Abstract
Protozoan parasites, such as Plasmodium, Leishmania, Toxoplasma, Cryptosporidium, and Trypanosoma, are causative agents of health-threatening diseases in both humans and animals, leading to significant health risks and socioeconomic losses globally. The development of effective therapeutic and prevention strategies for protozoan-caused diseases requires a full understanding of the pathogenesis and protective events occurring in infected hosts. Interferons (IFNs) are a family of cytokines with diverse biological effects in host antimicrobial defense and disease pathogenesis, including protozoan parasite infection. Type II IFN (IFN-γ) has been widely recognized as the essential defense cytokine in intracellular protozoan parasite infection, whereas recent studies also revealed the production and distinct function of type I and III IFNs in host defense against these parasites. Decoding the complex network of the IFN family in host–parasite interaction is critical for exploring potential new therapeutic strategies against intracellular protozoan parasite infection. Here, we review the complex effects of IFNs on the host defense against intracellular protozoan parasites and the crosstalk between distinct types of IFN signaling during infections.
1. Introduction
Interferon (IFN) was first discovered in supernatant from virus-infected cell cultures [1] and was shown to interfere with viral replication. In subsequent studies on IFN for its antiviral function, three groups of IFNs were discovered based on their distinct receptors and functions: type I (IFN-α/β/ε/κ/ω/τ/δ/ζ), which signals through the IFNAR1/2 receptor; type II (IFN-γ), which utilizes the IFNGR1/2 receptor; and type III (IFN-λs), which engages the IFNLR1 and IL-10R2 receptors [2,3,4]. Type I IFNs, encoded by distinct genes, include 13 partially homologous IFN-α subtypes in humans (14 in mice), a single IFNβ and several poorly described single gene products (IFN-ɛ, IFN-τ, IFN-κ, IFN-ω, IFN-δ and IFN-ζ) [5]. Most cell types possess the ability to produce and respond to type I IFNs, with autocrine and paracrine effects, in response to microbial stimuli such as exogenous nucleic acids [6]. IFN-γ, as the sole member of type II IFN, can only be released by immune cells, mainly T lymphocytes and natural killer (NK) cells, in response to special cytokines, such as IL-12 and IL-18, or microbial antigens through the recognition of pattern recognition receptors (PPRs) [7,8,9,10]. Type III IFNs encompass four subtypes, including IFN-λ1, IFN-λ2, IFN-λ3 and IFN-λ4, which are encoded by distinct genes closely located on human chromosome 19. The former three IFN-λs (IFN-λ1, IFN-λ2 and IFN-λ3) were simultaneously discovered by two different groups in 2003 and found to exhibit structural association with IL-10 and functional similarity with type I IFNs [2,3]. IFN-λ4 was discovered later, in 2013, through the sequencing of RNA samples derived from primary human hepatocytes treated with synthetic dsRNA mimicking hepatitis C virus infection (HCV). IFN-λ4 was identified to modulate the cell response to HCV treatment through the genetic variations within IFN-λ4 [11]. The three types of IFNs signal through distinct heterodimeric receptors, but similarly trigger relevant gene expression via the Janus kinase–signal transducer and activator of the transcription (JAK–STAT) signaling pathway (Figure 1). the type I and III IFN signaling cascades initiate from the activation of receptor-associated JAK1 and tyrosine kinase 2 (TYK2), which phosphorylates the cytosolic transcription factors, STAT1 and STAT2. The phosphorylated STAT1/STAT2 heterodimer interacts with transcription factor IFN regulatory factor 9 (IRF9) to form a tripartite signaling complex termed IFN-stimulated gene factor 3 (ISGF3) [12]. In contrast, IFN-γ activates IFNGR1/2-associated JAK1 and JAK2, leading to the phosphorylation and subsequent homodimerization of STAT1, which is the functional transcription factor in the IFN-γ signaling cascade and known as IFN-γ-activated factor (GAF) [13]. ISGF3 and GAF translocate to the nucleus and bind to specific motif IFN-stimulated response elements (ISRE) and IFN-γ-activation sites (GAS), respectively, in the promoter region of IFN-stimulated genes (ISGs), leading to the transcription of related ISGs [14]. The receptor for type III IFNs (IFNLR1/IL-10R2) is selectively expressed in epithelial cells, hepatocytes, and several immune cell types, including B lymphocytes, neutrophils, DCs, and macrophages [15], while receptors for type I and II IFNs (respectively IFNAR1/2 and IFNGR1/2) are ubiquitously expressed on nearly all nucleated cells [16].
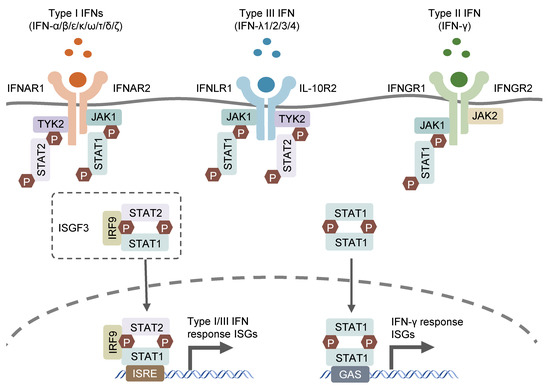
Figure 1.
Canonic IFN signaling pathways. IFNs bind to their specific receptors on cells surface and activate the subsequent JAK-STAT signaling pathways to prompt gene expression of their related ISGs.
It has generally been considered that IFN-γ is essential for anti-bacterial and anti-parasite immunity, whereas type I and type III IFNs are mainly produced and participate in anti-viral immunity [17,18,19,20,21]. Recent advances support that signaling triggered by all three IFN types results in important and distinct outcomes, including antimicrobial activity and immunomodulatory potential, which vary with respect to ISG profiles [14]. It is highly possible that the signaling pathways induced by different types of IFNs interact with each other as they employ similar signaling molecules, such as JAK and STAT1, and alter distinct but overlapping ISG expression profiles, leading to the final balance of the host response. It becomes clear that the host type I IFNs could function as a double-edged sword where they provide early resistance against acute viral infections but are detrimental to the host during certain bacterial infections and chronic viral infections [6], whereas type III IFNs may act similarly to type I IFNs in the host defense against viruses. An important role of this type of IFN has been identified in mucosal immune defense against other microbial pathogens in the intestinal and respiratory tracts, which is consistent with the distinct distribution of its receptor [17,18,19]. In addition, more than one type of IFNs can be induced by the same pathogens in the same host [15,20,21,22], which obvious complicates the situation since different types of IFNs might cause distinct effects on the pathogen–host interactions. The advances in IFN research have been reviewed extensively and will provide new insights into our understanding of the complex signaling network of various types of IFNs in the host defense against protozoan parasite infections [6,13,15,16].
2. IFN-γ and Intracellular Protozoan Parasites
IFN-γ is an important cytokine in both innate and adaptive immune responses during intracellular parasite infections (Figure 2). Elevated levels of IFN-γ is detected in both experimental animals and human patients following intracellular protozoal infections [23]. An extensive number of studies support a protective role for IFN-γ against the infection of intracellular protozoan parasites, including Plasmodium, Toxoplasma, Cryptosporidium, Trypanosoma, Leishmania, while a few studies also indicated that IFN-γ contributes to the pathogenesis of parasite infection (Table 1).
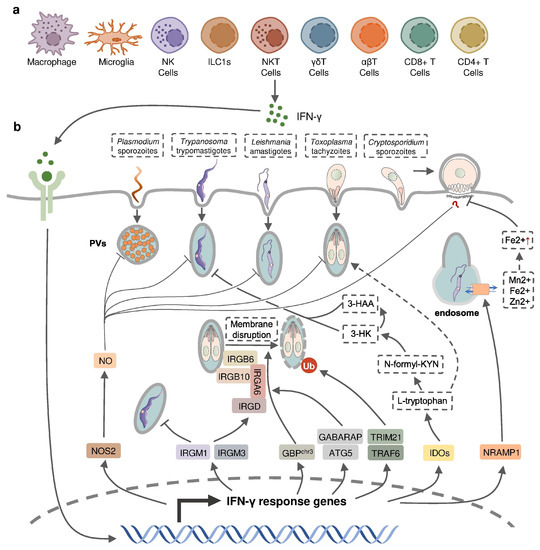
Figure 2.
IFN-γ-inducible cell-autonomous defense against intracellular protozoan parasite infections. (a) IFN-γ can be produced by multiple cell types, including immune cells, during infection and (b) acts on infected host cells to eliminate intracellular parasite by nitric oxide (NO) production, the disruption of parasitophorous vacuoles (PVs) through IFN-inducible GTPase, the restriction of ion assimilation by NRAMP1, and the inhibition of nutrient acquisition by IDOs.
2.1. IFN-γ Production in Protozoan Parasite Infection
IFN-γ production in intracellular protozoan-infected hosts is predominantly mediated by NK cells [24,25,26,27,28], and CD4+/CD8+ and Ag-specific T cells [29,30,31,32,33] in innate and adaptive immunity, respectively. Other types of immune cells have been reported to produce IFN-γ during protozoal infection (Figure 2); for example, natural killer T (NKT) cells were shown to secrete IFN-γ in the liver of P. yeolii-infected mice [34,35] and a significant proportion of γδ T cells and αβ T cells were reported to produce IFN-γ in the peripheral blood of Plasmodium-infected children [36,37,38]. CD11b+ CD45low microglia and CD11b+ CD45high blood-derived macrophages were identified as the major non-T, non-NK cells expressing IFN-γ in the brain of T. gondii-infected mice, whereas group 1 innate lymphoid cells (ILC1s) were identified to produce IFN-γ in the small intestine in response to the oral infection of T. gondii in addition to NK cells and T cells [39,40,41]. The production of IFN-γ by immune cells can be negatively regulated by anti-inflammatory Th2 cytokines such as IL-10 and IL-4 [42,43,44].

Table 1.
Effects of IFN-γ in host following intracellular protozoan parasite infections.
Table 1.
Effects of IFN-γ in host following intracellular protozoan parasite infections.
| Protective Effects | |||
| Parasite Species | Treatments and Findings | Effects | Ref. |
| P. falciparum | Recombinant IFN-γ in human hepatocyte cell culture | Hepatic schizont development ↓ | [45] |
| IFN-γ production in infected children | Occurrence of high-density and clinical episode of infection ↓ | [37] | |
| High IFN-γ level in infected patients | Occurrence of cerebral malaria ↓ | [46] | |
| P. berghei | Recombinant IFN-γ in rats, mice and human hepatocyte cell culture | Hepatic schizont development ↓ | [45] |
| Monoclonal IFN-γ neutralizing antibody in mice | Parasitemia ↑ | [47] | |
| Early IFN-γ production in infected mice | Occurrence of cerebral malaria ↓ | [48] | |
| P. vivax | Recombinant IFN-γ in chimpanzees | Parasitemia ↓ | [45] |
| P. Chabaudi | Recombinant IFN-γ in mice. | Parasitemia ↓ Intraerythrocytic parasites ↓ | [45] |
| IFN-γ−/− mice | Parasitemia ↑ | [49] | |
| P. cynomolgi | Recombinant IFN-γ in rhesus monkey | Hepatic schizont development ↓ | [45] |
| P. yoelii | Recombinant IFN-γ in mice | Parasitemia ↓ | [50] |
| IFN-γR−/− mice | Infection burden ↑ | [51] | |
| IFN-γ−/− mice | Parasitemia ↑ | [49] | |
| T. b. brucei | IFN-γ−/− mice | Parasitemia ↑ Survival time ↓ | [33] |
| T. b. rhodesiense | IFN-γ−/− mice | Parasitemia ↑ Survival time ↓ | [52] |
| L. major | IFN-γR−/− mice | Larger and progressing lesions | [53] |
| L. amazonesis | IFN-γ−/− mice | Devastating lesions in late infection stages | [54] |
| T. gondii | IFN-γ−/− mice | Survival ↓ Infection burden ↑ | [55] |
| C. parvum | Recombinant IFN-γ in intestinal enterocytes cell culture | Infection burden ↓ | [56] |
| IFN-γ−/− mice | Survival ↓ Occurrence of chronic infection ↑ | [57] | |
| Pathogenic Effects | |||
| Parasite Species | Treatments and Findings | Effects | Ref. |
| P. berghei | Late IFN-γ production in infected mice | Occurrence of cerebral malaria ↑ | [48] |
| Large amount of IFN-γ produced in infected mice | Susceptibility to cerebral malaria ↑ | [58,59] | |
| IFN-γR−/− mice | No cerebral malaria development | [60,61] | |
| IFN-γ−/− mice | No cerebral malaria development | [62,63] | |
| Monoclonal IFN-γ neutralizing antibody in mice | Occurrence of cerebral malaria ↓ Survival time ↑ | [64] | |
| P. yoelii, P. chabaudi, P. berghei | Overproduction of IFN-γ | Development of Tfh and GC B cell response ↓ Humoral immunity ↓ Autoimmune anemia ↑ | [65,66] [67,68] [69] |
| T. congolense | Overproducing IFN-γ in mice | Survival time ↓ | [70] |
| Reducing production of IFN-γ in mice | Survival time ↑ | [71] | |
| Monoclonal IFN-γ neutralizing antibody in mice | Susceptibility ↓ | [72] | |
2.2. IFN-γ in Host Defense against Protozoan Parasites
IFN-γ appears to be critical in controlling the infections of many intracellular parasites (Table 1). Exogenous IFN-γ was found to significantly diminish infections of Plasmodium in mice, rats, non-human primates, as well as in in vitro human hepatocytes, by inhibiting the parasite DNA replication during liver-stage development [45]. However, Plasmodium has also evolved a strategy to evade the host defense during liver-stage development by suppressing the expression of pro-inflammatory cytokines including IFN-γ in hepatic mononuclear cells [73]. Plasmodium-infected mice administrated with recombinant IFN-γ exhibited a suppressed blood-stage infection with the delayed onset of parasitemia, decreased levels of infected erythrocyte, and increased survival [50,74,75]. The positive effect of endogenous IFN-γ in the host defense against Plasmodium was determined from the increased susceptibility of rats treated with an anti-IFN-γ neutralizing antibody and in mice deficient in IFN-γ or the IFN-γ receptor [47,49,51]. Human studies also indicated a positive correlation between low IFN-γ production by live Plasmodium-stimulated peripheral blood mononuclear cells and the increased occurrence of symptomatic malaria as well as the risk of moderate-to-high-density P. falciparum reinfection [37]. Early IFN-γ production was shown to contribute to the protection against the development of murine cerebral malaria, the most severe neurological complication of Plasmodium infection, in P. berghei-infected mice and peripheral levels of IFN-γ were found to drop just before the onset of both human and murine cerebral malaria [46,48]. IFN-γ can be induced by malaria vaccines, as higher numbers of IFN-γ-producing T cells and increased IFN-γ level were detected in vaccine-treated subjects in several clinical trials [76,77,78]. Vaccination with chemically attenuated, asexual, blood-stage Plasmodium falciparum induces anti-parasitic cellular immune responses involving IFN-γ in Plasmodium-naïve volunteers [79]. Nevertheless, a subunit vaccine targeting Plasmodium falciparum circumsporozoite protein (CSP) activates the host immune responses dominated by parasite specific IgG antibody instead of IFN-γ [80]. Mice with a deficiency of IFN-γ or IFN-γ receptor have a higher susceptibility to L. major infection, accompanied by elevated Th2-type responses compared to the wild-type mice, but IFN-γ-deficient mice do not appear to succumb to L. amazonensis until 2 months post infection, suggesting that IFN-γ is induced at different stages of infection by diverse Leishmania species [53,54,81]. Similarly, IFN-γ or IFN-γ-receptor-deficient mice exhibited high susceptibility to infections by T. b. rhodesiense, T. b. brucei, and T. gondii [33,52,55,82]. While exogenous IFN-γ inhibits the development of C. parvum in cultured enterocytes without the need of immune effector cells, both IFN-γ-deficient and anti-IFN-γ-antibody-treated neonatal mice became more susceptible to C. parvum infection [56,57].
IFN-inducible cell-autonomous defense, including parasiticidal activity mediated by nitic oxide (NO), the disruption of parasitophorous vacuoles (PVs) related to IFN-inducible GTPase, the restriction of ion assimilation by natural resistance-associated macrophage protein 1 (NRAMP1), and the inhibition of nutrient acquisition by indoleamine 2,3-dioxygenases (IDOs), is critical for the restriction of parasite growth in infected cells and the elimination of the parasite inside the parasite-containing subcellular compartments [83] (Figure 2). Previous studies have underlined the role of nitric oxide synthase 2 (NOS2, iNOS)-NO in IFN-γ-mediated parasiticidal activity against intracellular protozoan parasites. IFN-γ-induced NO production showed the most evident parasiticidal activity against T. cruzi trypomastigotes and L. major amastigotes in IFN-γ-activated macrophages and P. falciparum as well as P. yoelii sporozoites in human and mouse hepatocytes, respectively [51,84,85,86]. Correspondingly, Nos2-deficient mice were highly susceptible to these pathogens [81,84,85]. NO production was reported to be induced by IFN-γ in hosts infected with T. gondii. Nevertheless, IFN-inducible NO might play a limited role and function at later time points in hosts infected with tachyzoites of the less virulent type II T. gondii, whereas it was essential in parasite control in virulent type I-T. gondii-strain-infected hosts [87]. C. parvum-infected mice had an increased level of NOS2 which was partially attributed to activated IFN-γ signaling [57]. A slightly longer infection period was observed in C. parvum-infected neonatal Nos2-deficient mice [88]. Nonetheless, several other studies with human enterocytes and mouse models indicated that the protective action of IFN-γ against C. parvum infection is independent of NO activity [56,89,90]. Thus, the precise mechanisms of NO-mediated antiprotozoal activity are still incompletely understood.
IFN-inducible immunity-related GTPases (IRGs) are a subfamily of IFN-γ-inducible GTPases characterized by a particular molecular weight between 21 to 47 kDa. They defend host cells against protozoa by targeting the PVs. IRGs can be divided into two groups—GKS-containing IRGs (IRGA, IRGB, IRGC or IRGD groups) and GMS-containing IRGs (IRGMs)—based on their canonical-lysine-containing (lysine, K) and non-canonical (methionine, M) G1 motifs (GxxG[K/M]S/T) within the conserved amino-terminal catalytic GTPase domain [91]. IRGM1, IRGM3 and IRGA6 enhance the IFN-γ-induced control of avirulent T. gondii strain in macrophages and astrocytes, which might account for the limited effect of NO in hosts infected with this parasite [92,93,94,95,96,97]. In host cells infected with avirulent T. gondii, IRGM proteins that act as guanine nucleotide dissociation inhibitors under an uninfected status are released from the endoplasmic reticulum which “turns on” the GKS-containing IRGs to target the PV [91]. IRGM1 also contributes to the elimination of T. cruzi infection in macrophages as IRGM1 KO macrophages displayed a defective intracellular killing of T. cruzi amastigotes [98]. A hierarchical model revealed the intrinsic order of IRGs when they are recruited to PV which indicated that IRGB6 and possibly IRGB10 bind to the vacuole before IRGA6, while IRGD is loaded last [99]. The recruitment of these molecules leads to vesiculation, membrane disruption, and sometimes necroptosis of the targeted PVs.
The other subfamily of IFN-γ-inducible GTPases, guanylate-binding proteins (GBPs), consists of a set of proteins with molecular weights between 65–73 kDa, comprising 7 and 11 members in humans and mice, respectively [100]. GBP genes are categorized into two clusters located on chromosome 3 and chromosome 5 in mice. Mice lacking GBPs on chromosome 3 (GBPchr3), including GBP1, GBP2, GBP3, GBP5 and GBP7, were highly susceptible to T. gondii infection due to the insufficient disruption of the PVs [101]. Moreover, macrophages lacking GBPchr3 showed defective loading of IRGB6 on the T. gondii PV membrane (PVM), suggesting that GBPs and IRGs coordinate with each other in PV targeting [101]. Compared with GBPchr3-deficient mice, mice deficient in GBP1 or GBP2 exhibited a milder decline of survival rates following T. gondii infection, indicating the complementary roles of each GBP on chromosome 3 in the host defense against parasite infection [102,103]. On the contrary, GBP1 was not recruited to T. cruzi compartments suggesting a protozoan specificity of GBP-mediated PV disruption [104]. The structural and biochemical cues of IFN-γ-inducible GTPase for targeting these molecules to the PV and whether membrane disruption is due to a direct effect of IRG activity, or a result of some intermediary molecules, remains unclear. Recent evidence suggests the C-terminal isoprenylation of GBP2 regulates the recruitment of GBP2 to the PVM by recognizing the ubiquitination on the PVM, which differentiates between the host membrane and the PVM [105]. E3 ligases such as TRAF6 and TRIM21 modulate ubiquitination of T. gondii PVM following IFN-γ treatment, whereas the dependent effect of these molecules on the IFN-γ-mediated elimination of T. gondii is controversial [106,107,108]. Interestingly, the distribution of GBPs in the host cytoplasm triggering the disruption of PVM is also regulated by ubiquitin-like autophagy proteins, such as autophagy-regulated gene 5 (ATG5) and GABARAP autophagy proteins, in an autophagy-independent fashion [109,110].
NRAMP1 is a highly hydrophobic integral membrane phosphoglycoprotein (~100 kD), expressed primarily in the late endosomal/early endosomal compartment of macrophages and polymorphonuclear leukocytes as a membrane transporter [111]. It has been shown to transport divalent ion cations, such as Mn2+, Fe2+ and Zn2+, to prevent the intracellular pathogens from these divalent metals essential for parasite survival [111,112]. NRAMP1 has been identified as an essential factor in the host defense against L. donovani, but the intrinsic mechanism remains unclear [113]. A previous study has revealed the correlation between cellular Fe2+ concentration and the IFN-γ-induced inhibition of C. parvum infection in intestinal enterocytes, but whether NRAMP1 is involved has not been investigated [56].
IDOs, IDO1 and IDO2, are both IFN-inducible, haem-containing oxidoreductases that degrade L-tryptophan to generate N-formylkynurenine (N-formyl-KYN) in the kynurenine pathway [114]. The removal of L-tryptophan restricted the growth of T. gondii in several IFN-γ-stimulated human cell lines including fibroblasts, lung epithelial cells, glioblastoma cells, retinal pigment epithelial cells, and macrophages [115,116,117,118,119,120,121]. Moreover, increased susceptibility to T. gondii occurred in mice with a double deficiency of IDO1 and IDO2 but not in IDO1-deficient mice, suggesting a significant role of both IDOs in the restriction of T. gondii infection in vivo [122]. IDOs may also control T. cruzi infection through the downstream kynurenine catabolites 3-hydroxykynurenine (3-HK) and 3-hydroxyanthranilic acid (3-HAA), which are likely to be harmful to T. cruzi amastigotes and trypomastigotes [123].
Additionally, IFN-γ could increase the expression of endothelial vascular cell adhesion molecule 1 (VCAM-1) to facilitate the recruitment of CD8+ T cells in the brain of mice chronically infected with T. gondii and enhance the cytotoxic potential of CD8+ T cells by inducing NO, which contributes to the host defense against parasites in the brain [40,124,125]. IFN-γ has been reported to modulate B-cell-mediated humoral immunity in Plasmodium infection via modulating the class-switching of antibody-producing B cells as IFN-γ-deficient mice produce less parasite-specific IgM, IgG3 and cytophilic IgG2a than wild-type mice [126].
2.3. IFN-γ in the Pathogenesis of Protozoan Infection
In contrast to the protective effect of IFN-γ, the response has also been reported to be involved in the pathogenesis of protozoan infection (Table 1). Although IFN-γ production as early as 24 h p.i. prevented the occurrence of experimental cerebral malaria in Plasmodium-infected mice, mice with late IFN-γ production at 3 to 4 days p.i. were found to develop severe experimental cerebral malaria [48]. IFN-γ mRNA accumulation was detected in mice susceptible to cerebral malaria [58]. The suppressed development of cerebral malaria was observed in Plasmodium-infected mice following the administration of anti-IFN-γ monoclonal antibody, treatment with IFN-γ-suppressive IL-10, inhibition of IFN-γ production, or deficiency of the IFN-γ receptor [59,60,62,64]. IFN-γ may mediate the development of experimental cerebral malaria through various mechanisms. IFN-γ, together with tumor necrosis factor (TNF) and lymphotoxin α, enhance the activation and apoptosis of the brain endothelium through the activation of endothelial cell and subsequently increased local binding of platelets [64,127,128]. IFN-γ is also necessary for the recruitment of CD8+ T cells in the brain by inducing the expression of canonical adhesion molecules, such as ICAM-1, CXCL9, and CXCL10. Accumulated CD8+ T cells mediate the immune responses against infected red blood cells sequestered in the brain and the lungs in susceptible mice, facilitating development of experimental CM [61,63]. The precise effect of IFN-γ in the development of cerebral malaria is still controversial. T. congolense highly susceptible BALB/c mice displayed significantly higher plasma IFN-γ levels compared to infected parasite-resistant C57BL/6 mice [72,129,130,131]. The IFN-γ-mediated accumulation and activation of erythrophagocytic myeloid cells led to acute anemia, liver injury, and a reduced survival time in T. brucei or T. congolense-infected BALB/c mice [70,71,132]. The overproduction of IFN-γ, induced by blocking IL-10R, shortened the survival time of both C57BL/6 and BALB/c mice following T. congolense infection [70]. T. congolense-susceptible BALB/c mice could switch to a relatively resistant-like phenotype by the neutralization of IFN-γ or by reducing the production of IFN-γ through the depletion of IL-12 during T. congolense infection [71,72]. Moreover, IFN-γ is also a crucial mediator in the humoral immunity that may exacerbate infection outcomes, leading to parasite-associated autoimmune disorders and chronic infection. Plasmodium DNA could induce autoreactive responses against erythrocytes by activating a population of B cells expressing CD11c and T-bet, in which IFN-γ acted as an essential factor, together with parasitic DNA to promote the expansion of autoreactive T-bet+ B cells, a major producer of autoantibodies promoting malarial anemia [69]. IFN-γ also contributed to the inhibition of T follicular helper cell differentiation during sever malaria infection, resulting in an impaired germ center B cell response and inefficient production of antibody-secreting plasma cells [65,66,67,68].
The precise role of IFN-γ in the host defense against protozoan parasite infection and in the pathogenesis of infection can be different during the infection of different pathogens, at different infectious stages, or in hosts with different intrinsic immune statues. Therefore, close attention to the alteration in IFN-γ levels and the IFN-γ-mediated immune response is necessary for timely adjustments of therapeutic strategies and predictions of prognosis of infection.
3. Type I IFNs and Intracellular Protozoan Parasites
Type I IFNs have been identified as critical immune regulators with diverse functions in infectious disease [6]. In contrast to supplying antiviral defenses for the host, these cytokines also exacerbate infection outcomes by suppressing the protective immune system or promoting proinflammatory events during infections, such as those caused by several viruses [133,134,135] and bacteria [136,137,138,139]. Recent studies support the double-edged functions of type I IFNs in the host defense against protozoan parasite infection.
3.1. Type I IFN Production in Protozoan Parasite Infection
The production of type I IFNs in intracellular protozoan infections involves multiple cell types through various signaling pathways (Figure 3). Hepatocytes produce type I IFNs during liver-stage P. yoelii and P. berghei infection in mice, but the molecular mechanisms involved in this induction remain controversial [34,140]. Leihl et al. demonstrated that both transcription factors interferon regulatory factor (IRF) 3 and IRF7 are critical mediators in a host type I IFN response and the specific cytosolic RNA sensor, melanoma differentiation-associated gene 5 (MDA5), recognized the pathogen-associated molecular pattern molecules (PAMPs) from P. berghei [140]. Miller et al. confirmed the involvement of IRF3 in host type I IFN production in a P. yoelii infection mouse model without the participation of IRF7 or all the known IRF3-related pattern recognition receptors (PPRs), including TLR3, TLR4, retinoic acid-inducible gene 1 (RIG-1) and MDA5 [34]. This inconsistency is probably due to the different Plasmodium species used in the infection models. During blood-stage Plasmodium infection, murine bone-marrow-derived dendritic cells (BMDCs) sensed Plasmodium RNA (P. yoelii, P. berghei, and P. falciparum) via the cytosolic RNA sensor MDA5 and subsequent adaptor protein mitochondrial antiviral signaling protein (MAVs) to activate the expression of type I IFN genes [140]. Plasmacytoid dendritic cells (pDCs; HLA-DR+ CD123+ CD304+) were identified as the major sources of type I IFNs in human blood-stage P. falciparum infection, whereas other peripheral blood mononuclear cells were also capable of producing type I IFNs [141]. Splenic red pulp macrophages generated significant quantities of type I IFNs in response to P. chabaudi infection in a TLR9-, MYD88-, and IRF7-dependent manner, in addition to plasmacytoid dendritic cells [142].
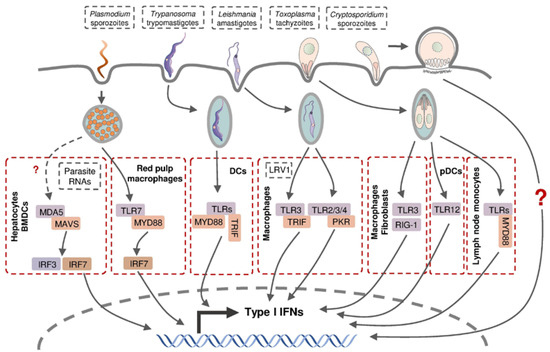
Figure 3.
Type I IFN production in host cells following intracellular parasite infection. Parasite-derived PAMPs can be recognized by intracellular PRRs and thus activate subsequent transcription factors and stimulate the expression of various type I IFN genes.
Murine macrophages and fibroblasts were reported to produce type I IFNs following infection by a few atypical T. gondii strains and the related parasite Neospora caninum, depending on endosomal TLRs, particularly TLR3, and cytoplasmic receptor RIG-1, respectively [143,144]. pDCs were able to produce low-levels of IFN-α in T. gondii-infected mice through the recognition of T. gondii profilin (TgPRF) by TLR12 [145]. Interestingly, several type I IFN-non-inducible T. gondii strains triggered type I IFN production in murine fibroblasts when they were heat-killed, but their co-infection with Neospora showed a strong inhibitory effect on type I IFN production suggesting T. gondii possesses type I IFN-inducing factors as well as the mechanism to suppress a host response to these factors [143,144]. A recent report demonstrated that IFN-β, a type I IFN, was produced by murine inflammatory monocytes in mesenteric lymph nodes following infection by T. gondii, whose induction is believed to be mediated through TLRs and Myd88 following the intake of the parasite [146].
As the major targeted cell type, macrophages were also found to produce type I IFNs in mice during infection with a non-metastatic Leishmania species L. major [147] and metastatic L. guyanensis [148], the latter of which was dependent on the TLR3-TRIF signaling pathway by recognizing parasite-derived factors, including dsRNA Leishmania RNA virus 1 (LRV1). Metastatic but non-LRV1-contained L. donovani are capable of engaging type I IFN induction via TLR2, TLR3, TLR4, and downstream protein kinase R (PKR) in murine peritoneal macrophages [149]. These findings indicate that the macrophages are capable of producing type I IFNs in response to infection with different Leishmania species through the recognition of various parasite-derived factors via distinct signaling pathways.
The presence of type I IFNs upon Trypanosoma infection was first observed in the serum from mice injected with T. cruzi intraperitoneally [150,151], which was later proved to be IFN-α protein [152]. T. cruzi-infected murine fibroblasts were shown to secrete IFN-β, consistent with the discovery of an early type I IFN response in vivo at the site of intradermal infection [153,154]. IFN-β production was detected in murine macrophages and DCs following T. cruzi infection, which was dependent on TLR-associated adaptor protein myeloid differentiation primary response 88 (MyD88) and TIR-domain-containing adapter-inducing interferon-β (TRIF) [155].
The induction of type I IFNs in Cryptosporidium-infected hosts was revealed much later than 2009, showing the production of IFN-α/β in C. parvum-infected enterocytes and DCs, but the intrinsic mechanism of the induction has not been determined [156,157,158].
3.2. Type I IFN in Host Defense against Protozoan Parasites
Type I IFN signaling was shown to be beneficial to the host defense against the infection of several protozoan parasites (Table 2). Ifnar-deficient mice, or mice with Ifnar conditionally knocked out on hepatocytes, failed to control P. yoelii and P. berghei replication in the liver and resulted in a higher parasite burden in the liver and blood of infected animals [34,140]. Type I IFNs facilitated the recruitment of NKT cells which is essential for the elimination of P. yoelii in the liver of infected mice [34]. Using Ifnar KO mice, type I IFN signaling induced by T. cruzi was shown to be required for controlling parasite growth during the acute phase of infection by activating the production of NO in infected spleen cells [159]. A study involving T. b. rhodesiense reported a beneficial effect of IFN-I during the acute infection phase as Ifnar KO mice displayed a delayed control of parasite burden and succumbed to infection earlier than wild-type controls [160]. The production of protective NOS2-drived NO induced by type I IFN signaling was also observed in L. major-infected mice, while L. major appears to have evolved strategies to evade this immune response [147]. L. major infection induces the expression of a translational repressor, macrophage eukaryotic translation initiation factor 4E binding protein 1 (4E-BP1), and subsequently represses type I IFN production [161,162]. Moreover, L. major produces casein kinase 1 (L-CK1) to phosphorylate IFNAR1 on Ser535, leading to the degradation of IFNAR1 and subsequent attenuation of type I IFN signaling during infection [163]. Toxoplasma has also evolved strategies for limiting both the induction of type I IFNs and the ability of type I IFNs to activate STAT1-dependent transcription. Mice lacking the receptor for type I IFN-1 exhibited higher parasite loads and decreased survival following T. gondii infection [164]. Type I IFN expression was also induced by C. parvum infection in both human and murine intestinal enterocytes in vivo and exogenous type I IFN pretreatment inhibited parasite development in both cell types. The treatment of neonatal severe combined immunodeficiency (SCID) mice with anti-IFN-α/β-neutralizing antibody before infection significantly increased oocyst reproduction [156]. However, type I IFN signaling does not protect against C. parvum infection in IFN-γ KO immunocompromised mice. The type I IFN response reduced oocyst shedding and symptoms but not the intestinal parasite burden following C. parvum infection in IFN-γ KO mice. Moreover, the C. parvum infection burden was significantly lower in Ifnar KO mice compared to wild-type mice, suggesting that type I IFN signaling may dampen the host defense against C. parvum [158].
Table 2.
Effects of type I IFNs in hosts following intracellular protozoan parasite infection.
3.3. Type I IFNs in the Pathogenesis of Protozoan Infection
Similar to IFN-γ, Type I IFN signaling may also play a role in the pathogenesis of protozoan infection (Table 2). Robust type I IFN signaling in highly activated neutrophils was found to be associated with increased levels of serum alanine and aspartate aminotransferases, indicating liver damage [165]. Plasmodium infection in the brain of mice lacking type I IFN receptor was reported to be resistant to otherwise lethal cerebral malaria [166]. A study using conventional DC-specific Ifnar1-deficient mice and mixed BM chimeras showed that type I IFN signaling induced by Plasmodium infection negatively impacted cDC function, limiting the ability of cDCs, particularly splenic CD8− cDCs, to prime IFN-γ–producing Th1 cells [167]. Type I IFNs suppressed the production of proinflammatory cytokines IL-17, IL-1β, IL-6, as well as IFN-γ, and promoted the development of immunosuppressive antigen-specific regulatory T cells (Tr1) to produce anti-proinflammatory cytokine IL-10 following P. falciparum infection [141]. One group, using Ifnar KO mice, showed that Ifnar KO mice could survive challenges with T. cruzi infection and were able to control parasite replication better than the wild-type mice [170]. Mice hyperresponsive for type I IFNs exhibited a significant defect in Th1 responses and IFN-γ production, suggesting that IFN-I plays a detrimental role in the early stages of disease. This corresponds to the finding that type I IFNs contribute to the downregulation of IFN-γ production and the loss of host resistance during chronic T. b. rhodesiense infection [160]. Type I IFNs were observed to suppress protective reactive oxygen species during human Leishmania infection which led to enhanced parasite burden [168]. L. amazonensis-, L. major-, or L. braziliensis-infected Ifnar KO mice developed attenuated cutaneous lesions and displayed a decreased parasite load, which is highly correlated to the robust recruitment of neutrophils, and promoted parasite-killing capability of neutrophils at early times post infection [169]. Hamsters challenged by a parasite strain harboring high Leishmania virus showed an increased production of IFN-β and higher susceptibility to Leishmania infection as well as more severe clinical symptoms [148]. Similar to that, in P. falciparum infection, type I IFNs induced by L. donovani amastigotes promoted the production of T-cell response suppressive IL-10, but through a distinct B-cell and endosomal-TLR dependent mechanism [171,172].
Current evidence suggests the roles of type I IFNs in host anti-protozoal defense and pathogenesis are controversial and depend on variation in the host species, parasite strains, parasite infection stage and location, and even parasite administration dose. The intrinsic mechanisms are mostly unclear. Future investigations need more precise methods to elucidate the exact spatial and temporal molecular activity inside different cell types from parasite-infected animals, and more studies are required to clarify the intrinsic mechanisms of immunomodulation and pathogenesis by type I IFNs.
4. Type III IFNs and Intracellular Protozoan Parasites
We know very little about the distinct role of type III IFNs in protozoal parasite infection as they are novel in this field. Type III IFNs were discovered as potent anti-viral cytokines similar to type I IFNs. Therefore, subsequent studies related to type III IFNs were mostly under the context of viral infections. However, recent studies revealed the production of type III IFNs in various bacterial and parasitic infections and assumed a potential immunomodulatory role in host defense against non-viral pathogens, even though current evidence is insufficient for a comprehensive conclusion [173].
A robust induction of type III IFN expression has been observed in patients infected with P. falciparum and in neonatal mice with C. parvum infection (Table 3). Transcriptome analysis identified IFN-λ as one of the top five differentially regulated cytokines in the blood of patients with febrile P. falciparum malaria. A subsequent study employing P. yoelii-infected Ifnlr-KO mice demonstrated that B-cell-intrinsic IFN-λ signals suppressed the acute antibody response, acute plasmablast response, and consequently impaired acute parasite clearance during a primary blood-stage malaria infection [174,175]. Clinical studies with P. falciparum-infected children in Kenya suggested a negative correlation between IFN-λ4 production and host immune protection against P. falciparum. Studies with both in vivo neonatal mice and in vitro intestinal epithelial cells showed that abundant IFN-λ3 was secreted in hosts in response to C. parvum infection [151,169]. Neutralization of IFN-λ3 exacerbated villus blunting and increased the fecal shedding of infectious C. parvum oocysts in neonatal mice, while prior supplementation of IFN-λ3 to intestinal epithelial cells reduced barrier disruption and enhanced cellular defense against C. parvum [176]. This C. parvum-induced type III IFN production has been attributed to the cell-intrinsic recognition mediated through TLR3 [158].
Table 3.
Effects of type III IFNs in hosts following intracellular protozoan parasite infection.
It is increasingly clear that although type I and III IFNs activate similar intracellular signaling cascades and gene expression in host cells, the two show distinct actions in infected hosts regarding the induction kinetics, tissue tropism, immunomodulatory effects, and even the ultimate impact on infection outcomes [177]. The unclear characteristics of type I and III IFN signaling in protozoan parasite infections prevent us from systematically understanding the commonalities and distinctions of their induction, regulation, and impacts during infection.
5. Crosstalk of IFNs
Protozoan parasites are able to induce the production of more than one type of IFN in the context of infection, in which different IFNs may act as similar protective effectors or play contrasting roles in the host defense against infection. Plasmodium liver-stage, as well as blood-stage, has been shown to induce both type I and II IFNs in the same host. A recent study using Ifnar−/− and Ifngr−/− mice determined that neither single type of IFNs established intact protection against P. chabaudi alone. The sum of the magnitude of the ISG response in the Ifnar1−/− and Ifngr1−/− animals was, on average, greater than the magnitude of the wild-type ISG response, suggesting the partial redundancy of the ISG response induced by these different IFNs [142]. However, revolutionary genetic studies revealed that some IFNs, including several subtypes of IFN-α as well as IFN-γ, have evolved under strong purifying selection to ensure their essential and nonredundant function in immunity to infection [178,179] (Figure 4). In some cases, type I IFNs act as an indirect enhancer of IFN-γ induction. For example, type I IFNs promoted the recruitment of NKT cells during liver-stage P. yoelii infection in mice to increase the production of IFN-γ [34]. Nevertheless, more evidence suggests that type I IFNs are potential negative regulators of protective IFN-γ signaling. Type I IFN signaling induced by P. falciparum, P. berghei or T. b. rhodesiense was reported to suppress Th1 cell development and subsequent IFN-γ production [141,160,167]. On the other hand, type I IFNs promote the production of anti-proinflammatory cytokine IL-10, an essential negative regulator of IFN-γ production in P. falciparum or L. donovani infections [141,172]. Fundamental questions can be asked based on these observations: why does the infected host produce different molecules playing a similar role “redundantly”? How do these distinct but closely related molecules interact with each other when induced in the same host? Does this interaction alter the infection outcome? The answers are complicated and remain open.
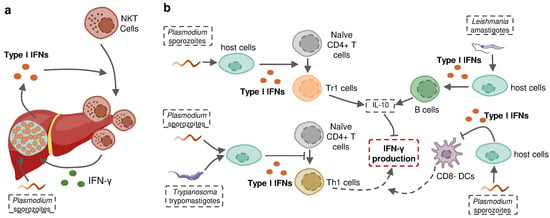
Figure 4.
Type I IFN signaling induced by protozoan parasite infection regulates IFN-γ production during infection. (a) Liver stage Plasmodium-induced type I IFNs recruit IFN-γ-producing NKT cells to the liver and sequentially enhance IFN-γ production. (b) Protozoan-parasite-induced type I IFNs suppress host IFN-γ production by promoting the development of Tr1 cells and stimulating B cells to increase host expression of IL-10, inhibiting the differentiation of naïve CD4+T cells towards Th1 cells and restricting the activity of CD8- DCs to suppress the priming of IFN-γ-producing Th1 cells.
6. Conclusions and Speculations
Type II IFN (IFN-γ) mainly acts as a protective effector in the host defense against many intracellular protozoal infections and contributes to the pathogenesis of parasite infection in some cases. The precise roles of type I and III IFNs in host anti-parasitic defense remain unclear and require future investigations. The three types of IFNs activate signaling pathways which are distinguishable but share essential components, resulting in distinct but overlapping transcriptional outcomes. Current evidence highlights the various effects of different IFN responses on host anti-parasitic defense and implicates an indirect, regulatory, impact of type I IFNs on type II IFN production by immune cells. The partially overlapping ISG responses induced by the three types of IFN signaling in infected cells indicates potential direct intracellular interactions among various types of IFN signaling pathways, which currently lacks attention from the research field. Future studies should address the regulatory mechanisms of type I/III IFNs on IFN-γ signaling and the temporal kinetics of these interactions between different IFN signaling pathways in host cells following infection by protozoan parasites. This will facilitate monitoring disease progression and direct the administration of therapeutic strategies to protozoan infections. A better understanding of IFN crosstalk in the context of protozoan parasite infections will also provide new insights into mechanisms of the pathogenesis and host immune activities of other diseases involving multiple IFN responses, and contribute to the future development of IFN-related therapies.
Funding
This research was funded by the National Institutes of Health, grant numbers AI116323, AI136877, AI141325, and AI156370.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Isaacs, A.; Lindenmann, J. Virus Interference. I. The Interferon. Proc. R. Soc. Lond. B Biol. Sci. 1957, 147, 258–267. [Google Scholar] [CrossRef] [PubMed]
- Sheppard, P.; Kindsvogel, W.; Xu, W.; Henderson, K.; Schlutsmeyer, S.; Whitmore, T.E.; Kuestner, R.; Garrigues, U.; Birks, C.; Roraback, J.; et al. IL-28, IL-29 and Their Class II Cytokine Receptor IL-28R. Nat. Immunol. 2003, 4, 63–68. [Google Scholar] [CrossRef] [PubMed]
- Kotenko, S.V.; Gallagher, G.; Baurin, V.V.; Lewis-Antes, A.; Shen, M.; Shah, N.K.; Langer, J.A.; Sheikh, F.; Dickensheets, H.; Donnelly, R.P. IFN-Λs Mediate Antiviral Protection through a Distinct Class II Cytokine Receptor Complex. Nat. Immunol. 2003, 4, 69–77. [Google Scholar] [CrossRef]
- Blouin, C.M.; Lamaze, C. Interferon Gamma Receptor: The Beginning of the Journey. Front. Immunol. 2013, 4, 267. [Google Scholar] [CrossRef] [PubMed]
- Pestka, S.; Krause, C.D.; Walter, M.R. Interferons, Interferon-like Cytokines, and Their Receptors. Immunol. Rev. 2004, 202, 8–32. [Google Scholar] [CrossRef]
- McNab, F.; Mayer-Barber, K.; Sher, A.; Wack, A.; O’Garra, A. Type I Interferons in Infectious Disease. Nat. Rev. Immunol. 2015, 15, 87–103. [Google Scholar] [CrossRef] [PubMed]
- Kaplanski, G. Interleukin-18: Biological Properties and Role in Disease Pathogenesis. Immunol. Rev. 2018, 281, 138–153. [Google Scholar] [CrossRef] [PubMed]
- Hemmi, H.; Takeuchi, O.; Kawai, T.; Kaisho, T.; Sato, S.; Sanjo, H.; Matsumoto, M.; Hoshino, K.; Wagner, H.; Takeda, K.; et al. A Toll-like Receptor Recognizes Bacterial DNA. Nature 2000, 408, 740–745. [Google Scholar] [CrossRef] [PubMed]
- Bosisio, D.; Polentarutti, N.; Sironi, M.; Bernasconi, S.; Miyake, K.; Webb, G.R.; Martin, M.U.; Mantovani, A.; Muzio, M. Stimulation of Toll-like Receptor 4 Expression in Human Mononuclear Phagocytes by Interferon-γ: A Molecular Basis for Priming and Synergism with Bacterial Lipopolysaccharide. Blood 2002, 99, 3427–3431. [Google Scholar] [CrossRef] [PubMed]
- Vignali, D.A.A.; Kuchroo, V.K. IL-12 Family Cytokines: Immunological Playmakers. Nat. Immunol. 2012, 13, 722–728. [Google Scholar] [CrossRef]
- Prokunina-Olsson, L.; Muchmore, B.; Tang, W.; Pfeiffer, R.M.; Park, H.; Dickensheets, H.; Hergott, D.; Porter-Gill, P.; Mumy, A.; Kohaar, I.; et al. A Variant Upstream of IFNL3 (IL28B) Creating a Novel Interferon Gene IFNL4 Is Associated with Impaired Clearance of Hepatitis C Virus. Nat. Genet. 2013, 45, 164–171. [Google Scholar] [CrossRef] [PubMed]
- Mesev, E.V.; LeDesma, R.A.; Ploss, A. Decoding Type I and III Interferon Signalling during Viral Infection. Nat. Microbiol. 2019, 4, 914–924. [Google Scholar] [CrossRef]
- Ivashkiv, L.B. IFNγ: Signalling, Epigenetics and Roles in Immunity, Metabolism, Disease and Cancer Immunotherapy. Nat. Rev. Immunol. 2018, 18, 545–558. [Google Scholar] [CrossRef] [PubMed]
- Schneider, W.M.; Chevillotte, M.D.; Rice, C.M. Interferon-Stimulated Genes: A Complex Web of Host Defenses. Annu. Rev. Immunol. 2014, 32, 513–545. [Google Scholar] [CrossRef] [PubMed]
- Ye, L.; Schnepf, D.; Staeheli, P. Interferon-λ Orchestrates Innate and Adaptive Mucosal Immune Responses. Nat. Rev. Immunol. 2019, 19, 614–625. [Google Scholar] [CrossRef] [PubMed]
- de Weerd, N.A.; Nguyen, T. The Interferons and Their Receptors—Distribution and Regulation. Immunol. Cell Biol. 2012, 90, 483–491. [Google Scholar] [CrossRef] [PubMed]
- Reuter, A.; Soubies, S.; Härtle, S.; Schusser, B.; Kaspers, B.; Staeheli, P.; Rubbenstroth, D. Antiviral Activity of Lambda Interferon in Chickens. J. Virol. 2014, 88, 2835–2843. [Google Scholar] [CrossRef] [PubMed]
- Sommereyns, C.; Paul, S.; Staeheli, P.; Michiels, T. IFN-Lambda (IFN-λ) Is Expressed in a Tissue-Dependent Fashion and Primarily Acts on Epithelial Cells In Vivo. PLoS Pathog. 2008, 4, e1000017. [Google Scholar] [CrossRef] [PubMed]
- Boisvert, M.; Shoukry, N.H. Type III Interferons in Hepatitis C Virus Infection. Front. Immunol. 2016, 7, 628. [Google Scholar] [CrossRef] [PubMed]
- Chesler, D.A.; Reiss, C.S. The Role of IFN-γ in Immune Responses to Viral Infections of the Central Nervous System. Cytokine Growth Factor Rev. 2002, 13, 441–454. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.; Brown, H.M.; Hwang, S. Direct Antiviral Mechanisms of Interferon-Gamma. Immune Netw. 2018, 18, e33. [Google Scholar] [CrossRef] [PubMed]
- Boxx, G.M.; Cheng, G. The Roles of Type I Interferon in Bacterial Infection. Cell Host Microbe 2016, 19, 760–769. [Google Scholar] [CrossRef] [PubMed]
- Ramharter, M.; Willheim, M.; Winkler, H.; Wahl, K.; Lagler, H.; Graninger, W.; Winkler, S. Cytokine Profile of Plasmodium Falciparum-Specific T Cells in Non-Immune Malaria Patients. Parasite Immunol. 2003, 25, 211–219. [Google Scholar] [CrossRef] [PubMed]
- Horowitz, A.; Riley, E.M. Activation of Human NK Cells by Plasmodium-Infected Red Blood Cells. Methods Mol. Biol. 2013, 923, 447–464. [Google Scholar] [CrossRef] [PubMed]
- Barakat, F.M.; McDonald, V.; Di Santo, J.P.; Korbel, D.S. Roles for NK Cells and an NK Cell-Independent Source of Intestinal Gamma Interferon for Innate Immunity to Cryptosporidium Parvum Infection. Infect. Immun. 2009, 77, 5044–5049. [Google Scholar] [CrossRef] [PubMed]
- Onyilagha, C.; Kuriakose, S.; Ikeogu, N.; Kung, S.K.P.; Uzonna, J.E. NK Cells Are Critical for Optimal Immunity to Experimental Trypanosoma Congolense Infection. J. Immunol. 2019, 203, 964–971. [Google Scholar] [CrossRef] [PubMed]
- Artavanis-Tsakonas, K.; Riley, E.M. Innate Immune Response to Malaria: Rapid Induction of IFN-γ from Human NK Cells by Live Plasmodium Falciparum-Infected Erythrocytes. J. Immunol. 2002, 169, 2956–2963. [Google Scholar] [CrossRef] [PubMed]
- Becker, I.; Salaiza, N.; Aguirre, M.; Delgado, J.; Carrillo-Carrasco, N.; Kobeh, L.G.; Ruiz, A.; Cervantes, R.; Torres, A.P.; Cabrera, N.; et al. Leishmania Lipophosphoglycan (LPG) Activates NK Cells through Toll-like Receptor-2. Mol. Biochem. Parasitol. 2003, 130, 65–74. [Google Scholar] [CrossRef] [PubMed]
- Filipe-Santos, O.; Pescher, P.; Breart, B.; Lippuner, C.; Aebischer, T.; Glaichenhaus, N.; Späth, G.F.; Bousso, P. A Dynamic Map of Antigen Recognition by CD4 T Cells at the Site of Leishmania Major Infection. Cell Host Microbe 2009, 6, 23–33. [Google Scholar] [CrossRef] [PubMed]
- Walther, M.; Jeffries, D.; Finney, O.C.; Njie, M.; Ebonyi, A.; Deininger, S.; Lawrence, E.; Ngwa-Amambua, A.; Jayasooriya, S.; Cheeseman, I.H.; et al. Distinct Roles for FOXP3+ and FOXP3− CD4+ T Cells in Regulating Cellular Immunity to Uncomplicated and Severe Plasmodium Falciparum Malaria. PLoS Pathog. 2009, 5, e1000364. [Google Scholar] [CrossRef] [PubMed]
- Ong’echa, J.M.O.; Lal, A.A.; Terlouw, D.J.; Ter Kuile, F.O.; Kariuki, S.K.; Udhayakumar, V.; Orago, A.S.S.; Hightower, A.W.; Nahlen, B.L.; Shi, Y.P. Association of Interferon-Gamma Responses to Pre-Erythrocytic Stage Vaccine Candidate Antigens of Plasmodium Falciparum in Young Kenyan Children with Improved Hemoglobin Levels: XV. Asembo Bay Cohort Project. Am. J. Trop. Med. Hyg. 2003, 68, 590–597. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Kang, H.; Kikuchi, T.; Suzuki, Y. Gamma Interferon Production, but Not Perforin-Mediated Cytolytic Activity, of T Cells Is Required for Prevention of Toxoplasmic Encephalitis in BALB/c Mice Genetically Resistant to the Disease. Infect. Immun. 2004, 72, 4432–4438. [Google Scholar] [CrossRef] [PubMed]
- Namangala, B.; Noël, W.; De Baetselier, P.; Brys, L.; Beschin, A. Relative Contribution of Interferon-γ and Interleukin-10 to Resistance to Murine African Trypanosomosis. J. Infect. Dis. 2001, 183, 1794–1800. [Google Scholar] [CrossRef] [PubMed]
- Miller, J.L.; Sack, B.K.; Baldwin, M.; Vaughan, A.M.; Kappe, S.H.I. Interferon-Mediated Innate Immune Responses against Malaria Parasite Liver Stages. Cell Rep. 2014, 7, 436–447. [Google Scholar] [CrossRef] [PubMed]
- Soulard, V.; Roland, J.; Sellier, C.; Gruner, A.C.; Leite-de-Moraes, M.; Franetich, J.-F.; Rénia, L.; Cazenave, P.-A.; Pied, S. Primary Infection of C57BL/6 Mice with Plasmodium Yoelii Induces a Heterogeneous Response of NKT Cells. Infect. Immun. 2007, 75, 2511–2522. [Google Scholar] [CrossRef]
- Hviid, L.; Kurtzhals, J.A.L.; Adabayeri, V.; Loizon, S.; Kemp, K.; Goka, B.Q.; Lim, A.; Mercereau-Puijalon, O.; Akanmori, B.D.; Behr, C. Perturbation and Proinflammatory Type Activation of Vδ1+ Γδ T Cells in African Children with Plasmodium Falciparum Malaria. Infect. Immun. 2001, 69, 3190–3196. [Google Scholar] [CrossRef]
- D’Ombrain, M.C.; Robinson, L.J.; Stanisic, D.I.; Taraika, J.; Bernard, N.; Michon, P.; Mueller, I.; Schofield, L. Association of Early Interferon-γ Production with Immunity to Clinical Malaria: A Longitudinal Study among Papua New Guinean Children. Clin. Infect. Dis. 2008, 47, 1380–1387. [Google Scholar] [CrossRef]
- Pamplona, A.; Silva-Santos, B. Γδ T Cells in Malaria: A Double-edged Sword. FEBS J. 2021, 288, 1118–1129. [Google Scholar] [CrossRef]
- Suzuki, Y.; Claflin, J.; Wang, X.; Lengi, A.; Kikuchi, T. Microglia and Macrophages as Innate Producers of Interferon-Gamma in the Brain Following Infection with Toxoplasma Gondii. Int. J. Parasitol. 2005, 35, 83–90. [Google Scholar] [CrossRef]
- Wang, X.; Suzuki, Y. Microglia Produce IFN-γ Independently from T Cells During Acute Toxoplasmosis in the Brain. J. Interferon Cytokine Res. 2007, 27, 599–605. [Google Scholar] [CrossRef]
- Klose, C.S.N.; Flach, M.; Möhle, L.; Rogell, L.; Hoyler, T.; Ebert, K.; Fabiunke, C.; Pfeifer, D.; Sexl, V.; Fonseca-Pereira, D.; et al. Differentiation of Type 1 ILCs from a Common Progenitor to All Helper-like Innate Lymphoid Cell Lineages. Cell 2014, 157, 340–356. [Google Scholar] [CrossRef] [PubMed]
- Torre, D.; Speranza, F.; Giola, M.; Matteelli, A.; Tambini, R.; Biondi, G. Role of Th1 and Th2 Cytokines in Immune Response to Uncomplicated Plasmodium Falciparum Malaria. Clin. Vaccine Immunol. 2002, 9, 348–351. [Google Scholar] [CrossRef] [PubMed]
- Loevenich, K.; Ueffing, K.; Abel, S.; Hose, M.; Matuschewski, K.; Westendorf, A.M.; Buer, J.; Hansen, W. DC-Derived IL-10 Modulates Pro-Inflammatory Cytokine Production and Promotes Induction of CD4+IL-10+ Regulatory T Cells during Plasmodium Yoelii Infection. Front. Immunol. 2017, 8, 152. [Google Scholar] [CrossRef] [PubMed]
- Ivanova, D.L.; Denton, S.L.; Fettel, K.D.; Sondgeroth, K.S.; Munoz Gutierrez, J.; Bangoura, B.; Dunay, I.R.; Gigley, J.P. Innate Lymphoid Cells in Protection, Pathology, and Adaptive Immunity During Apicomplexan Infection. Front. Immunol. 2019, 10, 196. [Google Scholar] [CrossRef]
- Doolan, D.L.; Hoffman, S.L. The Complexity of Protective Immunity against Liver-Stage Malaria. J. Immunol. 2000, 165, 1453–1462. [Google Scholar] [CrossRef]
- Prakash, D.; Fesel, C.; Jain, R.; Cazenave, P.-A.; Mishra, G.C.; Pied, S. Clusters of Cytokines Determine Malaria Severity in Plasmodium Falciparum–Infected Patients from Endemic Areas of Central India. J. Infect. Dis. 2006, 194, 198–207. [Google Scholar] [CrossRef]
- Schofield, L.; Villaquiran, J.; Ferreira, A.; Schellekens, H.; Nussenzweig, R.; Nussenzweig, V. γ Interferon, CD8+ T Cells and Antibodies Required for Immunity to Malaria Sporozoites. Nature 1987, 330, 664–666. [Google Scholar] [CrossRef]
- Mitchell, A.J.; Hansen, A.M.; Hee, L.; Ball, H.J.; Potter, S.M.; Walker, J.C.; Hunt, N.H. Early Cytokine Production Is Associated with Protection from Murine Cerebral Malaria. Infect. Immun. 2005, 73, 5645–5653. [Google Scholar] [CrossRef]
- van der Heyde, H.C.; Pepper, B.; Batchelder, J.; Cigel, F.; Weidanz, W.P. The Time Course of Selected Malarial Infections in Cytokine-Deficient Mice. Exp. Parasitol. 1997, 85, 206–213. [Google Scholar] [CrossRef]
- Shear, H.L.; Srinivasan, R.; Nolan, T.; Ng, C. Role of IFN-Gamma in Lethal and Nonlethal Malaria in Susceptible and Resistant Murine Hosts. J. Immunol. 1989, 143, 2038–2044. [Google Scholar] [CrossRef]
- Tsuji, M.; Miyahira, Y.; Nussenzweig, R.S.; Aguet, M.; Reichel, M.; Zavala, F. Development of Antimalaria Immunity in Mice Lacking IFN-Gamma Receptor. J. Immunol. 1995, 154, 5338–5344. [Google Scholar] [CrossRef] [PubMed]
- Hertz, C.J.; Filutowicz, H.; Mansfield, J.M. Resistance to the African Trypanosomes Is IFN-γ Dependent. J. Immunol. 1998, 161, 6775–6783. [Google Scholar] [CrossRef] [PubMed]
- Swihart, K.; Fruth, U.; Messmer, N.; Hug, K.; Behin, R.; Huang, S.; Del Giudice, G.; Aguet, M.; Louis, J.A. Mice from a Genetically Resistant Background Lacking the Interferon γ Receptor Are Susceptible to Infection with Leishmania Major but Mount a Polarized T Helper Cell 1-Type CD4+ T Cell Response. J. Exp. Med. 1995, 181, 961–971. [Google Scholar] [CrossRef] [PubMed]
- Pinheiro, R.O.; Rossi-Bergmann, B. Interferon-Gamma Is Required for the Late but Not Early Control of Leishmania Amazonensis Infection in C57Bl/6 Mice. Mem. Inst. Oswaldo Cruz 2007, 102, 79–82. [Google Scholar] [CrossRef] [PubMed]
- Scharton-Kersten, T.M.; Wynn, T.A.; Denkers, E.Y.; Bala, S.; Grunvald, E.; Hieny, S.; Gazzinelli, R.T.; Sher, A. In the Absence of Endogenous IFN-Gamma, Mice Develop Unimpaired IL-12 Responses to Toxoplasma Gondii While Failing to Control Acute Infection. J. Immunol. 1996, 157, 4045–4054. [Google Scholar] [CrossRef] [PubMed]
- Pollok, R.C.G.; Farthing, M.J.G.; Bajaj-Elliott, M.; Sanderson, I.R.; McDonald, V. Interferon Gamma Induces Enterocyte Resistance against Infection by the Intracellular Pathogen Cryptosporidium Parvum. Gastroenterology 2001, 120, 99–107. [Google Scholar] [CrossRef]
- Lacroix, S.; Mancassola, R.; Naciri, M.; Laurent, F. Cryptosporidium Parvum-Specific Mucosal Immune Response in C57BL/6 Neonatal and Gamma Interferon-Deficient Mice: Role of Tumor Necrosis Factor Alpha in Protection. Infect. Immun. 2001, 69, 1635–1642. [Google Scholar] [CrossRef]
- de Kossodo, S.; Grau, G.E. Profiles of Cytokine Production in Relation with Susceptibility to Cerebral Malaria. J. Immunol. 1993, 151, 4811–4820. [Google Scholar] [CrossRef]
- Kossodo, S.; Monso, C.; Juillard, P.; Velu, T.; Goldman, M.; Grau, G.E. Interleukin-10 Modulates Susceptibility in Experimental Cerebral Malaria. Immunology 1997, 91, 536–540. [Google Scholar] [CrossRef]
- Amani, V.; Vigário, A.M.; Belnoue, E.; Marussig, M.; Fonseca, L.; Mazier, D.; Rénia, L. Involvement of IFN-Gamma Receptor-Medicated Signaling in Pathology and Anti-Malarial Immunity Induced by Plasmodium Berghei Infection. Eur. J. Immunol. 2000, 30, 1646–1655. [Google Scholar] [CrossRef]
- Belnoue, E.; Potter, S.M.; Rosa, D.S.; Mauduit, M.; Grüner, A.C.; Kayibanda, M.; Mitchell, A.J.; Hunt, N.H.; Rénia, L. Control of Pathogenic CD8+ T Cell Migration to the Brain by IFN-γ during Experimental Cerebral Malaria. Parasite Immunol. 2008, 30, 544–553. [Google Scholar] [CrossRef]
- Yañez, D.M.; Manning, D.D.; Cooley, A.J.; Weidanz, W.P.; Heyde, H.C. van der Participation of Lymphocyte Subpopulations in the Pathogenesis of Experimental Murine Cerebral Malaria. J. Immunol. 1996, 157, 1620–1624. [Google Scholar] [CrossRef] [PubMed]
- Villegas-Mendez, A.; Greig, R.; Shaw, T.N.; de Souza, J.B.; Gwyer Findlay, E.; Stumhofer, J.S.; Hafalla, J.C.R.; Blount, D.G.; Hunter, C.A.; Riley, E.M.; et al. IFN-γ-Producing CD4+ T Cells Promote Experimental Cerebral Malaria by Modulating CD8+ T Cell Accumulation within the Brain. J. Immunol. 2012, 189, 968–979. [Google Scholar] [CrossRef] [PubMed]
- Grau, G.E.; Heremans, H.; Piguet, P.F.; Pointaire, P.; Lambert, P.H.; Billiau, A.; Vassalli, P. Monoclonal Antibody against Interferon Gamma Can Prevent Experimental Cerebral Malaria and Its Associated Overproduction of Tumor Necrosis Factor. Proc. Natl. Acad. Sci. USA 1989, 86, 5572–5574. [Google Scholar] [CrossRef] [PubMed]
- Zander, R.A.; Obeng-Adjei, N.; Guthmiller, J.J.; Kulu, D.I.; Li, J.; Ongoiba, A.; Traore, B.; Crompton, P.D.; Butler, N.S. PD-1 Co-Inhibitory and OX40 Co-Stimulatory Crosstalk Regulates Helper T Cell Differentiation and Anti-Plasmodium Humoral Immunity. Cell Host Microbe 2015, 17, 628–641. [Google Scholar] [CrossRef] [PubMed]
- Ryg-Cornejo, V.; Ioannidis, L.J.; Ly, A.; Chiu, C.Y.; Tellier, J.; Hill, D.L.; Preston, S.P.; Pellegrini, M.; Yu, D.; Nutt, S.L.; et al. Severe Malaria Infections Impair Germinal Center Responses by Inhibiting T Follicular Helper Cell Differentiation. Cell Rep. 2016, 14, 68–81. [Google Scholar] [CrossRef] [PubMed]
- Obeng-Adjei, N.; Portugal, S.; Tran, T.M.; Yazew, T.B.; Skinner, J.; Li, S.; Jain, A.; Felgner, P.L.; Doumbo, O.K.; Kayentao, K.; et al. Circulating Th1-Cell-Type Tfh Cells That Exhibit Impaired B Cell Help Are Preferentially Activated during Acute Malaria in Children. Cell Rep. 2015, 13, 425–439. [Google Scholar] [CrossRef]
- Guthmiller, J.J.; Graham, A.C.; Zander, R.A.; Pope, R.L.; Butler, N.S. Cutting Edge: IL-10 Is Essential for the Generation of Germinal Center B Cell Responses and Anti-Plasmodium Humoral Immunity. J. Immunol. 2017, 198, 617–622. [Google Scholar] [CrossRef]
- Rivera-Correa, J.; Guthmiller, J.J.; Vijay, R.; Fernandez-Arias, C.; Pardo-Ruge, M.A.; Gonzalez, S.; Butler, N.S.; Rodriguez, A. Plasmodium DNA-Mediated TLR9 Activation of T-Bet+ B Cells Contributes to Autoimmune Anaemia during Malaria. Nat. Commun. 2017, 8, 1282. [Google Scholar] [CrossRef]
- Shi, M.; Pan, W.; Tabel, H. Experimental African Trypanosomiasis: IFN-γ Mediates Early Mortality. Eur. J. Immunol. 2003, 33, 108–118. [Google Scholar] [CrossRef]
- Barkhuizen, M.; Magez, S.; Ryffel, B.; Brombacher, F. Interleukin-12p70 Deficiency Increases Survival and Diminishes Pathology in Trypanosoma Congolense Infection. J. Infect. Dis. 2008, 198, 1284–1291. [Google Scholar] [CrossRef] [PubMed]
- Uzonna, J.E.; Kaushik, R.S.; Gordon, J.R.; Tabel, H. Experimental Murine Trypanosoma Congolense Infections. I. Administration of Anti-IFN-γ Antibodies Alters Trypanosome-Susceptible Mice to a Resistant-Like Phenotype. J. Immunol. 1998, 161, 5507–5515. [Google Scholar] [CrossRef]
- Siddiqui, A.J.; Bhardwaj, J.; Goyal, M.; Prakash, K.; Adnan, M.; Alreshidi, M.M.; Patel, M.; Soni, A.; Redman, W. Immune Responses in Liver and Spleen against Plasmodium Yoelii Pre-Erythrocytic Stages in Swiss Mice Model. J. Adv. Res. 2020, 24, 29–41. [Google Scholar] [CrossRef] [PubMed]
- Clark, I.A.; Hunt, N.H.; Butcher, G.A.; Cowden, W.B. Inhibition of Murine Malaria (Plasmodium Chabaudi) in Vivo by Recombinant Interferon-Gamma or Tumor Necrosis Factor, and Its Enhancement by Butylated Hydroxyanisole. J. Immunol. 1987, 139, 3493–3496. [Google Scholar] [CrossRef] [PubMed]
- De Souza, J.B.; Williamson, K.H.; Otani, T.; Playfair, J.H. Early Gamma Interferon Responses in Lethal and Nonlethal Murine Blood-Stage Malaria. Infect. Immun. 1997, 65, 1593–1598. [Google Scholar] [CrossRef] [PubMed]
- Kester, K.E.; Cummings, J.F.; Ofori-Anyinam, O.; Ockenhouse, C.F.; Krzych, U.; Moris, P.; Schwenk, R.; Nielsen, R.A.; Debebe, Z.; Pinelis, E.; et al. Randomized, Double-Blind, Phase 2a Trial of Falciparum Malaria Vaccines RTS,S/AS01B and RTS,S/AS02A in Malaria-Naive Adults: Safety, Efficacy, and Immunologic Associates of Protection. J. Infect. Dis. 2009, 200, 337–346. [Google Scholar] [CrossRef]
- Chuang, I.; Sedegah, M.; Cicatelli, S.; Spring, M.; Polhemus, M.; Tamminga, C.; Patterson, N.; Guerrero, M.; Bennett, J.W.; McGrath, S.; et al. DNA Prime/Adenovirus Boost Malaria Vaccine Encoding P. Falciparum CSP and AMA1 Induces Sterile Protection Associated with Cell-Mediated Immunity. PLoS ONE 2013, 8, e55571. [Google Scholar] [CrossRef]
- Seder, R.A.; Chang, L.-J.; Enama, M.E.; Zephir, K.L.; Sarwar, U.N.; Gordon, I.J.; Holman, L.A.; James, E.R.; Billingsley, P.F.; Gunasekera, A.; et al. Protection Against Malaria by Intravenous Immunization with a Nonreplicating Sporozoite Vaccine. Science 2013, 341, 1359–1365. [Google Scholar] [CrossRef]
- Stanisic, D.I.; Fink, J.; Mayer, J.; Coghill, S.; Gore, L.; Liu, X.Q.; El-Deeb, I.; Rodriguez, I.B.; Powell, J.; Willemsen, N.M.; et al. Vaccination with Chemically Attenuated Plasmodium Falciparum Asexual Blood-Stage Parasites Induces Parasite-Specific Cellular Immune Responses in Malaria-Naïve Volunteers: A Pilot Study. BMC Med. 2018, 16, 184. [Google Scholar] [CrossRef]
- Chawla, B.; Mahajan, B.; Oakley, M.; Majam, V.F.; Belmonte, A.; Sedegah, M.; Shimp, R.L.; Kaslow, D.C.; Kumar, S. Antibody-Dependent, Gamma Interferon-Independent Sterilizing Immunity Induced by a Subunit Malaria Vaccine. Infect. Immun. 2019, 87, e00236-19. [Google Scholar] [CrossRef]
- Carneiro, M.B.H.; Lopes, M.E.D.M.; Vaz, L.G.; Sousa, L.M.A.; dos Santos, L.M.; de Souza, C.C.; Campos, A.C.D.A.; Gomes, D.A.; Gonçalves, R.; Tafuri, W.L.; et al. IFN-γ-Dependent Recruitment of CD4+ T Cells and Macrophages Contributes to Pathogenesis During Leishmania Amazonensis Infection. J. Interferon Cytokine Res. 2015, 35, 935–947. [Google Scholar] [CrossRef]
- Wu, H.; Liu, G.; Shi, M. Interferon Gamma in African Trypanosome Infections: Friends or Foes? Front. Immunol. 2017, 8, 1105. [Google Scholar] [CrossRef] [PubMed]
- MacMicking, J.D. Interferon-Inducible Effector Mechanisms in Cell-Autonomous Immunity. Nat. Rev. Immunol. 2012, 12, 367–382. [Google Scholar] [CrossRef] [PubMed]
- Hölscher, C.; Köhler, G.; Müller, U.; Mossmann, H.; Schaub, G.A.; Brombacher, F. Defective Nitric Oxide Effector Functions Lead to Extreme Susceptibility of Trypanosoma Cruzi-Infected Mice Deficient in Gamma Interferon Receptor or Inducible Nitric Oxide Synthase. Infect. Immun. 1998, 66, 1208–1215. [Google Scholar] [CrossRef] [PubMed]
- Mellouk, S.; Hoffman, S.L.; Liu, Z.Z.; de la Vega, P.; Billiar, T.R.; Nussler, A.K. Nitric Oxide-Mediated Antiplasmodial Activity in Human and Murine Hepatocytes Induced by Gamma Interferon and the Parasite Itself: Enhancement by Exogenous Tetrahydrobiopterin. Infect. Immun. 1994, 62, 4043–4046. [Google Scholar] [CrossRef] [PubMed]
- Green, S.J.; Meltzer, M.S.; Hibbs, J.B.; Nacy, C.A. Activated Macrophages Destroy Intracellular Leishmania Major Amastigotes by an L-Arginine-Dependent Killing Mechanism. J. Immunol. 1990, 144, 278–283. [Google Scholar] [CrossRef]
- Scharton-Kersten, T.M.; Yap, G.; Magram, J.; Sher, A. Inducible Nitric Oxide Is Essential for Host Control of Persistent but Not Acute Infection with the Intracellular Pathogen Toxoplasma Gondii. J. Exp. Med. 1997, 185, 1261–1274. [Google Scholar] [CrossRef]
- Leitch, G.J.; He, Q. Reactive Nitrogen and Oxygen Species Ameliorate Experimental Cryptosporidiosis in the Neonatal BALB/c Mouse Model. Infect. Immun. 1999, 67, 5885–5891. [Google Scholar] [CrossRef]
- Kolios, G.; Rooney, N.; Murphy, C.T.; Robertson, D.A.; Westwick, J. Expression of Inducible Nitric Oxide Synthase Activity in Human Colon Epithelial Cells: Modulation by T Lymphocyte Derived Cytokines. Gut 1998, 43, 56–63. [Google Scholar] [CrossRef]
- Hayward, A.R.; Chmura, K.; Cosyns, M. Interferon-Gamma Is Required for Innate Immunity to Cryptosporidium Parvum in Mice. J. Infect. Dis. 2000, 182, 1001–1004. [Google Scholar] [CrossRef]
- Hunn, J.P.; Koenen-Waisman, S.; Papic, N.; Schroeder, N.; Pawlowski, N.; Lange, R.; Kaiser, F.; Zerrahn, J.; Martens, S.; Howard, J.C. Regulatory Interactions between IRG Resistance GTPases in the Cellular Response to Toxoplasma Gondii. EMBO J. 2008, 27, 2495–2509. [Google Scholar] [CrossRef]
- Zhao, Y.; Ferguson, D.J.P.; Wilson, D.C.; Howard, J.C.; Sibley, L.D.; Yap, G.S. Virulent Toxoplasma Gondii Evade Immunity-Related GTPase (IRG)-Mediated Parasite Vacuole Disruption Within Primed Macrophages. J. Immunol. 2009, 182, 3775–3781. [Google Scholar] [CrossRef]
- Halonen, S.K.; Taylor, G.A.; Weiss, L.M. Gamma Interferon-Induced Inhibition of Toxoplasma Gondii in Astrocytes Is Mediated by IGTP. Infect. Immun. 2001, 69, 5573–5576. [Google Scholar] [CrossRef]
- Martens, S.; Parvanova, I.; Zerrahn, J.; Griffiths, G.; Schell, G.; Reichmann, G.; Howard, J.C. Disruption of Toxoplasma Gondii Parasitophorous Vacuoles by the Mouse P47-Resistance GTPases. PLoS Pathog. 2005, 1, e24. [Google Scholar] [CrossRef]
- Ling, Y.M.; Shaw, M.H.; Ayala, C.; Coppens, I.; Taylor, G.A.; Ferguson, D.J.P.; Yap, G.S. Vacuolar and Plasma Membrane Stripping and Autophagic Elimination of Toxoplasma Gondii in Primed Effector Macrophages. J. Exp. Med. 2006, 203, 2063–2071. [Google Scholar] [CrossRef]
- Zhao, Y.; Wilson, D.; Matthews, S.; Yap, G.S. Rapid Elimination of Toxoplasma Gondii by Gamma Interferon-Primed Mouse Macrophages Is Independent of CD40 Signaling. Infect. Immun. 2007, 75, 4799–4803. [Google Scholar] [CrossRef]
- Liesenfeld, O.; Parvanova, I.; Zerrahn, J.; Han, S.-J.; Heinrich, F.; Muñoz, M.; Kaiser, F.; Aebischer, T.; Buch, T.; Waisman, A.; et al. The IFN-γ-Inducible GTPase, Irga6, Protects Mice against Toxoplasma Gondii but Not against Plasmodium Berghei and Some Other Intracellular Pathogens. PLoS ONE 2011, 6, e20568. [Google Scholar] [CrossRef]
- Santiago, H.C.; Feng, C.G.; Bafica, A.; Roffe, E.; Arantes, R.M.; Cheever, A.; Taylor, G.; Vierira, L.Q.; Aliberti, J.; Gazzinelli, R.T.; et al. Mice Deficient in LRG-47 Display Enhanced Susceptibility to Trypanosoma Cruzi Infection Associated with Defective Hemopoiesis and Intracellular Control of Parasite Growth. J. Immunol. 2005, 175, 8165–8172. [Google Scholar] [CrossRef]
- Khaminets, A.; Hunn, J.P.; Könen-Waisman, S.; Zhao, Y.O.; Preukschat, D.; Coers, J.; Boyle, J.P.; Ong, Y.-C.; Boothroyd, J.C.; Reichmann, G.; et al. Coordinated Loading of IRG Resistance GTPases on to the Toxoplasma Gondii Parasitophorous Vacuole. Cell. Microbiol. 2010, 12, 939–961. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.-H.; Shenoy, A.R.; Kumar, P.; Bradfield, C.J.; MacMicking, J.D. IFN-Inducible GTPases in Host Defense. Cell Host Microbe 2012, 12, 432–444. [Google Scholar] [CrossRef]
- Yamamoto, M.; Okuyama, M.; Ma, J.S.; Kimura, T.; Kamiyama, N.; Saiga, H.; Ohshima, J.; Sasai, M.; Kayama, H.; Okamoto, T.; et al. A Cluster of Interferon-γ-Inducible P65 GTPases Plays a Critical Role in Host Defense against Toxoplasma Gondii. Immunity 2012, 37, 302–313. [Google Scholar] [CrossRef] [PubMed]
- Degrandi, D.; Kravets, E.; Konermann, C.; Beuter-Gunia, C.; Klümpers, V.; Lahme, S.; Rasch, E.; Mausberg, A.K.; Beer-Hammer, S.; Pfeffer, K. Murine Guanylate Binding Protein 2 (MGBP2) Controls Toxoplasma Gondii Replication. Proc. Natl. Acad. Sci. USA 2013, 110, 294–299. [Google Scholar] [CrossRef] [PubMed]
- Selleck, E.M.; Fentress, S.J.; Beatty, W.L.; Degrandi, D.; Pfeffer, K.; Virgin, H.W.; MacMicking, J.D.; Sibley, L.D. Guanylate-Binding Protein 1 (Gbp1) Contributes to Cell-Autonomous Immunity against Toxoplasma Gondii. PLoS Pathog. 2013, 9, e1003320. [Google Scholar] [CrossRef]
- Winter, S.V.; Niedelman, W.; Jensen, K.D.; Rosowski, E.E.; Julien, L.; Spooner, E.; Caradonna, K.; Burleigh, B.A.; Saeij, J.P.J.; Ploegh, H.L.; et al. Determinants of GBP Recruitment to Toxoplasma Gondii Vacuoles and the Parasitic Factors That Control It. PLoS ONE 2011, 6, e24434. [Google Scholar] [CrossRef]
- Selleck, E.M.; Orchard, R.C.; Lassen, K.G.; Beatty, W.L.; Xavier, R.J.; Levine, B.; Virgin, H.W.; Sibley, L.D. A Noncanonical Autophagy Pathway Restricts Toxoplasma Gondii Growth in a Strain-Specific Manner in IFN-γ-Activated Human Cells. mBio 2015, 6, e01157-15. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Sasai, M.; Ma, J.S.; Sakaguchi, N.; Ohshima, J.; Bando, H.; Saitoh, T.; Akira, S.; Yamamoto, M. P62 Plays a Specific Role in Interferon-γ-Induced Presentation of a Toxoplasma Vacuolar Antigen. Cell Rep. 2015, 13, 223–233. [Google Scholar] [CrossRef] [PubMed]
- Haldar, A.K.; Foltz, C.; Finethy, R.; Piro, A.S.; Feeley, E.M.; Pilla-Moffett, D.M.; Komatsu, M.; Frickel, E.-M.; Coers, J. Ubiquitin Systems Mark Pathogen-Containing Vacuoles as Targets for Host Defense by Guanylate Binding Proteins. Proc. Natl. Acad. Sci. USA 2015, 112, E5628–E5637. [Google Scholar] [CrossRef]
- Foltz, C.; Napolitano, A.; Khan, R.; Clough, B.; Hirst, E.M.; Frickel, E.-M. TRIM21 Is Critical for Survival of Toxoplasma Gondii Infection and Localises to GBP-Positive Parasite Vacuoles. Sci. Rep. 2017, 7, 5209. [Google Scholar] [CrossRef]
- Choi, J.; Park, S.; Biering, S.B.; Selleck, E.; Liu, C.Y.; Zhang, X.; Fujita, N.; Saitoh, T.; Akira, S.; Yoshimori, T.; et al. The Parasitophorous Vacuole Membrane of Toxoplasma Gondii Is Targeted for Disruption by Ubiquitin-like Conjugation Systems of Autophagy. Immunity 2014, 40, 924–935. [Google Scholar] [CrossRef]
- Sasai, M.; Sakaguchi, N.; Ma, J.S.; Nakamura, S.; Kawabata, T.; Bando, H.; Lee, Y.; Saitoh, T.; Akira, S.; Iwasaki, A.; et al. Essential Role for GABARAP Autophagy Proteins in Interferon-Inducible GTPase-Mediated Host Defense. Nat. Immunol. 2017, 18, 899–910. [Google Scholar] [CrossRef]
- Jabado, N.; Jankowski, A.; Dougaparsad, S.; Picard, V.; Grinstein, S.; Gros, P. Natural Resistance to Intracellular Infections. J. Exp. Med. 2000, 192, 1237–1248. [Google Scholar] [CrossRef] [PubMed]
- Kontoghiorghes, G.J.; Weinberg, E.D. Iron: Mammalian Defense Systems, Mechanisms of Disease, and Chelation Therapy Approaches. Blood Rev. 1995, 9, 33–45. [Google Scholar] [CrossRef] [PubMed]
- Vidal, S.; Tremblay, M.L.; Govoni, G.; Gauthier, S.; Sebastiani, G.; Malo, D.; Skamene, E.; Olivier, M.; Jothy, S.; Gros, P. The Ity/Lsh/Bcg Locus: Natural Resistance to Infection with Intracellular Parasites Is Abrogated by Disruption of the Nramp1 Gene. J. Exp. Med. 1995, 182, 655–666. [Google Scholar] [CrossRef] [PubMed]
- Savitz, J. The Kynurenine Pathway: A Finger in Every Pie. Mol. Psychiatry 2020, 25, 131–147. [Google Scholar] [CrossRef] [PubMed]
- Pfefferkorn, E.R. Interferon Gamma Blocks the Growth of Toxoplasma Gondii in Human Fibroblasts by Inducing the Host Cells to Degrade Tryptophan. Proc. Natl. Acad. Sci. USA 1984, 81, 908–912. [Google Scholar] [CrossRef] [PubMed]
- Däubener, W.; Spors, B.; Hucke, C.; Adam, R.; Stins, M.; Kim, K.S.; Schroten, H. Restriction of Toxoplasma Gondii Growth in Human Brain Microvascular Endothelial Cells by Activation of Indoleamine 2,3-Dioxygenase. Infect. Immun. 2001, 69, 6527–6531. [Google Scholar] [CrossRef]
- Nagineni, C.N.; Pardhasaradhi, K.; Martins, M.C.; Detrick, B.; Hooks, J.J. Mechanisms of Interferon-Induced Inhibition of Toxoplasma Gondii Replication in Human Retinal Pigment Epithelial Cells. Infect. Immun. 1996, 64, 4188–4196. [Google Scholar] [CrossRef]
- Murray, H.W.; Szuro-Sudol, A.; Wellner, D.; Oca, M.J.; Granger, A.M.; Libby, D.M.; Rothermel, C.D.; Rubin, B.Y. Role of Tryptophan Degradation in Respiratory Burst-Independent Antimicrobial Activity of Gamma Interferon-Stimulated Human Macrophages. Infect. Immun. 1989, 57, 845–849. [Google Scholar] [CrossRef]
- Däubener, W.; Remscheid, C.; Nockemann, S.; Pilz, K.; Seghrouchni, S.; Mackenzie, C.; Hadding, U. Anti-Parasitic Effector Mechanisms in Human Brain Tumor Cells: Role of Interferon-Gamma and Tumor Necrosis Factor-Alpha. Eur. J. Immunol. 1996, 26, 487–492. [Google Scholar] [CrossRef]
- Gupta, S.L.; Carlin, J.M.; Pyati, P.; Dai, W.; Pfefferkorn, E.R.; Murphy, M.J. Antiparasitic and Antiproliferative Effects of Indoleamine 2,3-Dioxygenase Enzyme Expression in Human Fibroblasts. Infect. Immun. 1994, 62, 2277–2284. [Google Scholar] [CrossRef]
- Heseler, K.; Spekker, K.; Schmidt, S.K.; MacKenzie, C.R.; Däubener, W. Antimicrobial and Immunoregulatory Effects Mediated by Human Lung Cells: Role of IFN-Gamma-Induced Tryptophan Degradation. FEMS Immunol. Med. Microbiol. 2008, 52, 273–281. [Google Scholar] [CrossRef] [PubMed]
- Divanovic, S.; Sawtell, N.M.; Trompette, A.; Warning, J.I.; Dias, A.; Cooper, A.M.; Yap, G.S.; Arditi, M.; Shimada, K.; DuHadaway, J.B.; et al. Opposing Biological Functions of Tryptophan Catabolizing Enzymes During Intracellular Infection. J. Infect. Dis. 2012, 205, 152–161. [Google Scholar] [CrossRef] [PubMed]
- Knubel, C.P.; Martínez, F.F.; Fretes, R.E.; Lujan, C.D.; Theumer, M.G.; Cervi, L.; Motrán, C.C. Indoleamine 2,3-dioxigenase (IDO) Is Critical for Host Resistance against Trypanosoma Cruzi. FASEB J. 2010, 24, 2689–2701. [Google Scholar] [CrossRef]
- Doolan, D.L.; Sedegah, M.; Hedstrom, R.C.; Hobart, P.; Yupin, C. Hoffman Circumventing Genetic Restriction of Protection against Malaria with Multigene DNA Immunization: CD8+ Cell-, Interferon Gamma-, and Nitric Oxide-Dependent Immunity. J. Exp. Med. 1996, 183, 1739–1746. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, Y.; Sa, Q.; Gehman, M.; Ochiai, E. Interferon-Gamma- and Perforin-Mediated Immune Responses for Resistance against Toxoplasma Gondii in the Brain. Expert Rev. Mol. Med. 2011, 13, e31. [Google Scholar] [CrossRef]
- Su, Z.; Stevenson, M.M. Central Role of Endogenous Gamma Interferon in Protective Immunity against Blood-Stage Plasmodium Chabaudi AS Infection. Infect. Immun. 2000, 68, 4399–4406. [Google Scholar] [CrossRef]
- Wassmer, S.C.; Combes, V.; Candal, F.J.; Juhan-Vague, I.; Grau, G.E. Platelets Potentiate Brain Endothelial Alterations Induced by Plasmodium Falciparum. Infect. Immun. 2006, 74, 645–653. [Google Scholar] [CrossRef] [PubMed]
- Wassmer, S.C.; Lépolard, C.; Traoré, B.; Pouvelle, B.; Gysin, J.; Grau, G.E. Platelets Reorient Plasmodium Falciparum-Infected Erythrocyte Cytoadhesion to Activated Endothelial Cells. J. Infect. Dis. 2004, 189, 180–189. [Google Scholar] [CrossRef]
- Uzonna, J.E.; Kaushik, R.S.; Gordon, J.R.; Tabel, H. Cytokines and Antibody Responses during Trypanosoma Congolense Infections in Two Inbred Mouse Strains That Differ in Resistance. Parasite Immunol. 1999, 21, 57–71. [Google Scholar] [CrossRef]
- Tabel, H.; Kaushik, R.S.; Uzonna, J.E. Susceptibility and Resistance to Trypanosoma Congolense Infections. Microbes Infect. 2000, 2, 1619–1629. [Google Scholar] [CrossRef]
- Tabel, H.; Wei, G.; Shi, M. T Cells and Immunopathogenesis of Experimental African Trypanosomiasis. Immunol. Rev. 2008, 225, 128–139. [Google Scholar] [CrossRef]
- Stijlemans, B.; Beschin, A.; Magez, S.; Van Ginderachter, J.A.; De Baetselier, P. Iron Homeostasis and Trypanosoma Brucei Associated Immunopathogenicity Development: A Battle/Quest for Iron. Biomed Res. Int. 2015, 2015, 819389. [Google Scholar] [CrossRef] [PubMed]
- Teijaro, J.R.; Ng, C.; Lee, A.M.; Sullivan, B.M.; Sheehan, K.C.F.; Welch, M.; Schreiber, R.D.; de la Torre, J.C.; Oldstone, M.B.A. Persistent LCMV Infection Is Controlled by Blockade of Type I Interferon Signaling. Science 2013, 340, 207–211. [Google Scholar] [CrossRef] [PubMed]
- Wilson, E.B.; Yamada, D.H.; Elsaesser, H.; Herskovitz, J.; Deng, J.; Cheng, G.; Aronow, B.J.; Karp, C.L.; Brooks, D.G. Blockade of Chronic Type I Interferon Signaling to Control Persistent LCMV Infection. Science 2013, 340, 202–207. [Google Scholar] [CrossRef]
- Arimori, Y.; Nakamura, R.; Yamada, H.; Shibata, K.; Maeda, N.; Kase, T.; Yoshikai, Y. Type I Interferon Limits Influenza Virus-Induced Acute Lung Injury by Regulation of Excessive Inflammation in Mice. Antivir. Res. 2013, 99, 230–237. [Google Scholar] [CrossRef]
- Teles, R.M.B.; Graeber, T.G.; Krutzik, S.R.; Montoya, D.; Schenk, M.; Lee, D.J.; Komisopoulou, E.; Kelly-Scumpia, K.; Chun, R.; Iyer, S.S.; et al. Type I Interferon Suppresses Type II Interferon-Triggered Human Anti-Mycobacterial Responses. Science 2013, 339, 1448–1453. [Google Scholar] [CrossRef]
- Antonelli, L.R.V.; Rothfuchs, A.G.; Gonçalves, R.; Roffê, E.; Cheever, A.W.; Bafica, A.; Salazar, A.M.; Feng, C.G.; Sher, A. Intranasal Poly-IC Treatment Exacerbates Tuberculosis in Mice through the Pulmonary Recruitment of a Pathogen-Permissive Monocyte/Macrophage Population. J. Clin. Investig. 2010, 120, 1674–1682. [Google Scholar] [CrossRef] [PubMed]
- Carrero, J.A.; Calderon, B.; Unanue, E.R. Type I Interferon Sensitizes Lymphocytes to Apoptosis and Reduces Resistance to Listeria Infection. J. Exp. Med. 2004, 200, 535–540. [Google Scholar] [CrossRef]
- Auerbuch, V.; Brockstedt, D.G.; Meyer-Morse, N.; O’Riordan, M.; Portnoy, D.A. Mice Lacking the Type I Interferon Receptor Are Resistant to Listeria Monocytogenes. J. Exp. Med. 2004, 200, 527–533. [Google Scholar] [CrossRef]
- Liehl, P.; Zuzarte-Luís, V.; Chan, J.; Zillinger, T.; Baptista, F.; Carapau, D.; Konert, M.; Hanson, K.K.; Carret, C.; Lassnig, C.; et al. Host-Cell Sensors for Plasmodium Activate Innate Immunity against Liver-Stage Infection. Nat. Med. 2014, 20, 47–53. [Google Scholar] [CrossRef]
- Montes de Oca, M.; Kumar, R.; Rivera, F.D.L.; Amante, F.H.; Sheel, M.; Faleiro, R.J.; Bunn, P.T.; Best, S.E.; Beattie, L.; Ng, S.S.; et al. Type I Interferons Regulate Immune Responses in Humans with Blood-Stage Plasmodium Falciparum Infection. Cell Rep. 2016, 17, 399–412. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.C.; Nelson, C.S.; Wilson, E.B.; Hou, B.; DeFranco, A.L.; DeRisi, J.L. Splenic Red Pulp Macrophages Produce Type I Interferons as Early Sentinels of Malaria Infection but Are Dispensable for Control. PLoS ONE 2012, 7, e48126. [Google Scholar] [CrossRef] [PubMed]
- Melo, M.B.; Nguyen, Q.P.; Cordeiro, C.; Hassan, M.A.; Yang, N.; McKell, R.; Rosowski, E.E.; Julien, L.; Butty, V.; Dardé, M.-L.; et al. Transcriptional Analysis of Murine Macrophages Infected with Different Toxoplasma Strains Identifies Novel Regulation of Host Signaling Pathways. PLOS Pathog. 2013, 9, e1003779. [Google Scholar] [CrossRef]
- Beiting, D.P.; Peixoto, L.; Akopyants, N.S.; Beverley, S.M.; Wherry, E.J.; Christian, D.A.; Hunter, C.A.; Brodsky, I.E.; Roos, D.S. Differential Induction of TLR3-Dependent Innate Immune Signaling by Closely Related Parasite Species. PLoS ONE 2014, 9, e88398. [Google Scholar] [CrossRef]
- Koblansky, A.A.; Jankovic, D.; Oh, H.; Hieny, S.; Sungnak, W.; Mathur, R.; Hayden, M.S.; Akira, S.; Sher, A.; Ghosh, S. Recognition of Profilin by Toll-like Receptor 12 Is Critical for Host Resistance to Toxoplasma Gondii. Immunity 2013, 38, 119–130. [Google Scholar] [CrossRef]
- Han, S.-J.; Melichar, H.J.; Coombes, J.L.; Chan, S.W.; Koshy, A.A.; Boothroyd, J.C.; Barton, G.M.; Robey, E.A. Internalization and TLR-Dependent Type I Interferon Production by Monocytes in Response to Toxoplasma Gondii. Immunol. Cell Biol. 2014, 92, 872–881. [Google Scholar] [CrossRef]
- Diefenbach, A.; Schindler, H.; Donhauser, N.; Lorenz, E.; Laskay, T.; MacMicking, J.; Röllinghoff, M.; Gresser, I.; Bogdan, C. Type 1 Interferon (IFNα/β) and Type 2 Nitric Oxide Synthase Regulate the Innate Immune Response to a Protozoan Parasite. Immunity 1998, 8, 77–87. [Google Scholar] [CrossRef]
- Ives, A.; Ronet, C.; Prevel, F.; Ruzzante, G.; Fuertes-Marraco, S.; Schutz, F.; Zangger, H.; Revaz-Breton, M.; Lye, L.-F.; Hickerson, S.M.; et al. Leishmania RNA Virus Controls the Severity of Mucocutaneous Leishmaniasis. Science 2011, 331, 775–778. [Google Scholar] [CrossRef]
- Dias, B.T.; Goundry, A.; Vivarini, A.C.; Costa, T.F.R.; Mottram, J.C.; Lopes, U.G.; Lima, A.P.C.A. Toll-Like Receptor- and Protein Kinase R-Induced Type I Interferon Sustains Infection of Leishmania Donovani in Macrophages. Front. Immunol. 2022, 13, 801182. [Google Scholar] [CrossRef]
- Sonnenfeld, G.; Kierszenbaum, F. Increased Serum Levels of an Interferon-like Activity during the Acute Period of Experimental Infection with Different Strains of Trypanosoma Cruzi. Am. J. Trop. Med. Hyg. 1981, 30, 1189–1191. [Google Scholar] [CrossRef]
- Kierszenbaum, F.; Sonnenfeld, G. Characterization of the Antiviral Activity Produced during Trypanosoma Cruzi Infection and Protective Effects of Exogenous Interferon against Experimental Chagas’ Disease. J. Parasitol. 1982, 68, 194–198. [Google Scholar] [CrossRef] [PubMed]
- Une, C.; Andersson, J.; Eloranta, M.-L.; Sunnemark, D.; Harris, R.A.; Örn, A. Enhancement of Natural Killer (NK) Cell Cytotoxicity and Induction of NK Cell-Derived Interferon-Gamma (IFN-γ) Display Different Kinetics during Experimental Infection with Trypanosoma Cruzi. Clin. Exp. Immunol. 2000, 121, 499–505. [Google Scholar] [CrossRef] [PubMed]
- de Avalos, S.V.; Blader, I.J.; Fisher, M.; Boothroyd, J.C.; Burleigh, B.A. Immediate/Early Response to Trypanosoma Cruzi Infection Involves Minimal Modulation of Host Cell Transcription. J. Biol. Chem. 2002, 277, 639–644. [Google Scholar] [CrossRef]
- Chessler, A.-D.C.; Unnikrishnan, M.; Bei, A.K.; Daily, J.P.; Burleigh, B.A. Trypanosoma Cruzi Triggers an Early Type I IFN Response In Vivo at the Site of Intradermal Infection. J. Immunol. 2009, 182, 2288–2296. [Google Scholar] [CrossRef]
- Koga, R.; Hamano, S.; Kuwata, H.; Atarashi, K.; Ogawa, M.; Hisaeda, H.; Yamamoto, M.; Akira, S.; Himeno, K.; Matsumoto, M.; et al. TLR-Dependent Induction of IFN-β Mediates Host Defense against Trypanosoma Cruzi. J. Immunol. 2006, 177, 7059–7066. [Google Scholar] [CrossRef]
- Barakat, F.M.; McDonald, V.; Foster, G.R.; Tovey, M.G.; Korbel, D.S. Cryptosporidium parvum Infection Rapidly Induces a Protective Innate Immune Response Involving Type I Interferon. J. Infect. Dis. 2009, 200, 1548–1555. [Google Scholar] [CrossRef]
- Heo, I.; Dutta, D.; Schaefer, D.A.; Iakobachvili, N.; Artegiani, B.; Sachs, N.; Boonekamp, K.E.; Bowden, G.; Hendrickx, A.P.A.; Willems, R.J.L.; et al. Modelling Cryptosporidium Infection in Human Small Intestinal and Lung Organoids. Nat. Microbiol. 2018, 3, 814–823. [Google Scholar] [CrossRef]
- Gibson, A.R.; Sateriale, A.; Dumaine, J.E.; Engiles, J.B.; Pardy, R.D.; Gullicksrud, J.A.; O’Dea, K.M.; Doench, J.G.; Beiting, D.P.; Hunter, C.A.; et al. A Genetic Screen Identifies a Protective Type III Interferon Response to Cryptosporidium That Requires TLR3 Dependent Recognition. PLoS Pathog. 2022, 18, e1010003. [Google Scholar] [CrossRef]
- Costa, V.M.A.; Torres, K.C.L.; Mendonça, R.Z.; Gresser, I.; Gollob, K.J.; Abrahamsohn, I.A. Type I IFNs Stimulate Nitric Oxide Production and Resistance to Trypanosoma Cruzi Infection. J. Immunol. 2006, 177, 3193–3200. [Google Scholar] [CrossRef]
- Lopez, R.; Demick, K.P.; Mansfield, J.M.; Paulnock, D.M. Type I IFNs Play a Role in Early Resistance, but Subsequent Susceptibility, to the African Trypanosomes. J. Immunol. 2008, 181, 4908–4917. [Google Scholar] [CrossRef]
- Colina, R.; Costa-Mattioli, M.; Dowling, R.J.O.; Jaramillo, M.; Tai, L.-H.; Breitbach, C.J.; Martineau, Y.; Larsson, O.; Rong, L.; Svitkin, Y.V.; et al. Translational Control of the Innate Immune Response through IRF-7. Nature 2008, 452, 323–328. [Google Scholar] [CrossRef] [PubMed]
- Jaramillo, M.; Gomez, M.A.; Larsson, O.; Shio, M.T.; Topisirovic, I.; Contreras, I.; Luxenburg, R.; Rosenfeld, A.; Colina, R.; McMaster, R.W.; et al. Leishmania Repression of Host Translation through MTOR Cleavage Is Required for Parasite Survival and Infection. Cell Host Microbe 2011, 9, 331–341. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Carvalho, L.P.; Bhattacharya, S.; Carbone, C.J.; Kumar, K.G.S.; Leu, N.A.; Yau, P.M.; Donald, R.G.K.; Weiss, M.J.; Baker, D.P.; et al. Mammalian Casein Kinase 1α and Its Leishmanial Ortholog Regulate Stability of IFNAR1 and Type I Interferon Signaling. Mol. Cell. Biol. 2009, 29, 6401–6412. [Google Scholar] [CrossRef] [PubMed]
- Rosowski, E.E.; Nguyen, Q.P.; Camejo, A.; Spooner, E.; Saeij, J.P.J. Toxoplasma Gondii Inhibits Gamma Interferon (IFN-γ)- and IFN-β-Induced Host Cell STAT1 Transcriptional Activity by Increasing the Association of STAT1 with DNA. Infect. Immun. 2014, 82, 706–719. [Google Scholar] [CrossRef]
- Rocha, B.C.; Marques, P.E.; Leoratti, F.M.d.S.; Junqueira, C.; Pereira, D.B.; Antonelli, L.R.d.V.; Menezes, G.B.; Golenbock, D.T.; Gazzinelli, R.T. Type I Interferon Transcriptional Signature in Neutrophils and High Frequency of Low-Density Granulocytes Are Associated with Tissue Damage in Malaria. Cell Rep. 2015, 13, 2829–2841. [Google Scholar] [CrossRef]
- Sharma, S.; DeOliveira, R.B.; Kalantari, P.; Parroche, P.; Goutagny, N.; Jiang, Z.; Chan, J.; Bartholomeu, D.C.; Lauw, F.; Hall, J.P.; et al. Innate Immune Recognition of an AT-Rich Stem-Loop DNA Motif in the Plasmodium Falciparum Genome. Immunity 2011, 35, 194–207. [Google Scholar] [CrossRef]
- Haque, A.; Best, S.E.; Montes de Oca, M.; James, K.R.; Ammerdorffer, A.; Edwards, C.L.; de Labastida Rivera, F.; Amante, F.H.; Bunn, P.T.; Sheel, M.; et al. Type I IFN Signaling in CD8– DCs Impairs Th1-Dependent Malaria Immunity. J. Clin. Investig. 2014, 124, 2483–2496. [Google Scholar] [CrossRef]
- Khouri, R.; Bafica, A.; Silva, M.D.P.P.; Noronha, A.; Kolb, J.-P.; Wietzerbin, J.; Barral, A.; Barral-Netto, M.; Van Weyenbergh, J. IFN-Beta Impairs Superoxide-Dependent Parasite Killing in Human Macrophages: Evidence for a Deleterious Role of SOD1 in Cutaneous Leishmaniasis. J. Immunol. 2009, 182, 2525–2531. [Google Scholar] [CrossRef]
- Xin, L.; Vargas-Inchaustegui, D.A.; Raimer, S.S.; Kelly, B.C.; Hu, J.; Zhu, L.; Sun, J.; Soong, L. Type I IFN Receptor Regulates Neutrophil Functions and Innate Immunity to Leishmania Parasites. J. Immunol. 2010, 184, 7047–7056. [Google Scholar] [CrossRef]
- Une, C.; Andersson, J.; Örn, A. Role of IFN-α/β and IL-12 in the Activation of Natural Killer Cells and Interferon-γ Production during Experimental Infection with Trypanosoma Cruzi. Clin. Exp. Immunol. 2003, 134, 195–201. [Google Scholar] [CrossRef]
- Bankoti, R.; Gupta, K.; Levchenko, A.; Stäger, S. Marginal Zone B Cells Regulate Antigen-Specific T Cell Responses during Infection. J. Immunol. 2012, 188, 3961–3971. [Google Scholar] [CrossRef]
- Silva-Barrios, S.; Smans, M.; Duerr, C.U.; Qureshi, S.T.; Fritz, J.H.; Descoteaux, A.; Stäger, S. Innate Immune B Cell Activation by Leishmania Donovani Exacerbates Disease and Mediates Hypergammaglobulinemia. Cell Rep. 2016, 15, 2427–2437. [Google Scholar] [CrossRef] [PubMed]
- Stifter, S.A.; Feng, C.G. Interfering with Immunity: Detrimental Role of Type I IFNs during Infection. J. Immunol. 2015, 194, 2455–2465. [Google Scholar] [CrossRef] [PubMed]
- Tran, T.M.; Jones, M.B.; Ongoiba, A.; Bijker, E.M.; Schats, R.; Venepally, P.; Skinner, J.; Doumbo, S.; Quinten, E.; Visser, L.G.; et al. Transcriptomic Evidence for Modulation of Host Inflammatory Responses during Febrile Plasmodium Falciparum Malaria. Sci. Rep. 2016, 6, 31291. [Google Scholar] [CrossRef] [PubMed]
- Hahn, W.O.; Pepper, M.; Liles, W.C. B Cell Intrinsic Expression of IFNλ Receptor Suppresses the Acute Humoral Immune Response to Experimental Blood-Stage Malaria. Virulence 2020, 11, 594–606. [Google Scholar] [CrossRef]
- Ferguson, S.H.; Foster, D.M.; Sherry, B.; Magness, S.T.; Nielsen, D.M.; Gookin, J.L. Interferon-Λ3 Promotes Epithelial Defense and Barrier Function Against Cryptosporidium Parvum Infection. Cell. Mol. Gastroenterol. Hepatol. 2019, 8, 1–20. [Google Scholar] [CrossRef]
- Lazear, H.M.; Schoggins, J.W.; Diamond, M.S. Shared and Distinct Functions of Type I and Type III Interferons. Immunity 2019, 50, 907–923. [Google Scholar] [CrossRef]
- Manry, J.; Laval, G.; Patin, E.; Fornarino, S.; Itan, Y.; Fumagalli, M.; Sironi, M.; Tichit, M.; Bouchier, C.; Casanova, J.-L.; et al. Evolutionary Genetic Dissection of Human Interferons. J. Exp. Med. 2011, 208, 2747–2759. [Google Scholar] [CrossRef]
- Manry, J.; Laval, G.; Patin, E.; Fornarino, S.; Tichit, M.; Bouchier, C.; Barreiro, L.B.; Quintana-Murci, L. Evolutionary Genetics Evidence of an Essential, Nonredundant Role of the IFN-γ Pathway in Protective Immunity. Hum. Mutat. 2011, 32, 633–642. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).