
Molecular Detection and Characterization of Theileria Infecting Wildebeest (Connochaetes taurinus) in the Maasai Mara National Reserve, Kenya

Abstract
:1. Introduction
2. Results
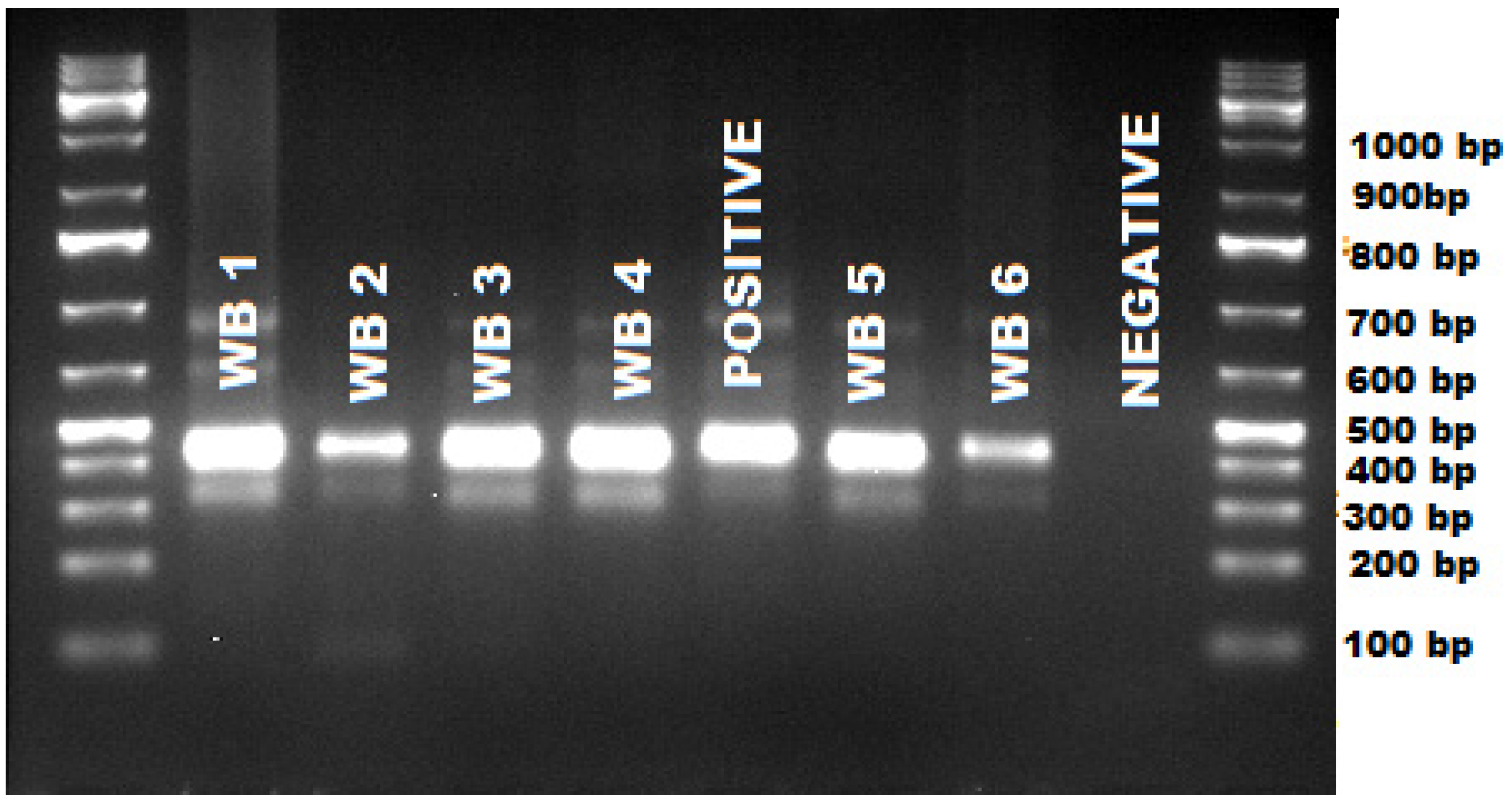

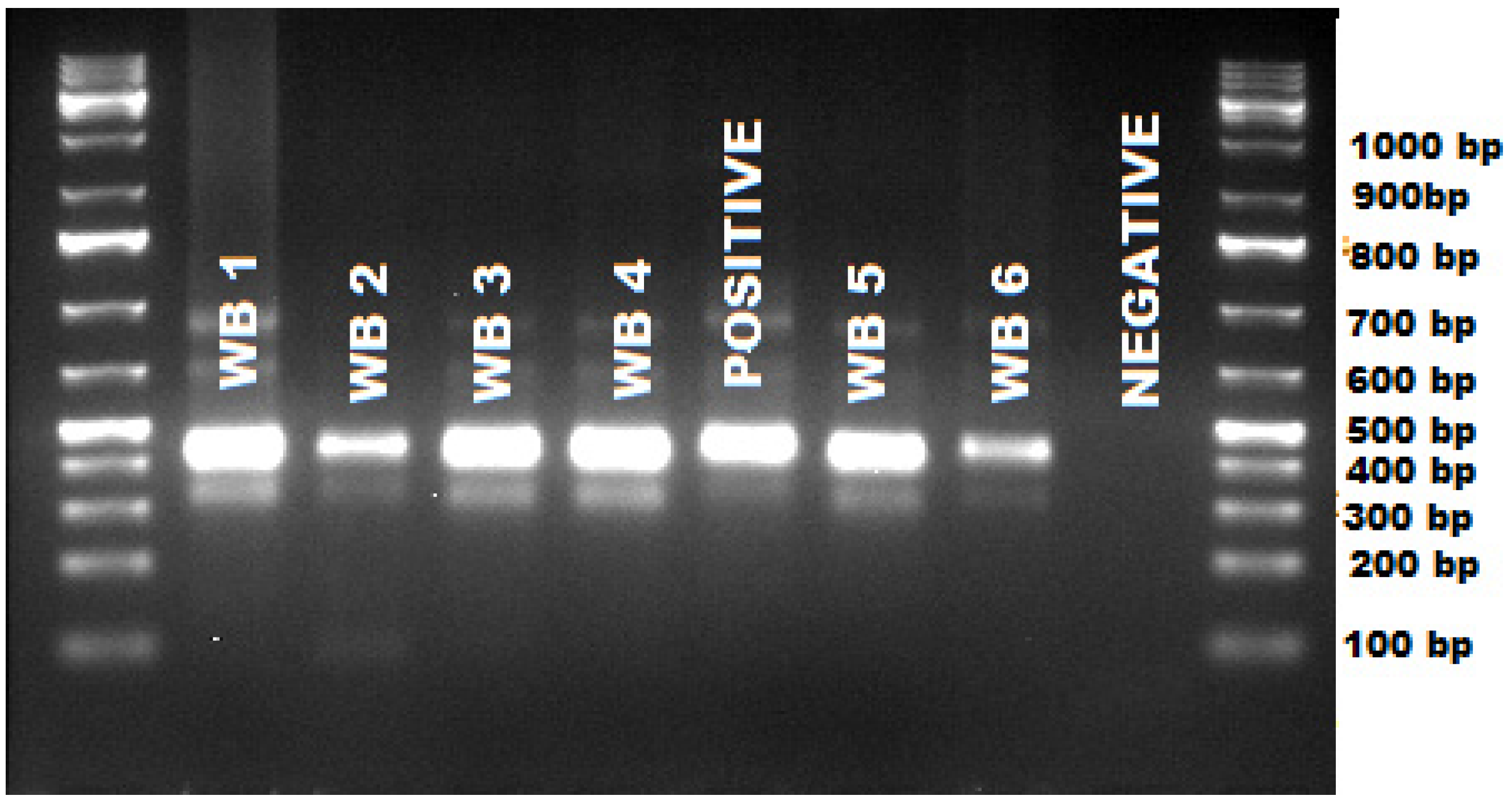{kind=link}
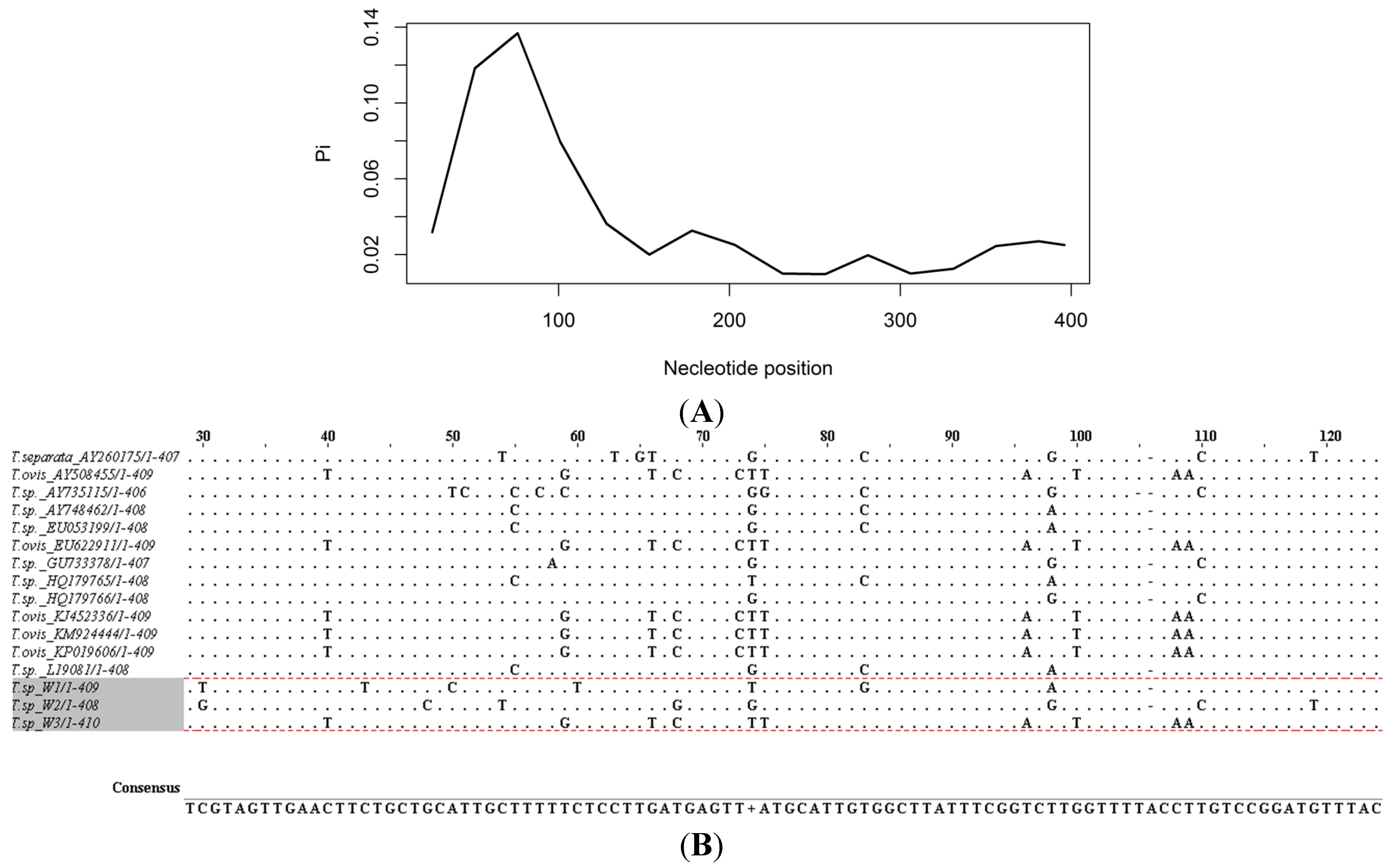
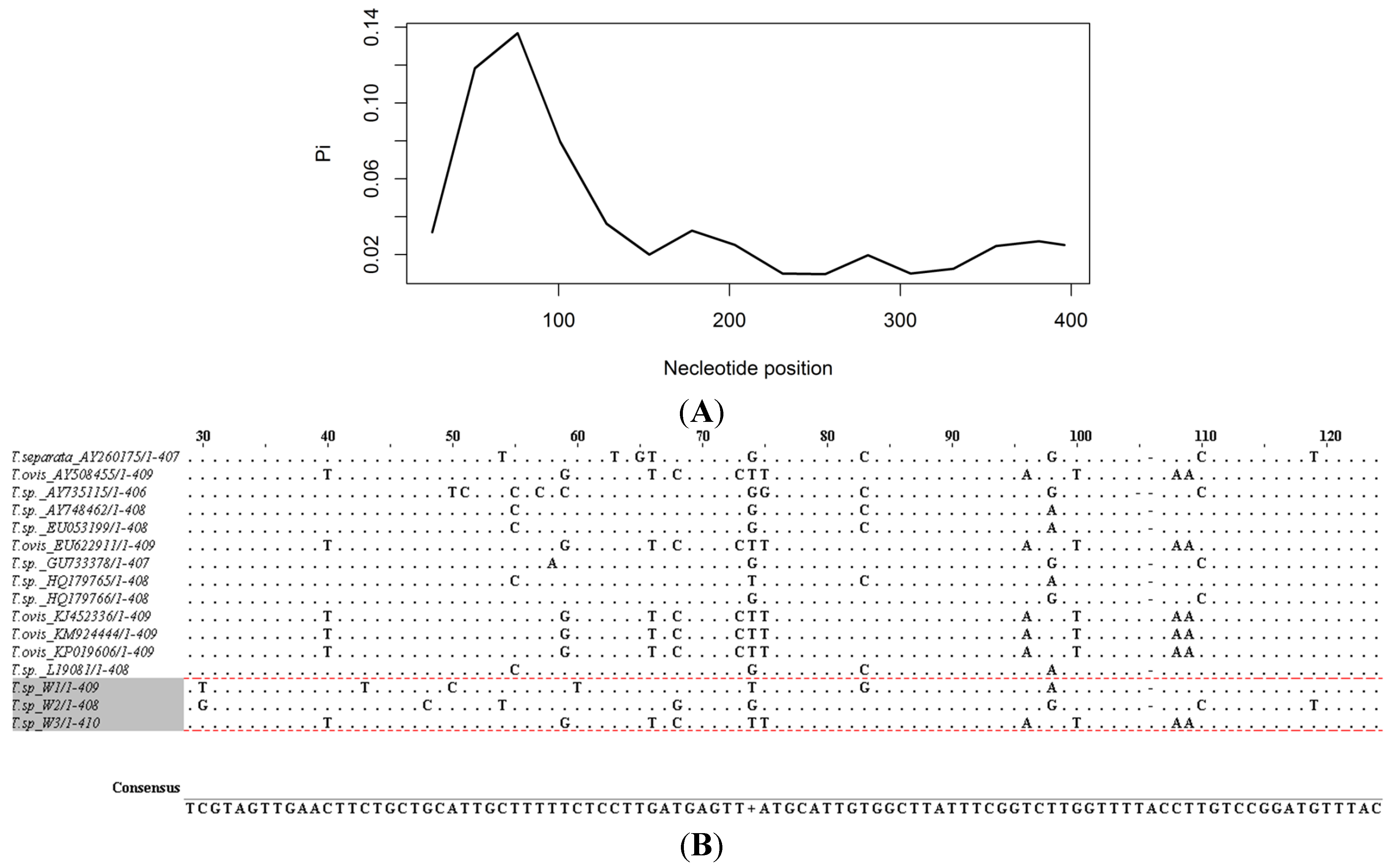{kind=link}
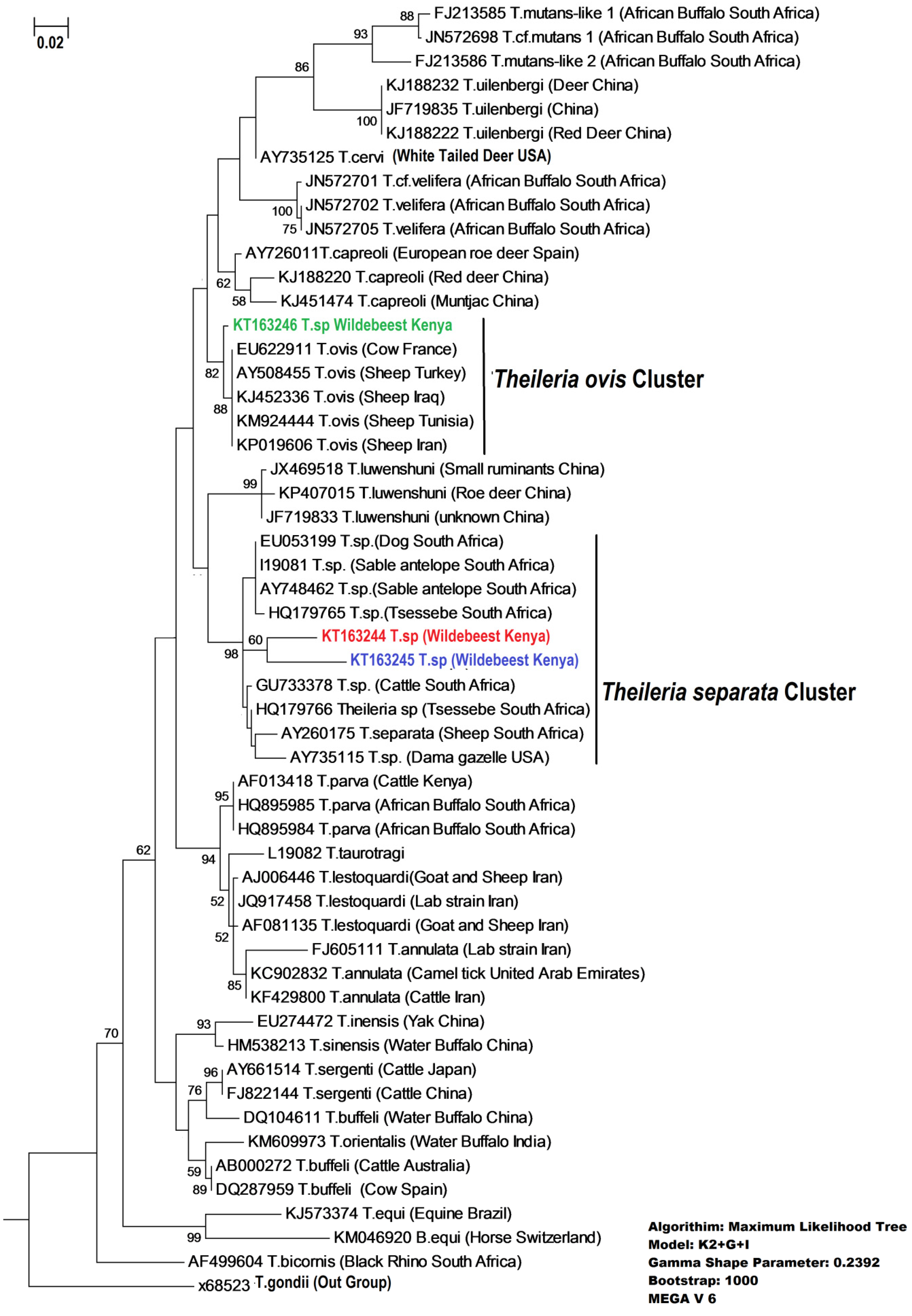
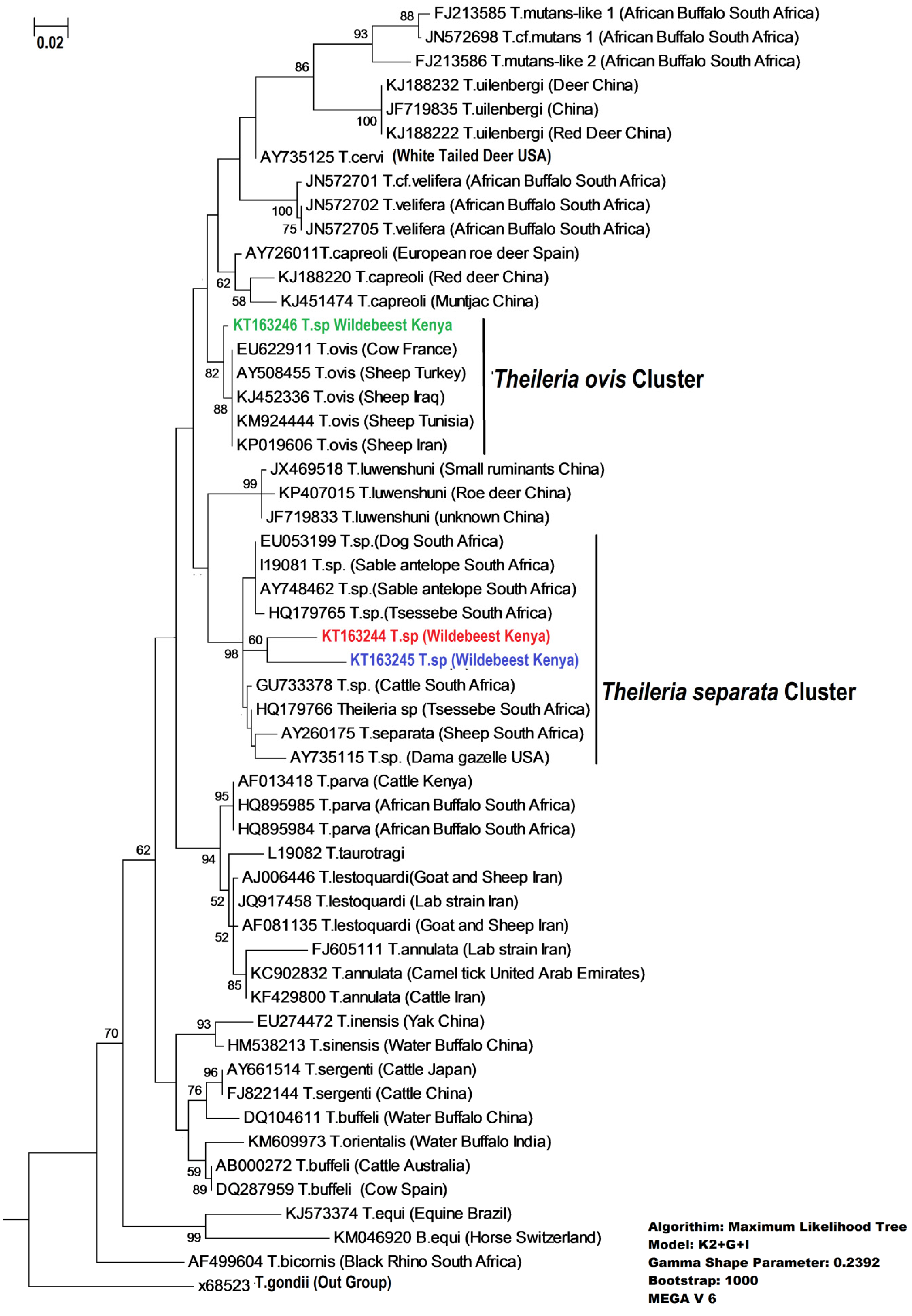{kind=link}
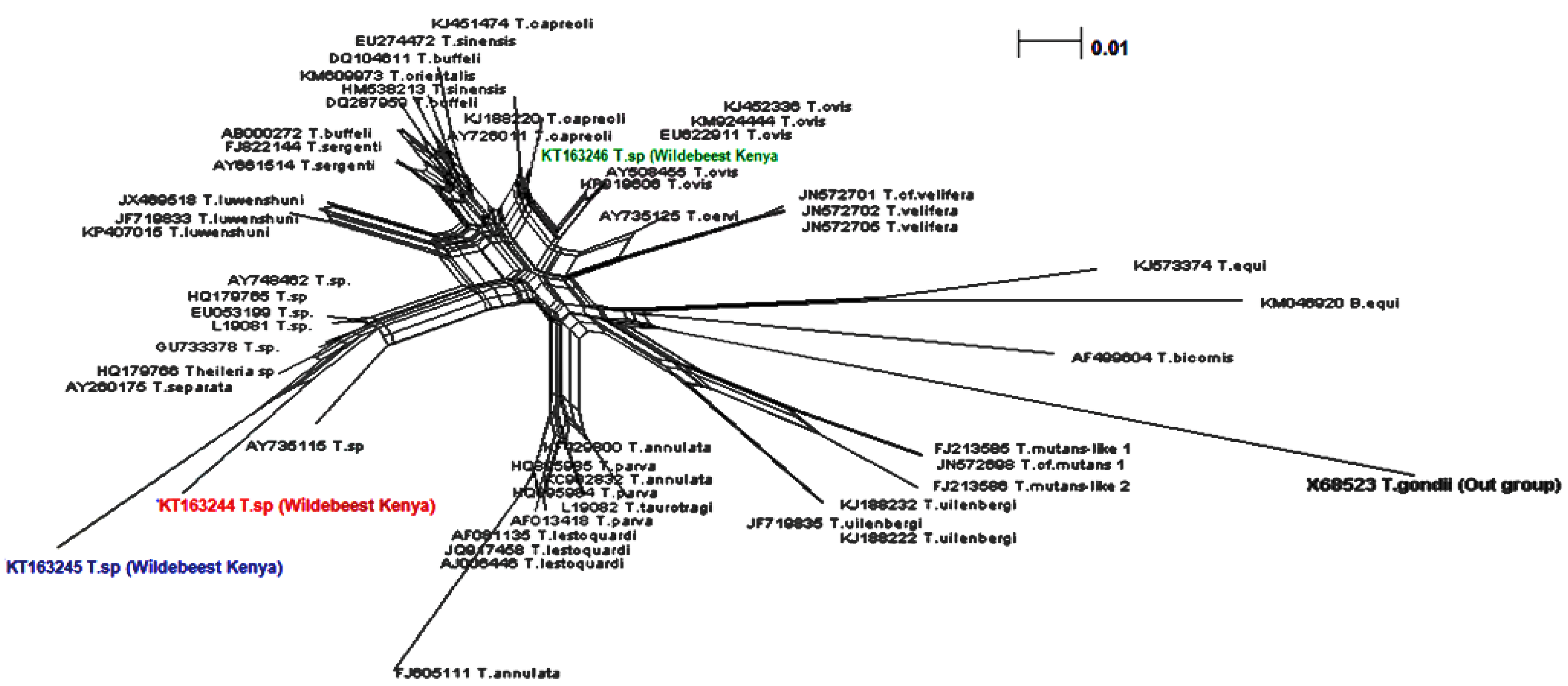
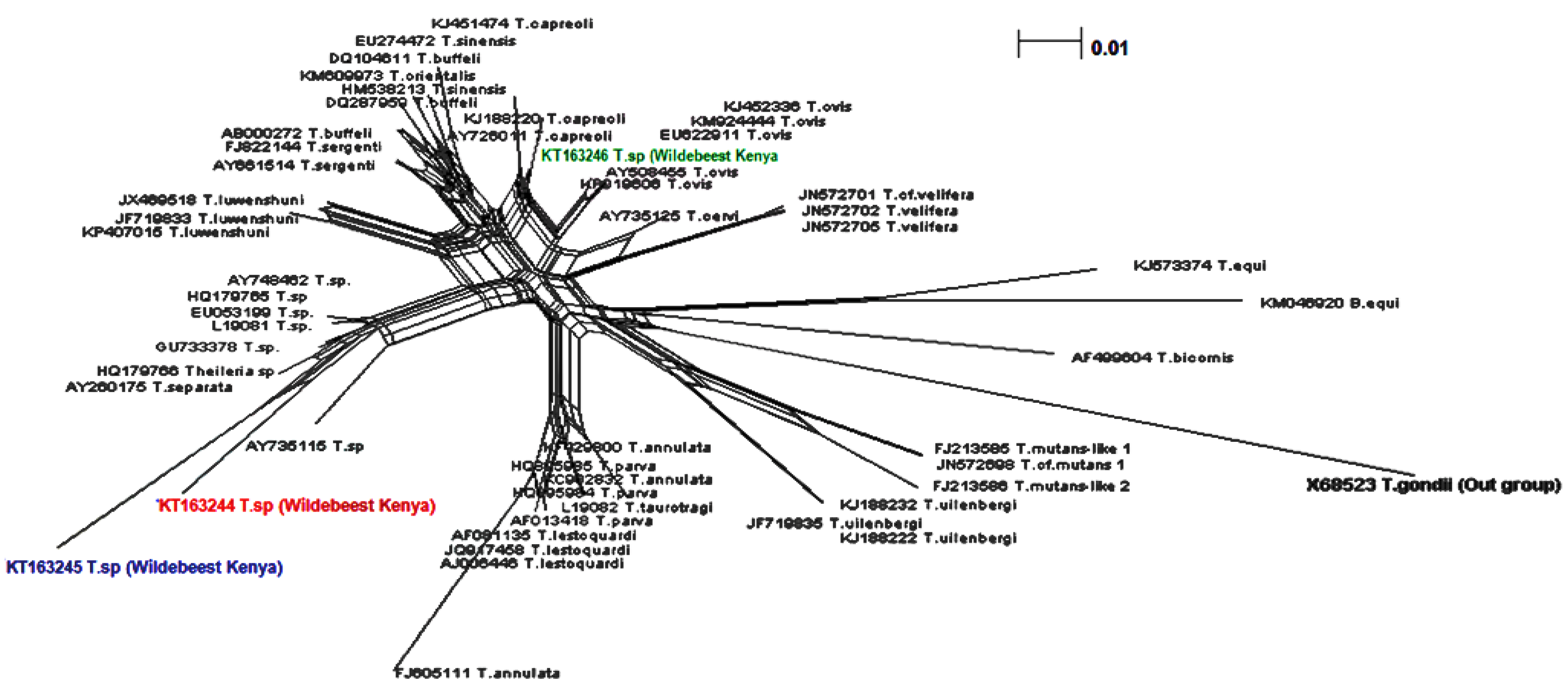{kind=link}
| Hap 1 | Hap 2 | Hap 3 | Theileria ovis | Theileria separata | |
|---|---|---|---|---|---|
| Hap 1 | 0.075 | 0.075 | 0.077 | 0.059 | |
| Hap 2 | 0.075 | 0.097 | 0.100 | 0.059 | |
| Hap 3 | 0.075 | 0.097 | 0.007 | 0.053 | |
| Theileria ovis | 0.077 | 0.100 | 0.007 | 0.056 | |
| Theileria separata | 0.059 | 0.05 | 0.053 | 0.056 |
3. Discussion
4. Materials and Methods
4.1. Ethics
4.2. The Study Area
4.3. Wildebeest Sampling
4.4. DNA Extraction and Amplification
4.5. Detection Purification and Sequencing of PCR Products
4.6. Sequence and Phylogenetic Analyses
5. Conclusions and Recommendations
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Jongejan, F.; Uilenberg, G. The global importance of ticks. Parasitology 2004, 129, S3–S14. [Google Scholar] [CrossRef] [PubMed]
- Uilenberg, G. International collaborative research: Significance of tick-borne hemoparasitic diseases to world animal health. Vet. Parasitol. 1995, 57, 19–41. [Google Scholar] [CrossRef]
- Mukhebi, A.; Perry, B.; Kruska, R. Estimated economics of theileriosis control in Africa. Prev. Vet. Med. 1992, 12, 73–85. [Google Scholar] [CrossRef]
- Bishop, R.; Musoke, A.; Morzaria, S.; Gardner, M.; Nene, V. Theileria: Intracellular protozoan parasites of wild and domestic ruminants transmitted by ixodid ticks. Parasitology 2004, 129, S271–S283. [Google Scholar] [CrossRef] [PubMed]
- Githaka, N.; Konnai, S.; Skilton, R.; Kariuki, E.; Kanduma, E.; Murata, S.; Ohashi, K. Genotypic variations in field isolates of Theileria species infecting giraffes (Giraffa camelopardalis tippelskirchi and Giraffa camelopardalis reticulata) in Kenya. Parasitol. Int. 2013, 62, 448–453. [Google Scholar] [CrossRef] [PubMed]
- Mans, B.J.; Pienaar, R.; Latif, A.A.; Potgieter, F.T. Diversity in the 18s ssu rRNA v4 hyper-variable region of Theileria spp. In Cape buffalo (Syncerus caffer) and cattle from Southern Africa. Parasitology 2011, 138, 766–779. [Google Scholar] [CrossRef] [PubMed]
- Young, A.; Brown, C.; Burridge, M.; Grootenhuis, J.; Kanhai, G.; Purnell, R.; Stagg, D. The incidence of theilerial parasites in East African buffalo (Syncerus caffer). Trop. Parasitol. 1978, 29, 281–288. [Google Scholar]
- Burridge, M.J. The role of wild mammals in the epidemiology of bovine theilerioses in East Africa. J. Wildl. Dis. 1975, 11, 68–75. [Google Scholar] [CrossRef] [PubMed]
- Otranto, D.; Cantacessi, C.; Pfeffer, M.; Dantas-Torres, F.; Brianti, E.; Deplazes, P.; Genchi, C.; Guberti, V.; Capelli, G. The role of wild canids and felids in spreading parasites to dogs and cats in Europe: Part 1: Protozoa and tick-borne agents. Vet. Parasitol. 2015, in press. [Google Scholar] [CrossRef] [PubMed]
- Hooge, J.; Howe, L.; Ezenwa, V.O. Identification of novel Theileria genotypes from grant’s gazelle. Int. J. Parasitol. 2015, 4, 239–243. [Google Scholar] [CrossRef] [PubMed]
- Githaka, N.; Konnai, S.; Bishop, R.; Odongo, D.; Lekolool, I.; Kariuki, E.; Gakuya, F.; Kamau, L.; Isezaki, M.; Murata, S.; et al. Identification and sequence characterization of novel Theileria genotypes from the waterbuck (Kobus defassa) in a Theileria parva-endemic area in Kenya. Vet. Parasitol. 2014, 202, 180–193. [Google Scholar] [CrossRef] [PubMed]
- Oosthuizen, M.C.; Allsopp, B.A.; Troskie, M.; Collins, N.E.; Penzhorn, B.L. Identification of novel Babesia and Theileria species in South African giraffe (Giraffa camelopardalis, Linnaeus, 1758) and roan antelope (Hippotragus equinus, Desmarest 1804). Vet. Parasitol. 2009, 163, 39–46. [Google Scholar] [CrossRef] [PubMed]
- Bhoora, R.; Buss, P.; Guthrie, A.J.; Penzhorn, B.L.; Collins, N.E. Genetic diversity of piroplasms in plains zebra (Equus quagga burchellii) and Cape mountain zebra (Equus zebra zebra) in south Africa. Vet. Parasitol. 2010, 174, 145–149. [Google Scholar] [CrossRef] [PubMed]
- Hawkins, E.; Kock, R.; McKeever, D.; Gakuya, F.; Musyoki, C.; Chege, S.M.; Mutinda, M.; Kariuki, E.; Davidson, Z.; Low, B.; et al. Prevalence of Theileria equi and Babesia caballi as well as the identification of associated ticks in sympatric Grevy’s zebras (Equus grevyi) and donkeys (Equus africanus asinus) in northern Kenya. J. Wildl. Dis. 2015, 51, 137–147. [Google Scholar] [CrossRef] [PubMed]
- Otiende, M.; Kivata, M.; Jowers, M.; Makumi, J.; Runo, S.; Obanda, V.; Gakuya, F.; Mutinda, M.; Kariuki, L.; Alasaad, S. Three novel haplotypes of Theileria bicornis in black and white rhinoceros in Kenya. Transbound. Emerg. Dis. 2014. [Google Scholar] [CrossRef] [PubMed]
- Holdo, R.M.; Holt, R.D.; Fryxell, J.M. Opposing rainfall and plant nutritional gradients best explain the wildebeest migration in the Serengeti. Am. Nat. 2009, 173, 431–445. [Google Scholar] [CrossRef] [PubMed]
- Williamson, D.; Williamson, J.; Ngwamotsoko, K. Wildebeest migration in the Kalahari. Afr. J. Ecol. 1988, 26, 269–280. [Google Scholar] [CrossRef]
- Cornell, S.; Isham, V.; Smith, G.; Grenfell, B. Spatial parasite transmission, drug resistance, and the spread of rare genes. Proc. Natl. Acad. Sci. 2003, 100, 7401–7405. [Google Scholar] [CrossRef] [PubMed]
- Morgan, E.; Medley, G.; Torgerson, P.; Shaikenov, B.; Milner-Gulland, E. Parasite transmission in a migratory multiple host system. Ecol. Model. 2007, 200, 511–520. [Google Scholar] [CrossRef]
- Cleaveland, S.; Kusiluka, L.; Ole Kuwai, J.; Bell, C.; Kazwala, R. Assessing the Impact of Malignant Catarrhal Fever in Ngorongoro District, Tanzania. Available online: http://www.eldis.org/fulltext/cape_new/MCF_Maasai_Tanzania.pdf (accessed on 11 August 2015).
- Hudson, P.J.; Dobson, A.P.; Newborn, D. Prevention of population cycles by parasite removal. Science 1998, 282, 2256–2258. [Google Scholar] [CrossRef] [PubMed]
- Albon, S.D.; Stien, A.; Irvine, R.J.; Langvatn, R.; Ropstad, E.; Halvorsen, O. The role of parasites in the dynamics of a reindeer population. Proc. R. Soc. B Biol. Sci. 2002, 269, 1625–1632. [Google Scholar] [CrossRef] [PubMed]
- Craig, B.H.; Pilkington, J.G.; Pemberton, J.M. Gastrointestinal nematode species burdens and host mortality in a feral sheep population. Parasitology 2006, 133, 485–496. [Google Scholar] [CrossRef] [PubMed]
- Grobler, J. Parasites and mortality of sable Hippotragus niger niger (Harris, 1838) in the matopos, zimbabwe. Koedoe 1981, 24, 119–123. [Google Scholar] [CrossRef]
- Gulland, F.M.D. The role of nematode parasites in soay sheep (Ovis aries L.) mortality during a population crash. Parasitology 1992, 105, 493–503. [Google Scholar] [CrossRef] [PubMed]
- Nijhof, A.M.; Pillay, V.; Steyl, J.; Prozesky, L.; Stoltsz, W.H.; Lawrence, J.A.; Penzhorn, B.L.; Jongejan, F. Molecular characterization of Theileria species associated with mortality in four species of African antelopes. J. Clin. Microbiol. 2005, 43, 5907–5911. [Google Scholar] [CrossRef] [PubMed]
- Wilson, D.; Bartsch, R.; Bigalke, R.; Thomas, S.E. Observations on mortality rates and disease in roan and sable antelope on nature reserves in the Transvaal. J. S. Afr. Wildl. Manag. Assoc. 1974, 4, 203–206. [Google Scholar]
- Govender, D.; Oosthuisen, M.; Penzhorn, B.L. Piroplasm parasites of white rhinoceroses (Ceratotherium simum) in the Kruger National Park, and their relation to anaemia. J. S. Afr. Vet. Assoc. 2011, 82, 36–40. [Google Scholar] [CrossRef] [PubMed]
- Lampen, F.; Bhoora, R.; Collins, N.E.; Penzhorn, B.L. Putative clinical piroplasmosis in a burchell’s zebra (Equus quagga burchelli): Clinical communication. J. S. Afr. Vet. Assoc. 2009, 80, 257–260. [Google Scholar] [CrossRef] [PubMed]
- Nijhof, A.M.; Penzhorn, B.L.; Lynen, G.; Mollel, J.O.; Morkel, P.; Bekker, C.P.J.; Jongejan, F. Babesia bicornis sp. nov. and Theileria bicornis sp. nov.: Tick-borne parasites associated with mortality in the black rhinoceros (Diceros bicornis). J. Clin. Microbiol. 2003, 41, 2249–2254. [Google Scholar] [CrossRef] [PubMed]
- Wilson, D.E.; Hirst, S.M. Ecology and factors limiting roan and sable antelope populations in South Africa. Wildl. Monogr. 1977, 54, 3–111. [Google Scholar]
- Criado-Fornelio, A.; Gónzalez-del-Rı́, M.; Buling-Sarana, A.; Barba-Carretero, J. The “expanding universe” of piroplasms. Vet. Parasitol. 2004, 119, 337–345. [Google Scholar] [CrossRef] [PubMed]
- Nagore, D.; Garcı́a-Sanmartı́n, J.; Garcı́a-Pérez, A.L.; Juste, R.A.; Hurtado, A. Detection and identification of equine Theileria and Babesia species by reverse line blotting: Epidemiological survey and phylogenetic analysis. Vet. Parasitol. 2004, 123, 41–54. [Google Scholar] [CrossRef] [PubMed]
- Oura, C.; Tait, A.; Asiimwe, B.; Lubega, G.; Weir, W. Haemoparasite prevalence and Theileria parva strain diversity in Cape buffalo (Syncerus caffer) in Uganda. Vet. Parasitol. 2011, 175, 212–219. [Google Scholar] [CrossRef] [PubMed]
- Schnittger, L.; Yin, H.; Gubbels, M.; Beyer, D.; Niemann, S.; Jongejan, F.; Ahmed, J. Phylogeny of sheep and goat Theileria and Babesia parasites. Parasitol. Res. 2003, 91, 398–406. [Google Scholar] [PubMed]
- Razmi, G.; Yaghfoori, S. Molecular surveillance of Theileria ovis, Theileria lestoquardi and Theileria annulata infection in sheep and ixodid ticks in Iran. Onderstepoort J. Vet. Res. 2013, 80, 01–05. [Google Scholar] [CrossRef] [PubMed]
- Serneels, S.; Lambin, E.F. Impact of land-use changes on the wildebeest migration in the northern part of the Serengeti-Mara ecosystem. J. Biogeogr. 2001, 28, 391–407. [Google Scholar] [CrossRef]
- Homewood, K.; Lambin, E.F.; Coast, E.; Kariuki, A.; Kikula, I.; Kivelia, J.; Said, M.; Serneels, S.; Thompson, M. Long-term changes in Serengeti-Mara wildebeest and land cover: Pastoralism, population, or policies? Proc. Natl. Acad. Sci. 2001, 98, 12544–12549. [Google Scholar] [CrossRef] [PubMed]
- Barnard, B.J.H.; van der Lugt, J.; Mushi, E.Z. Malignant catarrhal fever. In Infectious Diseases of Livestock with Special Reference to Southern Africa; Coetzer, J., Thomson, G., Tustin, R., Eds.; Oxford University Press Southern Africa: Cape Town, South Africa; Oxford, UK, 1994; Volume 1, pp. 947–957. [Google Scholar]
- Oura, C.A.L.; Tait, A.; Asiimwe, B.; Lubega, G.W.; Weir, W. Theileria parva genetic diversity and haemoparasite prevalence in cattle and wildlife in and around Lake Mburo National Park in Uganda. Parasitol. Res. 2011, 108, 1365–1374. [Google Scholar] [CrossRef] [PubMed]
- Elisa, M.; Gwakisa, P.; Sibeko, K.; Oosthuizen, M.; Geysen, D. Molecular characterization of Theileria parva field strains derived from cattle and buffalo sympatric populations of northern Tanzania. Am. J. Res. Commun. 2014, 2, 10–22. [Google Scholar]
- Mijele, D.; Obanda, V.; Omondi, P.; Soriguer, R.C.; Gakuya, F.; Otiende, M.; Hongo, P.; Alasaad, S. Spatio-temporal distribution of injured elephants in Masai Mara and the putative negative and positive roles of the local community. PLoS ONE 2013, 8, e71179. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. Mega6: Molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. Muscle: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [PubMed]
- Gouy, M.; Guindon, S.; Gascuel, O. Seaview version 4: A multiplatform graphical user interface for sequence alignment and phylogenetic tree building. Mol. Biol. Evol. 2010, 27, 221–224. [Google Scholar] [CrossRef] [PubMed]
- Librado, P.; Rozas, J. DnaSP v5: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics 2009, 25, 1451–1452. [Google Scholar] [CrossRef] [PubMed]
- Benson, D.A.; Karsch-Mizrachi, I.; Lipman, D.J.; Ostell, J.; Sayers, E.W. Genbank. Nucleic Acids Res. 2009, 37, D26–D31. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Tamura, K.; Nei, M. Estimation of the number of nucleotide substitutions in the control region of mitochondrial DNA in humans and chimpanzees. Mol. Biol. Evol. 1993, 10, 512–526. [Google Scholar] [PubMed]
- Felsenstein, J. Confidence limits on phylogenies: An approach using the bootstrap. Evolution 1985, 39, 783–791. [Google Scholar] [CrossRef]
- Huson, D.H.; Scornavacca, C. Dendroscope 3: An interactive tool for rooted phylogenetic trees and networks. Syst. Biol. 2012, 61, 1061–1067. [Google Scholar] [CrossRef] [PubMed]
- Huson, D.H.; Bryant, D. Application of phylogenetic networks in evolutionary studies. Mol. Biol. Evol. 2006, 23, 254–267. [Google Scholar] [CrossRef] [PubMed]
- Waterhouse, A.M.; Procter, J.B.; Martin, D.M.A.; Clamp, M.; Barton, G.J. Jalview version 2—A multiple sequence alignment editor and analysis workbench. Bioinformatics 2009, 25, 1189–1191. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wamuyu, L.; Obanda, V.; Kariuki, D.; Gakuya, F.; Makanda, M.; Otiende, M.; Ommeh, S. Molecular Detection and Characterization of Theileria Infecting Wildebeest (Connochaetes taurinus) in the Maasai Mara National Reserve, Kenya. Pathogens 2015, 4, 626-638. https://doi.org/10.3390/pathogens4030626
Wamuyu L, Obanda V, Kariuki D, Gakuya F, Makanda M, Otiende M, Ommeh S. Molecular Detection and Characterization of Theileria Infecting Wildebeest (Connochaetes taurinus) in the Maasai Mara National Reserve, Kenya. Pathogens. 2015; 4(3):626-638. https://doi.org/10.3390/pathogens4030626
Chicago/Turabian StyleWamuyu, Lucy, Vincent Obanda, Daniel Kariuki, Francis Gakuya, Moni Makanda, Moses Otiende, and Sheila Ommeh. 2015. "Molecular Detection and Characterization of Theileria Infecting Wildebeest (Connochaetes taurinus) in the Maasai Mara National Reserve, Kenya" Pathogens 4, no. 3: 626-638. https://doi.org/10.3390/pathogens4030626
APA StyleWamuyu, L., Obanda, V., Kariuki, D., Gakuya, F., Makanda, M., Otiende, M., & Ommeh, S. (2015). Molecular Detection and Characterization of Theileria Infecting Wildebeest (Connochaetes taurinus) in the Maasai Mara National Reserve, Kenya. Pathogens, 4(3), 626-638. https://doi.org/10.3390/pathogens4030626