2, 129–134. [Google Scholar] [CrossRef] [PubMed]
- Chin, A.C.; Parkos, C.A. Pathobiology of neutrophil transepithelial migration: Implications in mediating epithelial injury. Annu. Rev. Pathol. 2007, 2, 111–143. [Google Scholar] [PubMed]
- Madara, J.L.; Patapoff, T.W.; Gillece-Castro, B.; Colgan, S.P.; Parkos, C.A.; Delp, C.; Mrsny, R.J. 5'-adenosine monophosphate is the neutrophil-derived paracrine factor that elicits chloride secretion from t84 intestinal epithelial cell monolayers. J. Clin. Investig. 1993, 91, 2320–2325. [Google Scholar] [CrossRef] [PubMed]
- Madara, J.L.; Parkos, C.; Colgan, S.; MacLeod, R.J.; Nash, S.; Matthews, J.; Delp, C.; Lencer, W. Cl- secretion in a model intestinal epithelium induced by a neutrophil-derived secretagogue. J. Clin. Investig. 1992, 89, 1938–1944. [Google Scholar] [CrossRef] [PubMed]
- Weissmuller, T.; Campbell, E.L.; Rosenberger, P.; Scully, M.; Beck, P.L.; Furuta, G.T.; Colgan, S.P. Pmns facilitate translocation of platelets across human and mouse epithelium and together alter fluid homeostasis via epithelial cell-expressed ecto-ntpdases. J. Clin. Investig. 2008, 118, 3682–3692. [Google Scholar] [CrossRef] [PubMed]
- Cotton, J.A.; Motta, J.P.; Schenck, L.P.; Hirota, S.A.; Beck, P.L.; Buret, A.G. Giardia duodenalis infection reduces granulocyte infiltration in an in vivo model of bacterial toxin-induced colitis and attenuates inflammation in human intestinal tissue. PLoS ONE 2014. [Google Scholar] [CrossRef] [PubMed]
- Cotton, J.A.; Bhargava, A.; Ferraz, J.G.; Yates, R.M.; Beck, P.L.; Buret, A.G. Giardia duodenalis cathepsin b proteases degrade intestinal epithelial interleukin-8 and attenuate interleukin-8-induced neutrophil chemotaxis. Infect. Immun. 2014, 82, 2772–2787. [Google Scholar] [CrossRef] [PubMed]
- Bhargava, A.; Cotton, J.A.; Dixon, B.R.; Gedamu, L.; Yates, R.M.; Buret, A.G. Giardia duodenalis surface cysteine proteases induce cleavage of the intestinal epithelial cytoskeletal protein villin via myosin light chain kinase. PLoS ONE 2015. [Google Scholar] [CrossRef] [PubMed]
- Turk, V.; Stoka, V.; Vasiljeva, O.; Renko, M.; Sun, T.; Turk, B.; Turk, D. Cysteine cathepsins: From structure, function and regulation to new frontiers. Biochim. Biophys. Acta 2012, 1824, 68–88. [Google Scholar] [CrossRef] [PubMed]
- Aurrecoechea, C.; Brestelli, J.; Brunk, B.P.; Carlton, J.M.; Dommer, J.; Fischer, S.; Gajria, B.; Gao, X.; Gingle, A.; Grant, G.; et al. Giardiadb and trichdb: Integrated genomic resources for the eukaryotic protist pathogens Giardia lamblia and trichomonas vaginalis. Nucleic Acids Res. 2009, 37, D526–D530. [Google Scholar] [CrossRef] [PubMed]
- Sajid, M.; McKerrow, J.H. Cysteine proteases of parasitic organisms. Mol. Biochem. Parasitol. 2002, 120, 1–21. [Google Scholar] [CrossRef]
- DuBois, K.N.; Abodeely, M.; Sakanari, J.; Craik, C.S.; Lee, M.; McKerrow, J.H.; Sajid, M. Identification of the major cysteine protease of Giardia and its role in encystation. J. Biol. Chem. 2008, 283, 18024–18031. [Google Scholar] [CrossRef] [PubMed]
- Dou, Z.; Coppens, I.; Carruthers, V.B. Non-canonical maturation of two papain-family proteases in toxoplasma gondii. J. Biol. Chem. 2013, 288, 3523–3534. [Google Scholar] [CrossRef] [PubMed]
- Sievers, F.; Wilm, A.; Dineen, D.; Gibson, T.J.; Karplus, K.; Li, W.; Lopez, R.; McWilliam, H.; Remmert, M.; Söding, J.; et al. Fast, scalable generation of high-quality protein multiple sequence alignments using clustal omega. Mol. Syst. Biol. 2011, 7, 539. [Google Scholar] [CrossRef] [PubMed]
- Wampfler, P.B.; Faso, C.; Hehl, A.B. The cre/loxp system in Giardia lamblia: Genetic manipulations in a binucleate tetraploid protozoan. Int. J. Parasitol. 2014, 44, 497–506. [Google Scholar] [CrossRef] [PubMed]
- McKerrow, J.H.; Caffrey, C.; Kelly, B.; Loke, P.; Sajid, M. Proteases in parasitic diseases. Annu. Rev. Pathol. 2006, 1, 497–536. [Google Scholar] [CrossRef] [PubMed]
- Que, X.; Kim, S.H.; Sajid, M.; Eckmann, L.; Dinarello, C.A.; McKerrow, J.H.; Reed, S.L. A surface amebic cysteine proteinase inactivates interleukin-18. Infect. Immun. 2003, 71, 1274–1280. [Google Scholar] [CrossRef] [PubMed]
- Reed, S.L.; Ember, J.A.; Herdman, D.S.; DiScipio, R.G.; Hugli, T.E.; Gigli, I. The extracellular neutral cysteine proteinase of entamoeba histolytica degrades anaphylatoxins c3a and c5a. J. Immunol. 1995, 155, 266–274. [Google Scholar] [PubMed]
- Mumy, K.L.; McCormick, B.A. Events at the host-microbial interface of the gastrointestinal tract. Ii. Role of the intestinal epithelium in pathogen-induced inflammation. Am. J. Physiol. Gastrointest. Liver Physiol. 2005, 288, G854–859. [Google Scholar] [CrossRef] [PubMed][Green Version]
- McCormick, B.A.; Colgan, S.P.; Delp-Archer, C.; Miller, S.I.; Madara, J.L. Salmonella typhimurium attachment to human intestinal epithelial monolayers: Transcellular signalling to subepithelial neutrophils. J. Cell Biol. 1993, 123, 895–907. [Google Scholar] [CrossRef] [PubMed]
- McCormick, B.A.; Miller, S.I.; Carnes, D.; Madara, J.L. Transepithelial signaling to neutrophils by salmonellae: A novel virulence mechanism for gastroenteritis. Infect. Immun. 1995, 63, 2302–2309. [Google Scholar] [PubMed]
- Fisher, B.S.; Estrano, C.E.; Cole, J.A. Modeling long-term host cell-Giardia lamblia interactions in an in vitro co-culture system. PLoS ONE 2013. [Google Scholar] [CrossRef] [PubMed]
- Zareie, M.; McKay, D.M.; Kovarik, G.G.; Perdue, M.H. Monocyte/macrophages evoke epithelial dysfunction: Indirect role of tumor necrosis factor-alpha. Am. J. Physiol. 1998, 275, C932–C939. [Google Scholar] [PubMed]
- Kostman, R. Infantile genetic agranulocytosis. A review with presentation of ten new cases. Acta Paediatr. Scand. 1975, 64, 362–368. [Google Scholar] [PubMed]
- Zeidler, C.; Germeshausen, M.; Klein, C.; Welte, K. Clinical implications of ela2-, hax1-, and g-csf-receptor (csf3r) mutations in severe congenital neutropenia. Br. J. Haematol. 2009, 144, 459–467. [Google Scholar] [CrossRef] [PubMed]
- Kuijpers, T.; Lutter, R. Inflammation and repeated infections in cgd: Two sides of a coin. Cell. Mol. Life Sci. 2012, 69, 7–15. [Google Scholar] [CrossRef] [PubMed]
- Jarchum, I.; Liu, M.; Shi, C.; Equinda, M.; Pamer, E.G. Critical role for myd88-mediated neutrophil recruitment during clostridium difficile colitis. Infect. Immun. 2012, 80, 2989–2996. [Google Scholar] [CrossRef] [PubMed]
- Hasegawa, M.; Yamazaki, T.; Kamada, N.; Tawaratsumida, K.; Kim, Y.G.; Nunez, G.; Inohara, N. Nucleotide-binding oligomerization domain 1 mediates recognition of clostridium difficile and induces neutrophil recruitment and protection against the pathogen. J. Immunol. 2011, 186, 4872–4880. [Google Scholar] [CrossRef] [PubMed]
- Spehlmann, M.E.; Dann, S.M.; Hruz, P.; Hanson, E.; McCole, D.F.; Eckmann, L. Cxcr2-dependent mucosal neutrophil influx protects against colitis-associated diarrhea caused by an attaching/effacing lesion-forming bacterial pathogen. J. Immunol. 2009, 183, 3332–3343. [Google Scholar] [CrossRef] [PubMed]
- Stecher, B.; Hardt, W.D. The role of microbiota in infectious disease. Trends Microbiol. 2008, 16, 107–114. [Google Scholar] [CrossRef] [PubMed]
- Stecher, B.; Robbiani, R.; Walker, A.W.; Westendorf, A.M.; Barthel, M.; Kremer, M.; Chaffron, S.; Macpherson, A.J.; Buer, J.; Parkhill, J.; et al. Salmonella enterica serovar Typhimurium exploits inflammation to compete with the intestinal microbiota. PLoS Biol. 2007, 5, 2177–2189. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.Z.; Jellbauer, S.; Poe, A.J.; Ton, V.; Pesciaroli, M.; Kehl-Fie, T.E.; Restrepo, N.A.; Hosking, M.P.; Edwards, R.A.; Battistoni, A.; et al. Zinc sequestration by the neutrophil protein calprotectin enhances Salmonella growth in the inflamed gut. Cell Host Microbe 2012, 11, 227–239. [Google Scholar] [CrossRef] [PubMed]
- Winter, S.E.; Thiennimitr, P.; Winter, M.G.; Butler, B.P.; Huseby, D.L.; Crawford, R.W.; Russell, J.M.; Bevins, C.L.; Adams, L.G.; Tsolis, R.M.; et al. Gut inflammation provides a respiratory electron acceptor for Salmonella. Nature 2010, 467, 426–429. [Google Scholar] [CrossRef] [PubMed]
- Thiennimitr, P.; Winter, S.E.; Winter, M.G.; Xavier, M.N.; Tolstikov, V.; Huseby, D.L.; Sterzenbach, T.; Tsolis, R.M.; Roth, J.R.; Baumler, A.J. Intestinal inflammation allows Salmonella to use ethanolamine to compete with the microbiota. Proc. Natl. Acad. Sci. USA 2011, 108, 17480–17485. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Liu, B.; Sartor, R.B.; Jobin, C. Phosphatidylinositol 3-kinase-gamma signaling promotes campylobacter jejuni-induced colitis through neutrophil recruitment in mice. J. Immunol. 2013, 190, 357–365. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.A.; Ma, C.; Knodler, L.A.; Valdez, Y.; Rosenberger, C.M.; Deng, W.; Finlay, B.B.; Vallance, B.A. Toll-like receptor 4 contributes to colitis development but not to host defense during citrobacter rodentium infection in mice. Infect. Immun. 2006, 74, 2522–2536. [Google Scholar] [CrossRef] [PubMed]
- Kuhl, A.A.; Kakirman, H.; Janotta, M.; Dreher, S.; Cremer, P.; Pawlowski, N.N.; Loddenkemper, C.; Heimesaat, M.M.; Grollich, K.; Zeitz, M.; et al. Aggravation of different types of experimental colitis by depletion or adhesion blockade of neutrophils. Gastroenterology 2007, 133, 1882–1892. [Google Scholar] [CrossRef] [PubMed]
- Campbell, E.L.; Bruyninckx, W.J.; Kelly, C.J.; Glover, L.E.; McNamee, E.N.; Bowers, B.E.; Bayless, A.J.; Scully, M.; Saeedi, B.J.; Golden-Mason, L.; et al. Transmigrating neutrophils shape the mucosal microenvironment through localized oxygen depletion to influence resolution of inflammation. Immunity 2014, 40, 66–77. [Google Scholar] [CrossRef] [PubMed]
- Zindl, C.L.; Lai, J.F.; Lee, Y.K.; Maynard, C.L.; Harbour, S.N.; Ouyang, W.; Chaplin, D.D.; Weaver, C.T. Il-22-producing neutrophils contribute to antimicrobial defense and restitution of colonic epithelial integrity during colitis. Proc. Natl. Acad. Sci. USA 2013, 110, 12768–12773. [Google Scholar] [CrossRef] [PubMed]
- Morris, S.M., Jr. Arginine: Beyond protein. Am. J. Clin. Nutr. 2006, 83, 508S–512S. [Google Scholar] [PubMed]
- Das, P.; Lahiri, A.; Lahiri, A.; Chakravortty, D. Modulation of the arginase pathway in the context of microbial pathogenesis: A metabolic enzyme moonlighting as an immune modulator. PLoS Pathog. 2010. [Google Scholar] [CrossRef] [PubMed]
- Schofield, P.J.; Costello, M.; Edwards, M.R.; O’Sullivan, W.J. The arginine dihydrolase pathway is present in Giardia intestinalis. Int. J. Parasitol. 1990, 20, 697–699. [Google Scholar] [CrossRef]
- Stadelmann, B.; Merino, M.C.; Persson, L.; Svard, S.G. Arginine consumption by the intestinal parasite Giardia intestinalis reduces proliferation of intestinal epithelial cells. PLoS ONE 2012. [Google Scholar] [CrossRef] [PubMed]
- Tako, E.A.; Hassimi, M.F.; Li, E.; Singer, S.M. Transcriptomic analysis of the host response to Giardia duodenalis infection reveals redundant mechanisms for parasite control. MBio 2013, 4, e00660–e00613. [Google Scholar] [CrossRef] [PubMed]
- Andersen, Y.S.; Gillin, F.D.; Eckmann, L. Adaptive immunity-dependent intestinal hypermotility contributes to host defense against Giardia spp. Infect. Immun. 2006, 74, 2473–2476. [Google Scholar] [CrossRef] [PubMed]
- Li, E.; Zhou, P.; Singer, S.M. Neuronal nitric oxide synthase is necessary for elimination of Giardia lamblia infections in mice. J. Immunol. 2006, 176, 516–521. [Google Scholar] [CrossRef] [PubMed]
- Ringqvist, E.; Palm, J.E.; Skarin, H.; Hehl, A.B.; Weiland, M.; Davids, B.J.; Reiner, D.S.; Griffiths, W.J.; Eckmann, L.; Gillin, F.D.; et al. Release of metabolic enzymes by Giardia in response to interaction with intestinal epithelial cells. Mol. Biochem. Parasitol. 2008, 159, 85–91. [Google Scholar] [CrossRef] [PubMed]
- Ringqvist, E.; Avesson, L.; Soderbom, F.; Svard, S.G. Transcriptional changes in Giardia during host-parasite interactions. Int. J. Parasitol. 2011, 41, 277–285. [Google Scholar] [CrossRef] [PubMed]
- Lundberg, J.O.; Weitzberg, E.; Gladwin, M.T. The nitrate-nitrite-nitric oxide pathway in physiology and therapeutics. Nat. Rev. Drug Discov. 2008, 7, 156–167. [Google Scholar] [CrossRef] [PubMed]
- Salzman, A.; Denenberg, A.G.; Ueta, I.; O’Connor, M.; Linn, S.C.; Szabo, C. Induction and activity of nitric oxide synthase in cultured human intestinal epithelial monolayers. Am. J. Physiol. 1996, 270, G565–G573. [Google Scholar] [PubMed]
- Witthoft, T.; Eckmann, L.; Kim, J.M.; Kagnoff, M.F. Enteroinvasive bacteria directly activate expression of inos and no production in human colon epithelial cells. Am. J. Physiol. 1998, 275, G564–G571. [Google Scholar] [PubMed]
- Clark, I.A.; Rockett, K.A. Nitric oxide and parasitic disease. Adv. Parasitol. 1996, 37, 1–56. [Google Scholar] [PubMed]
- Fang, F.C. Perspectives series: Host/pathogen interactions. Mechanisms of nitric oxide-related antimicrobial activity. J. Clin. Investig. 1997, 99, 2818–2825. [Google Scholar] [CrossRef] [PubMed]
- James, S.L. Role of nitric oxide in parasitic infections. Microbiol. Rev. 1995, 59, 533–547. [Google Scholar] [PubMed]
- Stadelmann, B.; Hanevik, K.; Andersson, M.K.; Bruserud, O.; Svard, S.G. The role of arginine and arginine-metabolizing enzymes during Giardia—Host cell interactions in vitro. BMC Microbiol. 2013. [Google Scholar] [CrossRef] [PubMed]
- Mokrzycka, M.; Kolasa, A.; Kosierkiewicz, A.; Wiszniewska, B. Inducible nitric oxide synthase in duodenum of children with Giardia lamblia infection. Folia Histochem. Cytobiol. 2010, 48, 191–196. [Google Scholar] [CrossRef] [PubMed]
- MacMicking, J.D.; Nathan, C.; Hom, G.; Chartrain, N.; Fletcher, D.S.; Trumbauer, M.; Stevens, K.; Xie, Q.W.; Sokol, K.; Hutchinson, N.; et al. Altered responses to bacterial infection and endotoxic shock in mice lacking inducible nitric oxide synthase. Cell 1995, 81, 641–650. [Google Scholar] [CrossRef]
- McCafferty, D.M.; Mudgett, J.S.; Swain, M.G.; Kubes, P. Inducible nitric oxide synthase plays a critical role in resolving intestinal inflammation. Gastroenterology 1997, 112, 1022–1027. [Google Scholar] [CrossRef] [PubMed]
- McCafferty, D.M.; Miampamba, M.; Sihota, E.; Sharkey, K.A.; Kubes, P. Role of inducible nitric oxide synthase in trinitrobenzene sulphonic acid induced colitis in mice. Gut 1999, 45, 864–873. [Google Scholar] [CrossRef] [PubMed]
- Beck, P.L.; Xavier, R.; Wong, J.; Ezedi, I.; Mashimo, H.; Mizoguchi, A.; Mizoguchi, E.; Bhan, A.K.; Podolsky, D.K. Paradoxical roles of different nitric oxide synthase isoforms in colonic injury. Am. J. Physiol. Gastrointest. Liver Physiol. 2004, 286, G137–G147. [Google Scholar] [CrossRef] [PubMed]
- Krieglstein, C.F.; Cerwinka, W.H.; Laroux, F.S.; Salter, J.W.; Russell, J.M.; Schuermann, G.; Grisham, M.B.; Ross, C.R.; Granger, D.N. Regulation of murine intestinal inflammation by reactive metabolites of oxygen and nitrogen: Divergent roles of superoxide and nitric oxide. J. Exp. Med. 2001, 194, 1207–1218. [Google Scholar] [CrossRef] [PubMed]
- Resta-Lenert, S.; Barrett, K.E. Enteroinvasive bacteria alter barrier and transport properties of human intestinal epithelium: Role of inos and cox-2. Gastroenterology 2002, 122, 1070–1087. [Google Scholar] [CrossRef] [PubMed]
- Skinn, A.C.; MacNaughton, W.K. Nitric oxide inhibits camp-dependent cftr trafficking in intestinal epithelial cells. Am. J. Physiol. Gastrointest. Liver Physiol. 2005, 289, G739–G744. [Google Scholar] [PubMed]
- Ashida, H.; Ogawa, M.; Kim, M.; Mimuro, H.; Sasakawa, C. Bacteria and host interactions in the gut epithelial barrier. Nat. Chem. Biol. 2012, 8, 36–45. [Google Scholar] [CrossRef] [PubMed]
- Ashida, H.; Mimuro, H.; Ogawa, M.; Kobayashi, T.; Sanada, T.; Kim, M.; Sasakawa, C. Cell death and infection: A double-edged sword for host and pathogen survival. J. Cell Biol. 2011, 195, 931–942. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; Ashida, H.; Ogawa, M.; Yoshikawa, Y.; Mimuro, H.; Sasakawa, C. Bacterial interactions with the host epithelium. Cell Host Microbe 2010, 8, 20–35. [Google Scholar] [CrossRef] [PubMed]
- Buret, A.G.; Bhargava, A. Modulatory mechanisms of enterocyte apoptosis by viral, bacterial and parasitic pathogens. Crit. Rev. Microbiol. 2014, 40, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Carneiro, L.A.; Travassos, L.H.; Soares, F.; Tattoli, I.; Magalhaes, J.G.; Bozza, M.T.; Plotkowski, M.C.; Sansonetti, P.J.; Molkentin, J.D.; Philpott, D.J.; et al. Shigella induces mitochondrial dysfunction and cell death in nonmyleoid cells. Cell Host Microbe 2009, 5, 123–136. [Google Scholar] [CrossRef] [PubMed]
- Paesold, G.; Guiney, D.G.; Eckmann, L.; Kagnoff, M.F. Genes in the Salmonella pathogenicity island 2 and the Salmonella virulence plasmid are essential for Salmonella-induced apoptosis in intestinal epithelial cells. Cell. Microbiol. 2002, 4, 771–781. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.M.; Eckmann, L.; Savidge, T.C.; Lowe, D.C.; Witthoft, T.; Kagnoff, M.F. Apoptosis of human intestinal epithelial cells after bacterial invasion. J. Clin. Investig. 1998, 102, 1815–1823. [Google Scholar] [CrossRef] [PubMed]
- Clark, C.S.; Maurelli, A.T. Shigella flexneri inhibits staurosporine-induced apoptosis in epithelial cells. Infect. Immun. 2007, 75, 2531–2539. [Google Scholar] [CrossRef] [PubMed]
- Faherty, C.S.; Maurelli, A.T. Spa15 of shigella flexneri is secreted through the type iii secretion system and prevents staurosporine-induced apoptosis. Infect. Immun. 2009, 77, 5281–5290. [Google Scholar] [CrossRef] [PubMed]
- Alm, K.; Oredsson, S. Cells and polyamines do it cyclically. Essays Biochem. 2009, 46, 63–76. [Google Scholar] [CrossRef] [PubMed]
- Solaymani-Mohammadi, S.; Singer, S.M. Regulation of intestinal epithelial cell cytoskeletal remodeling by cellular immunity following gut infection. Mucosal Immunol. 2013, 6, 369–378. [Google Scholar] [CrossRef] [PubMed]
- Reuther, J.Y.; Baldwin, A.S., Jr. Apoptosis promotes a caspase-induced amino-terminal truncation of ikappabalpha that functions as a stable inhibitor of nf-kappab. J. Biol. Chem. 1999, 274, 20664–20670. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.W.; Kim, B.J.; Chung, C.W.; Jo, D.G.; Kim, I.K.; Song, Y.H.; Kwon, Y.K.; Woo, H.N.; Jung, Y.K. Caspase cleavage product lacking amino-terminus of ikappabalpha sensitizes resistant cells to tnf-alpha and trail-induced apoptosis. J. Cell. Biochem. 2002, 85, 334–345. [Google Scholar] [CrossRef] [PubMed]
- Kang, K.H.; Lee, K.H.; Kim, M.Y.; Choi, K.H. Caspase-3-mediated cleavage of the nf-kappa b subunit p65 at the nh2 terminus potentiates naphthoquinone analog-induced apoptosis. J.Biol. Chem. 2001, 276, 24638–24644. [Google Scholar] [CrossRef] [PubMed]
- Iwasaki, A.; Medzhitov, R. Toll-like receptor control of the adaptive immune responses. Nat. Immunol. 2004, 5, 987–995. [Google Scholar] [CrossRef] [PubMed]
- Reis e Sousa, C. Dendritic cells in a mature age. Nat. Rev. Immunol. 2006, 6, 476–483. [Google Scholar] [CrossRef] [PubMed]
- Rescigno, M.; Urbano, M.; Valzasina, B.; Francolini, M.; Rotta, G.; Bonasio, R.; Granucci, F.; Kraehenbuhl, J.P.; Ricciardi-Castagnoli, P. Dendritic cells express tight junction proteins and penetrate gut epithelial monolayers to sample bacteria. Nat. Immunol. 2001, 2, 361–367. [Google Scholar] [CrossRef] [PubMed]
- Chieppa, M.; Rescigno, M.; Huang, A.Y.; Germain, R.N. Dynamic imaging of dendritic cell extension into the small bowel lumen in response to epithelial cell tlr engagement. J. Exp. Med. 2006, 203, 2841–2852. [Google Scholar] [CrossRef] [PubMed]
- Kamda, J.D.; Singer, S.M. Phosphoinositide 3-kinase-dependent inhibition of dendritic cell interleukin-12 production by Giardia lamblia. Infect. Immun. 2009, 77, 685–693. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.Y.; Kim, J.; Noh, H.J.; Kim, H.P.; Park, S.J. Giardia lamblia binding immunoglobulin protein triggers maturation of dendritic cells via activation of tlr4-myd88-p38 and erk1/2 mapks. Parasite Immunol. 2014, 36, 627–646. [Google Scholar] [CrossRef] [PubMed]
- Obendorf, J.; Renner Viveros, P.; Fehlings, M.; Klotz, C.; Aebischer, T.; Ignatius, R. Increased expression of cd25, cd83, and cd86, and secretion of il-12, il-23, and il-10 by human dendritic cells incubated in the presence of toll-like receptor 2 ligands and Giardia duodenalis. Parasit Vectors 2013. [Google Scholar] [CrossRef] [PubMed]
- Banik, S.; Renner Viveros, P.; Seeber, F.; Klotz, C.; Ignatius, R.; Aebischer, T. Giardia duodenalis arginine deiminase modulates the phenotype and cytokine secretion of human dendritic cells by depletion of arginine and formation of ammonia. Infect. Immun. 2013, 81, 2309–2317. [Google Scholar] [CrossRef] [PubMed]
- Grit, G.H.; Devriendt, B.; van Coppernolle, S.; Geurden, T.; Hope, J.; Vercruysse, J.; Cox, E.; Geldhof, P.; Claerebout, E. Giardia duodenalis stimulates partial maturation of bovine dendritic cells associated with altered cytokine secretion and induction of t-cell proliferation. Parasite Immunol. 2014, 36, 157–169. [Google Scholar] [CrossRef] [PubMed]
- Gordon, S. Alternative activation of macrophages. Nat. Rev. Immunol. 2003, 3, 23–35. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Sica, A.; Sozzani, S.; Allavena, P.; Vecchi, A.; Locati, M. The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol. 2004, 25, 677–686. [Google Scholar] [CrossRef] [PubMed]
- Mills, C.D.; Kincaid, K.; Alt, J.M.; Heilman, M.J.; Hill, A.M. M-1/m-2 macrophages and the th1/th2 paradigm. J. Immunol. 2000, 164, 6166–6173. [Google Scholar] [CrossRef] [PubMed]
- Martinez, F.O.; Gordon, S. The m1 and m2 paradigm of macrophage activation: Time for reassessment. F1000Prime Rep. 2014. [Google Scholar] [CrossRef] [PubMed]
- Maloney, J.; Keselman, A.; Li, E.; Singer, S.M. Macrophages expressing arginase 1 and nitric oxide synthase 2 accumulate in the small intestine during Giardia lamblia infection. Microbes Infect. 2015, 17, 462–467. [Google Scholar] [CrossRef] [PubMed]
- Dorman, S.E.; Holland, S.M. Interferon-gamma and interleukin-12 pathway defects and human disease. Cytokine Growth Factor Rev. 2000, 11, 321–333. [Google Scholar] [CrossRef]
- Jouanguy, E.; Doffinger, R.; Dupuis, S.; Pallier, A.; Altare, F.; Casanova, J.L. Il-12 and ifn-gamma in host defense against mycobacteria and Salmonella in mice and men. Curr. Opin. Immunol. 1999, 11, 346–351. [Google Scholar] [CrossRef]
- Lathrop, S.K.; Binder, K.A.; Starr, T.; Cooper, K.G.; Chong, A.; Carmody, A.B.; Steele-Mortimer, O. Replication of Salmonella enterica serovar Typhimurium in human monocyte-derived macrophages. Infect. Immun. 2015, 83, 2661–2671. [Google Scholar] [CrossRef] [PubMed]
- Pesce, J.T.; Ramalingam, T.R.; Mentink-Kane, M.M.; Wilson, M.S.; El Kasmi, K.C.; Smith, A.M.; Thompson, R.W.; Cheever, A.W.; Murray, P.J.; Wynn, T.A. Arginase-1-expressing macrophages suppress th2 cytokine-driven inflammation and fibrosis. PLoS Pathog. 2009. [Google Scholar] [CrossRef] [PubMed]
- Salgame, P.; Yap, G.S.; Gause, W.C. Effect of helminth-induced immunity on infections with microbial pathogens. Nat. Immunol. 2013, 14, 1118–1126. [Google Scholar] [CrossRef] [PubMed]
- Boef, A.G.; May, L.; van Bodegom, D.; van Lieshout, L.; Verweij, J.J.; Maier, A.B.; Westendorp, R.G.; Eriksson, U.K. Parasitic infections and immune function: Effect of helminth infections in a malaria endemic area. Immunobiology 2013, 218, 706–711. [Google Scholar] [CrossRef] [PubMed]
- Mengesha, B.; Endris, M.; Takele, Y.; Mekonnen, K.; Tadesse, T.; Feleke, A.; Diro, E. Prevalence of malnutrition and associated risk factors among adult visceral leishmaniasis patients in northwest ethiopia: A cross sectional study. BMC Res. Notes 2014, 7, 75. [Google Scholar] [CrossRef] [PubMed]
- Adamu, H.; Wegayehu, T.; Petros, B. High prevalence of diarrhoegenic intestinal parasite infections among non-art HIV patients in fitche hospital, ethiopia. PLoS ONE 2013. [Google Scholar] [CrossRef] [PubMed]
- Boaitey, Y.A.; Nkrumah, B.; Idriss, A.; Tay, S.C. Gastrointestinal and urinary tract pathogenic infections among HIV seropositive patients at the komfo anokye teaching hospital in ghana. BMC Res Notes 2012. [Google Scholar] [CrossRef] [PubMed]
- Sanyaolu, A.O.; Oyibo, W.A.; Fagbenro-Beyioku, A.F.; Gbadegeshin, A.H.; Iriemenam, N.C. Comparative study of entero-parasitic infections among hiv sero-positive and sero-negative patients in lagos, nigeria. Acta Trop. 2011, 120, 268–272. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cotton, J.A.; Amat, C.B.; Buret, A.G. Disruptions of Host Immunity and Inflammation by Giardia Duodenalis: Potential Consequences for Co-Infections in the Gastro-Intestinal Tract. Pathogens 2015, 4, 764-792. https://doi.org/10.3390/pathogens4040764
Cotton JA, Amat CB, Buret AG. Disruptions of Host Immunity and Inflammation by Giardia Duodenalis: Potential Consequences for Co-Infections in the Gastro-Intestinal Tract. Pathogens. 2015; 4(4):764-792. https://doi.org/10.3390/pathogens4040764
Chicago/Turabian StyleCotton, James A., Christina B. Amat, and Andre G. Buret. 2015. "Disruptions of Host Immunity and Inflammation by Giardia Duodenalis: Potential Consequences for Co-Infections in the Gastro-Intestinal Tract" Pathogens 4, no. 4: 764-792. https://doi.org/10.3390/pathogens4040764
APA StyleCotton, J. A., Amat, C. B., & Buret, A. G. (2015). Disruptions of Host Immunity and Inflammation by Giardia Duodenalis: Potential Consequences for Co-Infections in the Gastro-Intestinal Tract. Pathogens, 4(4), 764-792. https://doi.org/10.3390/pathogens4040764