
Deploying Metagenomics to Characterize Microbial Pathogens During Outbreak of Acute Febrile Illness Among Children in Tanzania

 , , , , and
, , , , and
Abstract
1. Introduction
2. Materials and Methods
2.1. Study Design and Setting
2.2. Study Participants and Inclusion Criteria
2.3. Laboratory Procedures
2.3.1. Total Nucleic Acid Extraction, cDNA Synthesis and Microbial Enrichment
2.3.2. Genomic Library Preparation and Sequencing
2.4. Sequence Data Analysis
2.5. Statistical Analysis
3. Results
3.1. Demographic and Clinical Presentations
3.2. Genomic Data Quality Metrics
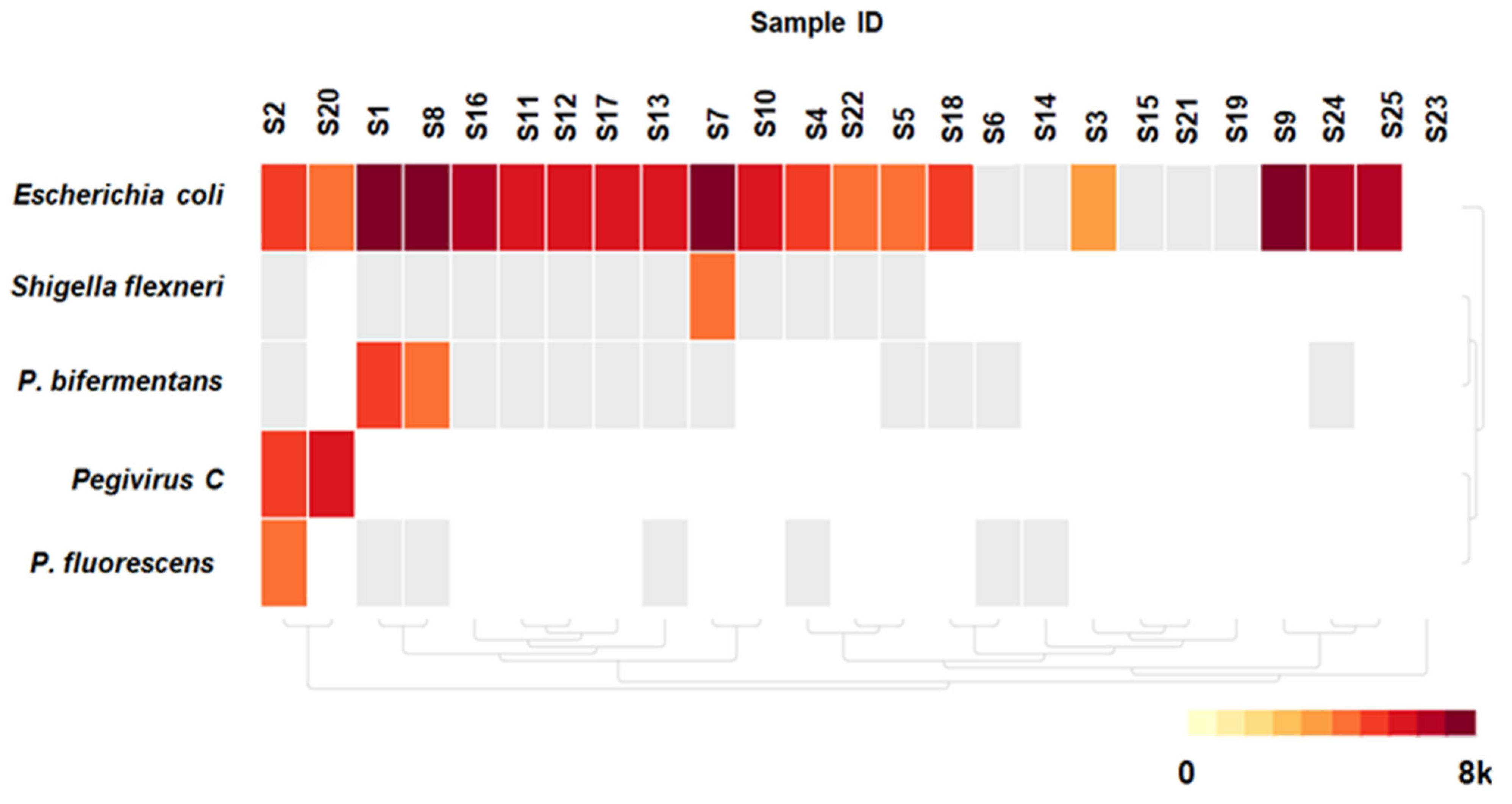
3.3. Detected Microorganisms
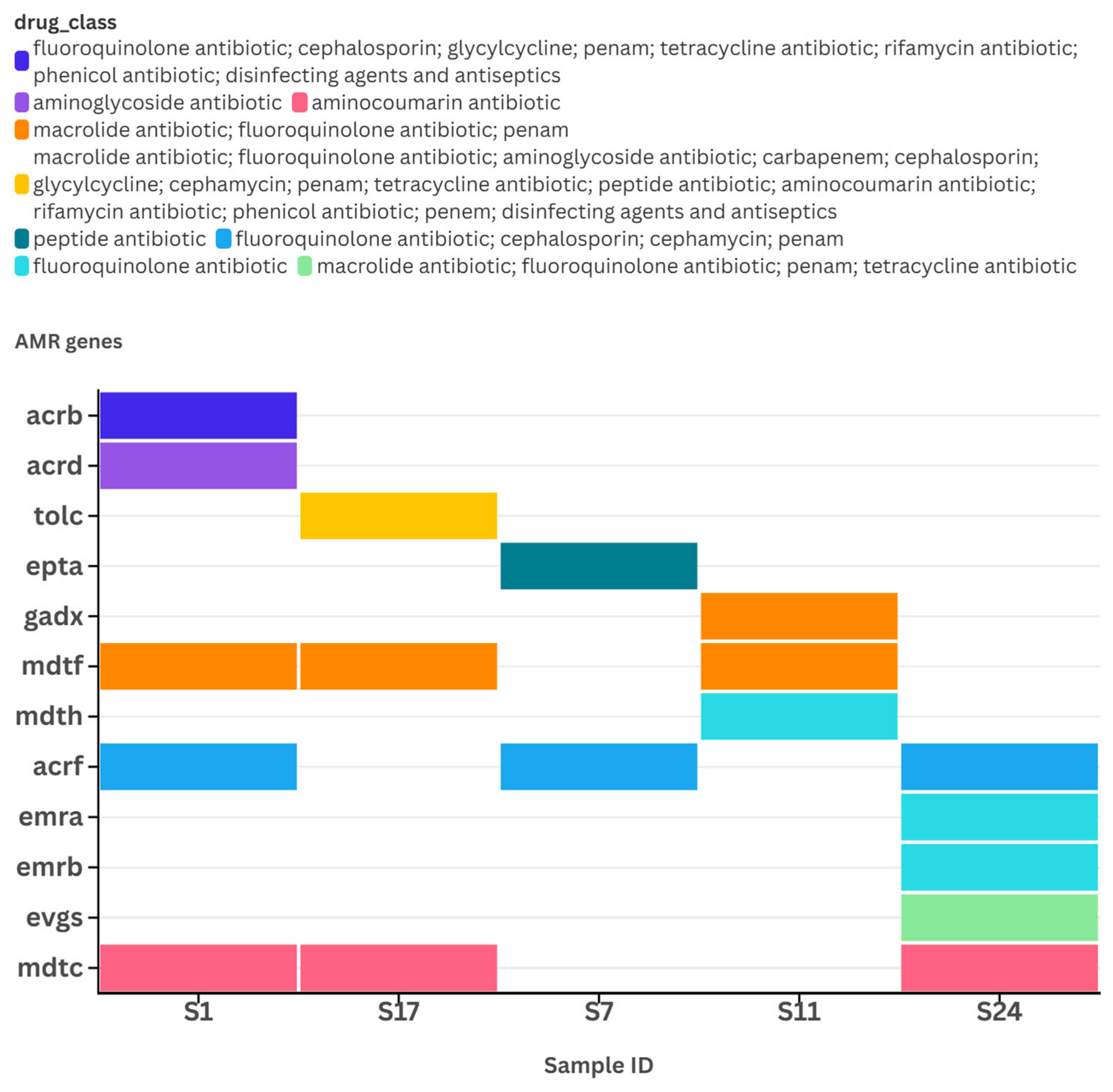
3.4. Reads Quality Metrics and Detection of Antimicrobial Resistance (AMR) Genes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AFI | Acute febrile illness |
| AMR | Antimicrobial resistance |
| cDNA | Complementary DNA |
| CZ-ID | Chan–Zuckerberg mNGS bioinformatics pipeline |
| mNGS | Metagenomic next-generation sequencing |
| PCR | Polymerase chain reaction |
| rPM | Reads per million |
References
- Ngom, R.; Gueye, A.S.; Lassieur, S.; Oloo, S.; Shahid, R.; Mize, V.; Okot, C.L.; Waogodo, J.C.; Fall, I.S. Five decades of infectious diseases outbreaks in the African region (1970–2018) a geographic snapshot. Soc. Sci. Humanit. Open 2023, 8, 100625. [Google Scholar] [CrossRef]
- Omosigho, P.O.; Okesanya, O.J.; Olaleke, N.O.; Eshun, G.; Lucero-Prisno, D.E. Multiple burden of infectious disease outbreaks: Implications for Africa healthcare system. J. Taibah Univ. Med. Sci. 2023, 18, 1446–1448. [Google Scholar] [CrossRef]
- Crump, J.A.; Morrissey, A.B.; Nicholson, W.L.; Massung, R.F.; Stoddard, R.A.; Galloway, R.L.; Ooi, E.E.; Maro, V.P.; Saganda, W.; Kinabo, G.D.; et al. Etiology of Severe Non-malaria Febrile Illness in Northern Tanzania: A Prospective Cohort Study. PLoS Neglected Trop. Dis. 2013, 7, e2324. [Google Scholar] [CrossRef]
- Maze, M.J.; Bassat, Q.; Feasey, N.A.; Mandomando, I.; Musicha, P.; Crump, J.A. The epidemiology of febrile illness in sub-Saharan Africa: Implications for diagnosis and management. Clin. Microbiol. Infect. 2018, 24, 808–814. [Google Scholar] [CrossRef]
- Crump, J.A.; Ramadhani, H.O.; Morrissey, A.B.; Saganda, W.; Mwako, M.S.; Yang, L.-Y.; Chow, S.-C.; Morpeth, S.C.; Reyburn, H.; Njau, B.N.; et al. Invasive bacterial and fungal infections among hospitalized HIV-infected and HIV-uninfected adults and adolescents in northern Tanzania. Clin. Infect. Dis. 2011, 52, 341–348. [Google Scholar] [CrossRef]
- Kajeguka, D.C.; Kaaya, R.D.; Mwakalinga, S.; Ndossi, R.; Ndaro, A.; Chilongola, J.O.; Mosha, F.W.; Schiøler, K.L.; Kavishe, R.A.; Alifrangis, M.; et al. Prevalence of dengue and chikungunya virus infections in north-eastern Tanzania: A cross sectional study among participants presenting with malaria-like symptoms. BMC Infect. Dis. 2016, 16, 183. [Google Scholar] [CrossRef]
- Vairo, F.; Mboera, L.E.G.; Nardo, P.D.; Oriyo, N.M.; Meschi, S.; Rumisha, S.F.; Colavita, F.; Mhina, A.; Carletti, F.; Mwakapeje, E.; et al. Clinical, Virologic, and Epidemiologic Characteristics of Dengue Outbreak, Dar es Salaam, Tanzania, 2014. Emerg. Infect. Dis. 2016, 22, 895–899. [Google Scholar] [CrossRef]
- Nachega, J.B.; Nsanzimana, S.; Rawat, A.; Wilson, L.A.; Rosenthal, P.J.; Siedner, M.J.; Varma, J.K.; Kilmarx, P.H.; Mutesa, L.; Tanner, M.; et al. Advancing detection and response capacities for emerging and re-emerging pathogens in Africa. Lancet Infect. Dis. 2022, 23, e185–e189. [Google Scholar] [CrossRef]
- Peters, R.P.H.; Agtmael, M.A.V.; Danner, S.A.; Savelkoul, P.H.M.; Vandenbroucke-Grauls, C.M.J.E. New developments in the diagnosis of bloodstream infections. Lancet Infect. Dis. 2004, 4, 751–760. [Google Scholar] [CrossRef]
- Faron, M.L.; Buchan, B.W.; Ledeboer, N.A. Matrix-assisted laser desorption ionization-time of flight mass spectrometry for use with positive blood cultures: Methodology, performance, and optimization. J. Clin. Microbiol. 2017, 55, 3328–3338. [Google Scholar] [CrossRef]
- Moore, C.C.; Jacob, S.T.; Banura, P.; Zhang, J.; Stroup, S.; Boulware, D.R.; Scheld, W.M.; Houpt, E.R.; Liu, J. Etiology of Sepsis in Uganda Using a Quantitative Polymerase Chain Reaction-based TaqMan Array Card. Clin. Infect. Dis. 2018, 68, 266–272. [Google Scholar] [CrossRef] [PubMed]
- Foox, J.; Tighe, S.W.; Nicolet, C.M.; Zook, J.M.; Byrska-Bishop, M.; Clarke, W.E.; Khayat, M.M.; Mahmoud, M.; Laaguiby, P.K.; Herbert, Z.T.; et al. Performance assessment of DNA sequencing platforms in the ABRF Next-Generation Sequencing Study. Nat. Biotechnol. 2021, 39, 1129–1140. [Google Scholar] [CrossRef] [PubMed]
- Culligan, E.P.; Sleator, R.D.; Marchesi, J.R.; Hill, C. Metagenomics and novel gene discovery: Promise and potential for novel therapeutics. Virulence 2014, 5, 399–412. [Google Scholar] [CrossRef]
- Hayuma, P.M.; Wang, C.W.; Liheluka, E.; Baraka, V.; Madebe, R.A.; Minja, D.T.R.; Alifrangis, M.; Lusingu, J.P.A. Prevalence of asymptomatic malaria, submicroscopic parasitaemia and anaemia in Korogwe District, north-eastern Tanzania. Malar J. 2021, 20, 424. [Google Scholar] [CrossRef]
- GCCA Korogwe, Tanga, TZ Climate Zone, Monthly Averages, Historical Weather Data. Available online: https://tcktcktck.org/tanzania/tanga/korogwe (accessed on 12 March 2023).
- Butler, D.; Mozsary, C.; Meydan, C.; Foox, J.; Rosiene, J.; Shaiber, A.; Danko, D.; Afshinnekoo, E.; MacKay, M.; Sedlazeck, F.J.; et al. Shotgun transcriptome, spatial omics, and isothermal profiling of SARS-CoV-2 infection reveals unique host responses, viral diversification, and drug interactions. Nat Commun. 2021, 12, 1660. [Google Scholar] [CrossRef]
- Feehery, G.R.; Yigit, E.; Oyola, S.O.; Langhorst, B.W.; Schmidt, V.T.; Stewart, F.J.; Dimalanta, E.T.; Amaral-Zettler, L.A.; Davis, T.; Quail, M.A.; et al. A Method for Selectively Enriching Microbial DNA from Contaminating Vertebrate Host DNA. PLoS ONE 2013, 8, e76096. [Google Scholar] [CrossRef]
- Kalantar, K.L.; Carvalho, T.; De Bourcy, C.F.A.; Dimitrov, B.; Dingle, G.; Egger, R.; Han, J.; Holmes, O.B.; Juan, Y.-F.; King, R.; et al. IDseq-An open source cloud-based pipeline and analysis service for metagenomic pathogen detection and monitoring. GigaScience 2020, 9, giaa111. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef]
- Wu, T.D.; Nacu, S. Fast and SNP-tolerant detection of complex variants and splicing in short reads. Bioinformatics 2010, 26, 873. [Google Scholar] [CrossRef]
- Ye, Y.; Choi, J.H.; Tang, H. RAPSearch: A fast protein similarity search tool for short reads. BMC Bioinform. 2011, 12, 159. [Google Scholar] [CrossRef] [PubMed]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [PubMed]
- Ministry of Health, United Republic of Tanzania. National Multi-Sectoral Cholera Prevention and Control Plan (2023–2027). Available online: https://www.moh.go.tz/storage/app/uploads/public/677/be0/2c8/677be02c88d8a451323386.pdf (accessed on 20 April 2025).
- Moreno, K.M.F.; de Andrade, V.A.; de Melo Iani, F.C.; Fonseca, V.; Lima, M.T.; de Castro Barbosa, E.; Tomé, L.M.R.; Guimarães, N.R.; Fritsch, H.M.; Adelino, T.; et al. Exploring Microorganisms Associated to Acute Febrile Illness and Severe Neurological Disorders of Unknown Origin: A Nanopore Metagenomics Approach. Genes 2024, 15, 922. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, J.F.; Laubscher, F.; Held, J.; Eckerle, I.; Docquier, M.; Grobusch, M.P.; Mordmüller, B.; Kaiser, L.; Cordey, S. Unbiased metagenomic next-generation sequencing of blood from hospitalized febrile children in Gabon. Emerg. Microbes Infect. 2020, 9, 1242–1244. [Google Scholar] [CrossRef]
- Cordey, S.; Laubscher, F.; Hartley, M.A.; Junier, T.; Keitel, K.; Docquier, M.; Guex, N.; Iseli, C.; Vieille, G.; Le Mercier, P.; et al. Blood virosphere in febrile Tanzanian children. Emerg. Microbes Infect. 2021, 10, 982–993. [Google Scholar] [CrossRef]
- Grundy, B.S.; Parikh, H.; Jacob, S.; Banura, P.; Moore, C.C.; Liu, J.; Houpt, E.R. Pathogen Detection Using Metagenomic Next-Generation Sequencing of Plasma Samples from Patients with Sepsis in Uganda. Microbiol. Spectr. 2023, 11, e0431222. [Google Scholar] [CrossRef]
- Yu, Y.; Wan, Z.; Wang, J.H.; Yang, X.; Zhang, C. Review of human pegivirus: Prevalence, transmission, pathogenesis, and clinical implication. Virulence 2022, 13, 324–341. [Google Scholar] [CrossRef]
- Li, X.Z.; Plésiat, P.; Nikaido, H. The challenge of efflux-mediated antibiotic resistance in Gram-negative bacteria. Clin. Microbiol. Rev. 2015, 28, 337–418. [Google Scholar] [CrossRef]
- Olaitan, A.O.; Morand, S.; Rolain, J.M. Mechanisms of polymyxin resistance: Acquired and intrinsic resistance in bacteria. Front Microbiol. 2014, 5, 643. [Google Scholar] [CrossRef]
- Neema, C.; Nyambura, M.; Janneth, M.; Eliudi, E.; Edwin, S.; Pascale, O.; Egyir, B. Surveillance of antimicrobial resistance in human health in Tanzania: 2016–2021. Afr. J. Lab. Med. 2023, 12, 2053. [Google Scholar] [CrossRef]
- Davies, J.; Davies, D. Origins and evolution of antibiotic resistance. Microbiol. Mol. Biol. Rev. 2010, 74, 414–433. [Google Scholar] [CrossRef] [PubMed]
- Ulomi, W.J.; Mgaya, F.X.; Kimera, Z.; Matee, M.I. Determination of Sulphonamides and Tetracycline Residues in Liver Tissues of Broiler Chicken Sold in Kinondoni and Ilala Municipalities, Dar es Salaam, Tanzania. Antibiotics 2022, 11, 1222. [Google Scholar] [CrossRef] [PubMed]
- Horumpende, P.G.; Sonda, T.B.; van Zwetselaar, M.; Antony, M.L.; Tenu, F.F.; Mwanziva, C.E.; Shao, E.R.; Mshana, S.E.; Mmbaga, B.T.; Chilongola, J.O.; et al. Prescription and non-prescription antibiotic dispensing practices in part I and part II pharmacies in Moshi Municipality, Kilimanjaro Region in Tanzania: A simulated clients approach. PLoS ONE 2018, 13, e0207465. [Google Scholar] [CrossRef] [PubMed]
- Sonda, T.B.; Horumpende, P.G.; Kumburu, H.H.; van Zwetselaar, M.; Mshana, S.E.; Alifrangis, M.; Lund, O.; Aarestrup, F.M.; Chilongola, J.O.; Mmbaga, B.T.; et al. Ceftriaxone use in a tertiary care hospital in Kilimanjaro, Tanzania: A need for a hospital antibiotic stewardship programme. PLoS ONE 2019, 14, e0220261. [Google Scholar] [CrossRef]
- United Republic of Tanzania, Ministry of Health, Ministry of Livestock and Fisheries. The National Action Plan on Antimicrobial Resistance 2023–2028. Available online: https://cdn.who.int/media/docs/default-source/antimicrobial-resistance/amr-spc-npm/nap-library/the-united-republic-of-tanzania---national-action-plan-2023--2028.pdf (accessed on 20 April 2025).
- United Republic of Tanzania, Prime Minister’s Office. National One Health Strategic Plan 2022–2027. Available online: https://www.pmo.go.tz/uploads/documents/sw-1677564782-National%20One%20Health%20Strategic%20Plan%202022%20-2027.pdf (accessed on 12 March 2023).
- Hendriksen, R.S.; Munk, P.; Njage, P.; Van Bunnik, B.; Mcnally, L.; Lukjancenko, O.; Röder, T.; Nieuwenhuijse, D.; Pedersen, S.K.; Kjeldgaard, J.; et al. Global monitoring of antimicrobial resistance based on metagenomics analyses of urban sewage. Nat. Commun. 2019, 10, 1124. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristic | Frequency | Microbial Detection by mNGS | ||
|---|---|---|---|---|
| Yes | No | |||
| Age in years | n (%) | n (%) | n (%) | * p-value |
| 1–12 | 13 (52) | 11 (57.9) | 2 (33.3) | 0.378 |
| ≥13 | 12 (48) | 8 (42.1) | 4 (66.7) | |
| Mean age [SD] | 11.6 [5] | |||
| Sex | ||||
| Male | 13 (52) | 10 (52.6) | 3 (50) | >0.99 |
| Female | 12 (48) | 9 (47.4) | 3 (50) | |
| Body temperature in °C | ||||
| 38–39 (Mild fever) | 16 (64) | 13 (68.4) | 3 (50) | 0.335 |
| >39–40 (Moderate fever) | 7 (28) | 4 (21.1) | 3 (50) | |
| >40 (High grade fever) | 2 (8) | 2 (10.5) | 0 (00) | |
| Mean temperature [SD] | 39 [0.5] | |||
| Lactic acid in mmol/L | ||||
| Normal (0.5–2.2) | 2 (8) | 1 (5.3) | 1 (16.7) | 0.430 |
| Abnormal (>2.2) | 23 (92) | 18 (94.7) | 5 (83.3) | |
| Mean lactate [SD] | 4.7 [2] | |||
| Hemoglobin (Hb) in g/dL | ||||
| >11.5 | 16 (64) | 13 (68.4) | 3 (50) | 0.233 |
| 10–11.5 | 6 (24) | 5 (26.3) | 1 (16.7) | |
| 8–<10 | 3 (12) | 1 (5.3) | 2 (33.3) | |
| Mean Hb [SD] | 12 [2] | |||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mziray, S.R.; Githinji, G.; de Laurent, Z.R.; Mbelele, P.M.; Mohammed, K.S.; Wadugu, B.D.; Grundy, B.S.; Heysell, S.K.; Mpagama, S.G.; Chilongola, J.O. Deploying Metagenomics to Characterize Microbial Pathogens During Outbreak of Acute Febrile Illness Among Children in Tanzania. Pathogens 2025, 14, 601. https://doi.org/10.3390/pathogens14060601
Mziray SR, Githinji G, de Laurent ZR, Mbelele PM, Mohammed KS, Wadugu BD, Grundy BS, Heysell SK, Mpagama SG, Chilongola JO. Deploying Metagenomics to Characterize Microbial Pathogens During Outbreak of Acute Febrile Illness Among Children in Tanzania. Pathogens. 2025; 14(6):601. https://doi.org/10.3390/pathogens14060601
Chicago/Turabian StyleMziray, Shabani Ramadhani, George Githinji, Zaydah R. de Laurent, Peter M. Mbelele, Khadija S. Mohammed, Boaz D. Wadugu, Brian S. Grundy, Scott K. Heysell, Stellah G. Mpagama, and Jaffu O. Chilongola. 2025. "Deploying Metagenomics to Characterize Microbial Pathogens During Outbreak of Acute Febrile Illness Among Children in Tanzania" Pathogens 14, no. 6: 601. https://doi.org/10.3390/pathogens14060601
APA StyleMziray, S. R., Githinji, G., de Laurent, Z. R., Mbelele, P. M., Mohammed, K. S., Wadugu, B. D., Grundy, B. S., Heysell, S. K., Mpagama, S. G., & Chilongola, J. O. (2025). Deploying Metagenomics to Characterize Microbial Pathogens During Outbreak of Acute Febrile Illness Among Children in Tanzania. Pathogens, 14(6), 601. https://doi.org/10.3390/pathogens14060601