
Screening the Pathogen Box to Discover and Characterize New Cruzain and TbrCatL Inhibitors

 and
and
Abstract
1. Introduction
2. Materials and Methods
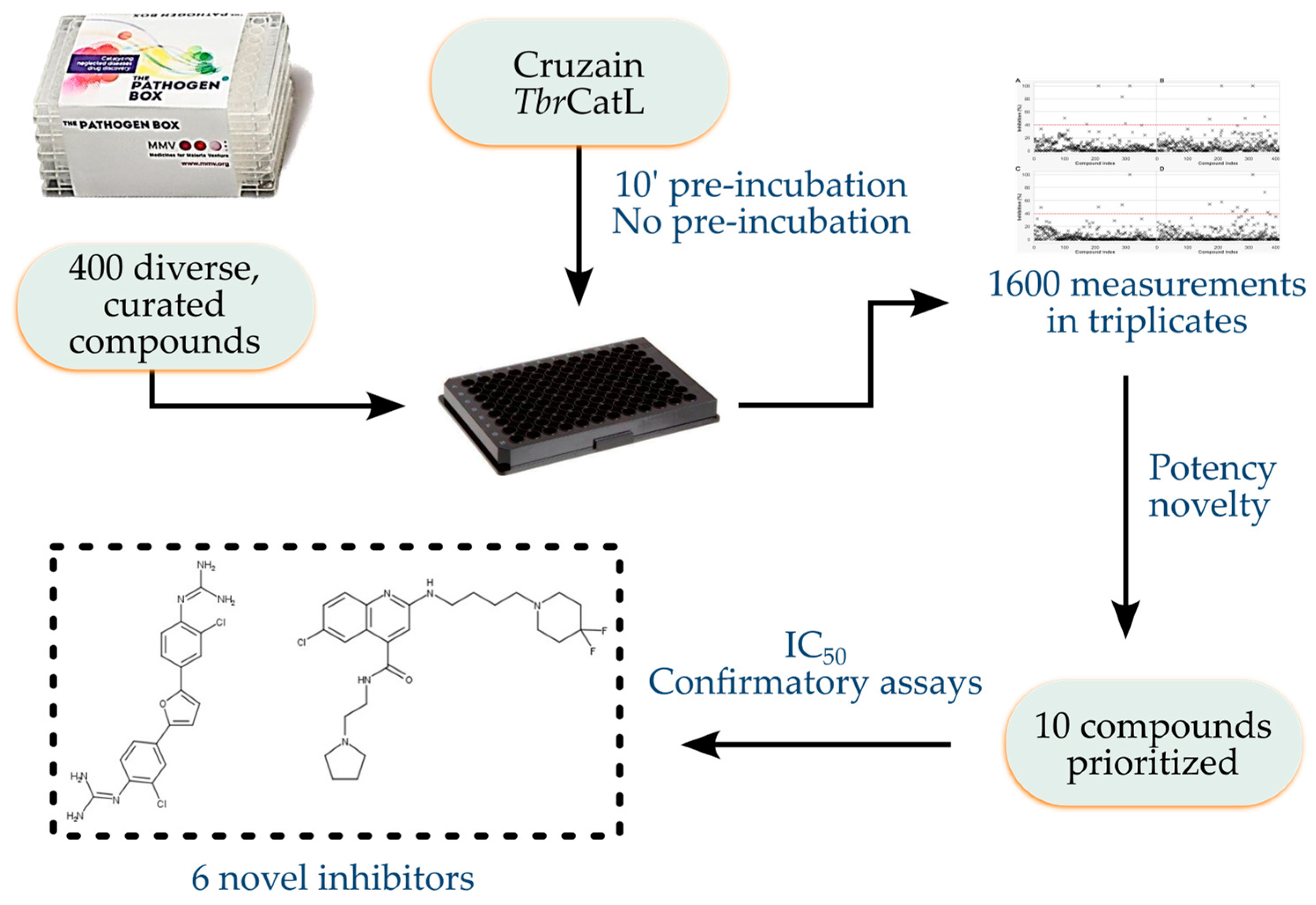
2.1. Data on the PB Collection
2.2. PB Collection Samples
2.3. Assays against Cruzain and TbrCatL
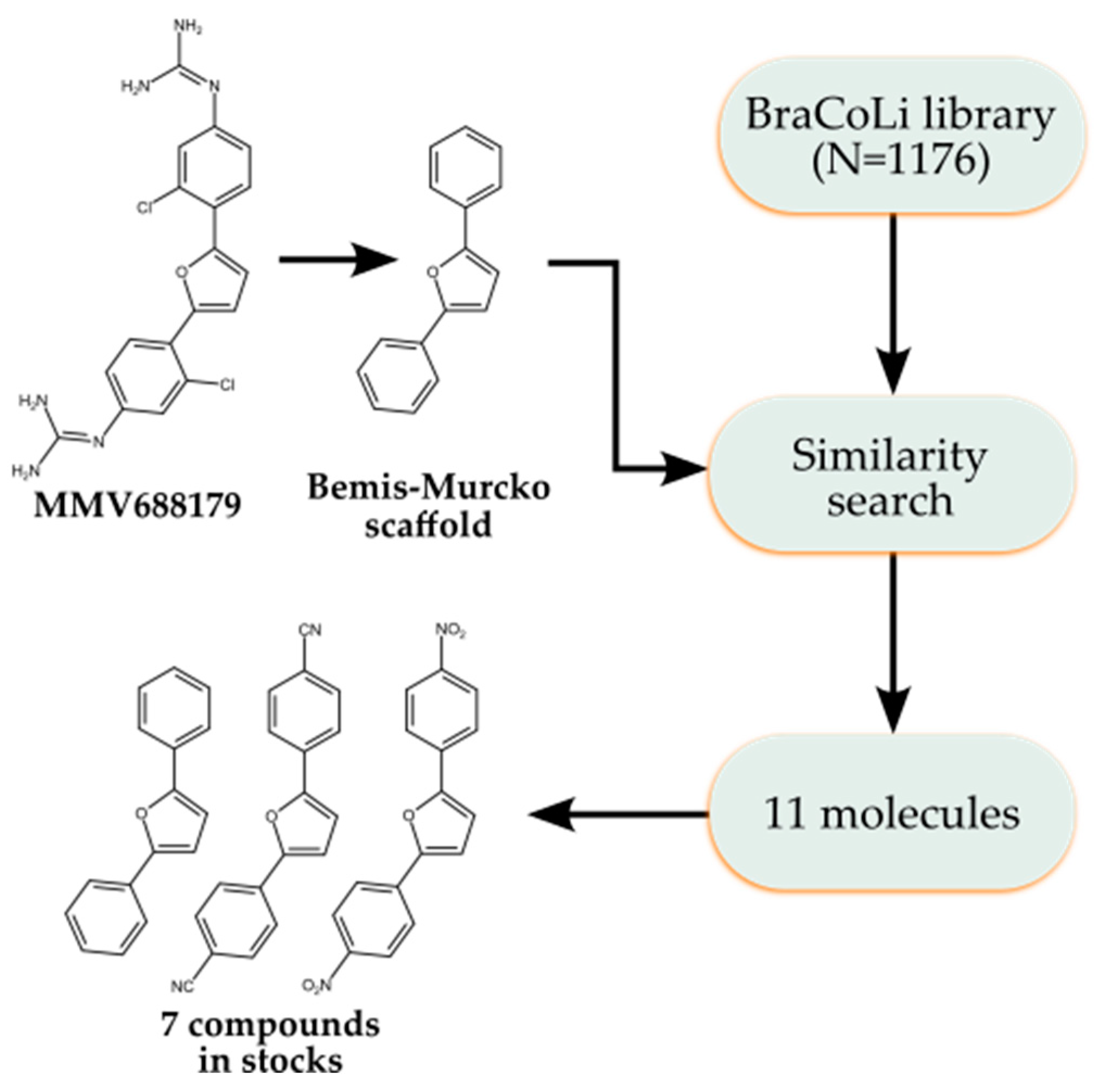
2.4. Analogue Search
2.5. Synthesis and Characterization
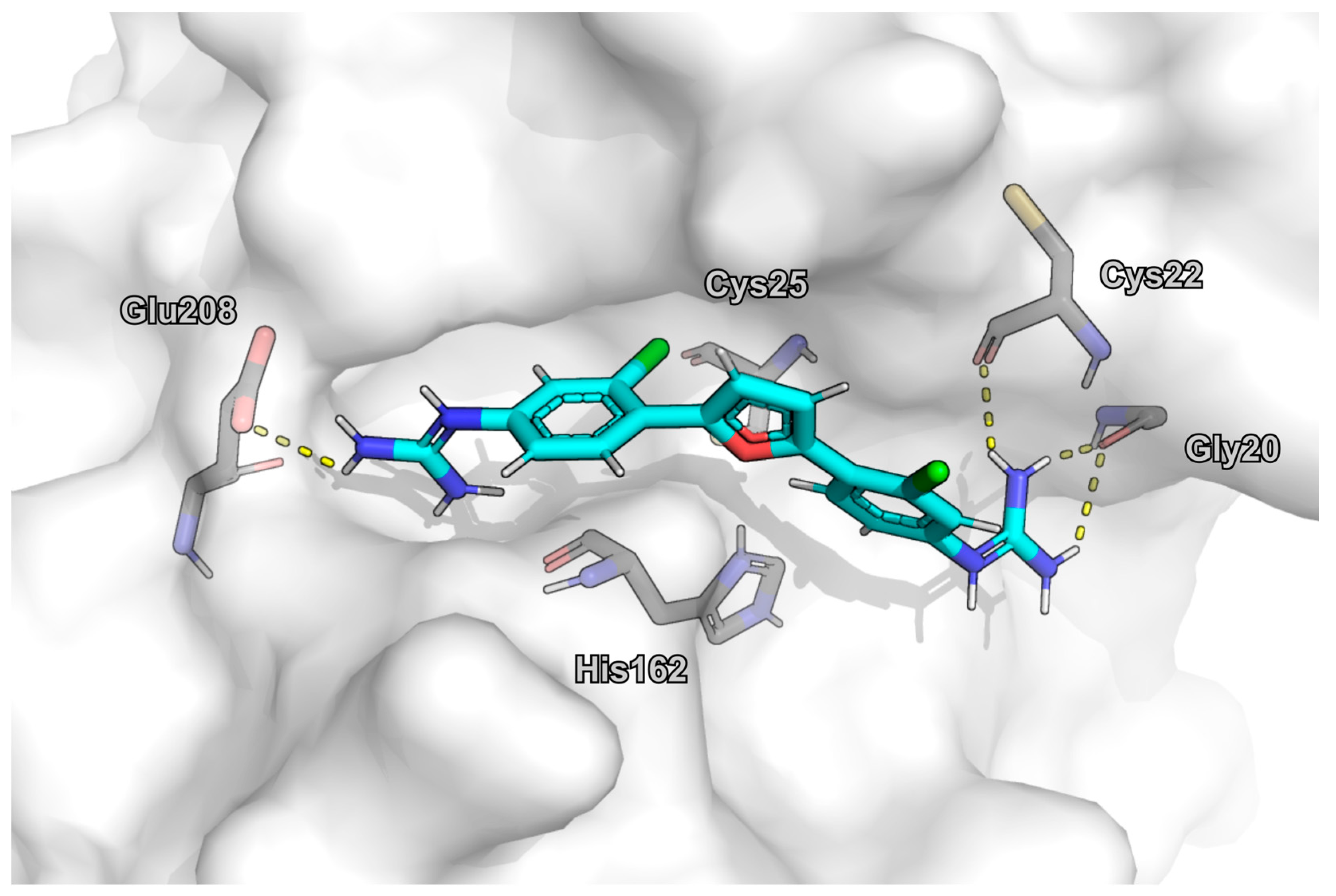
2.6. Molecular Docking
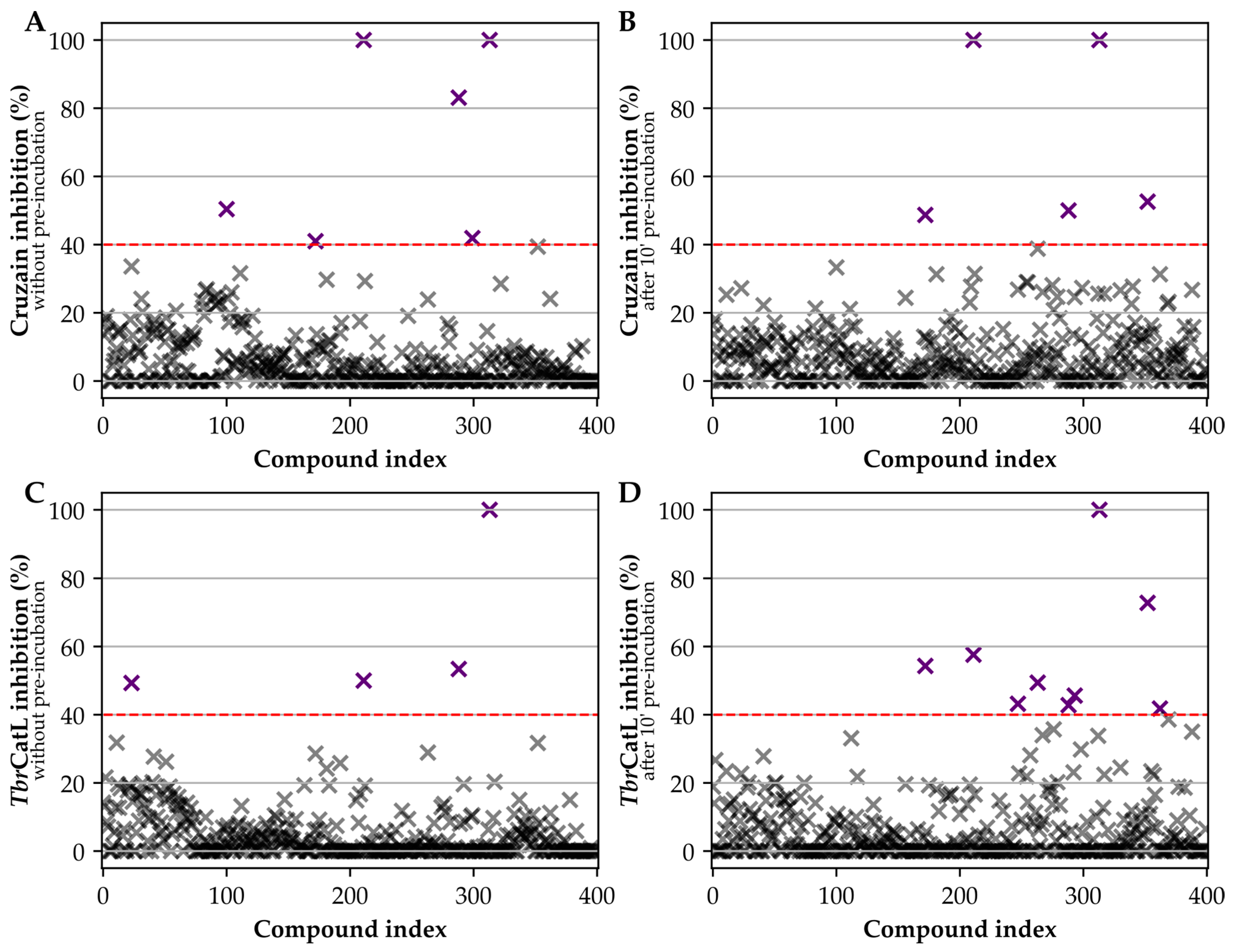
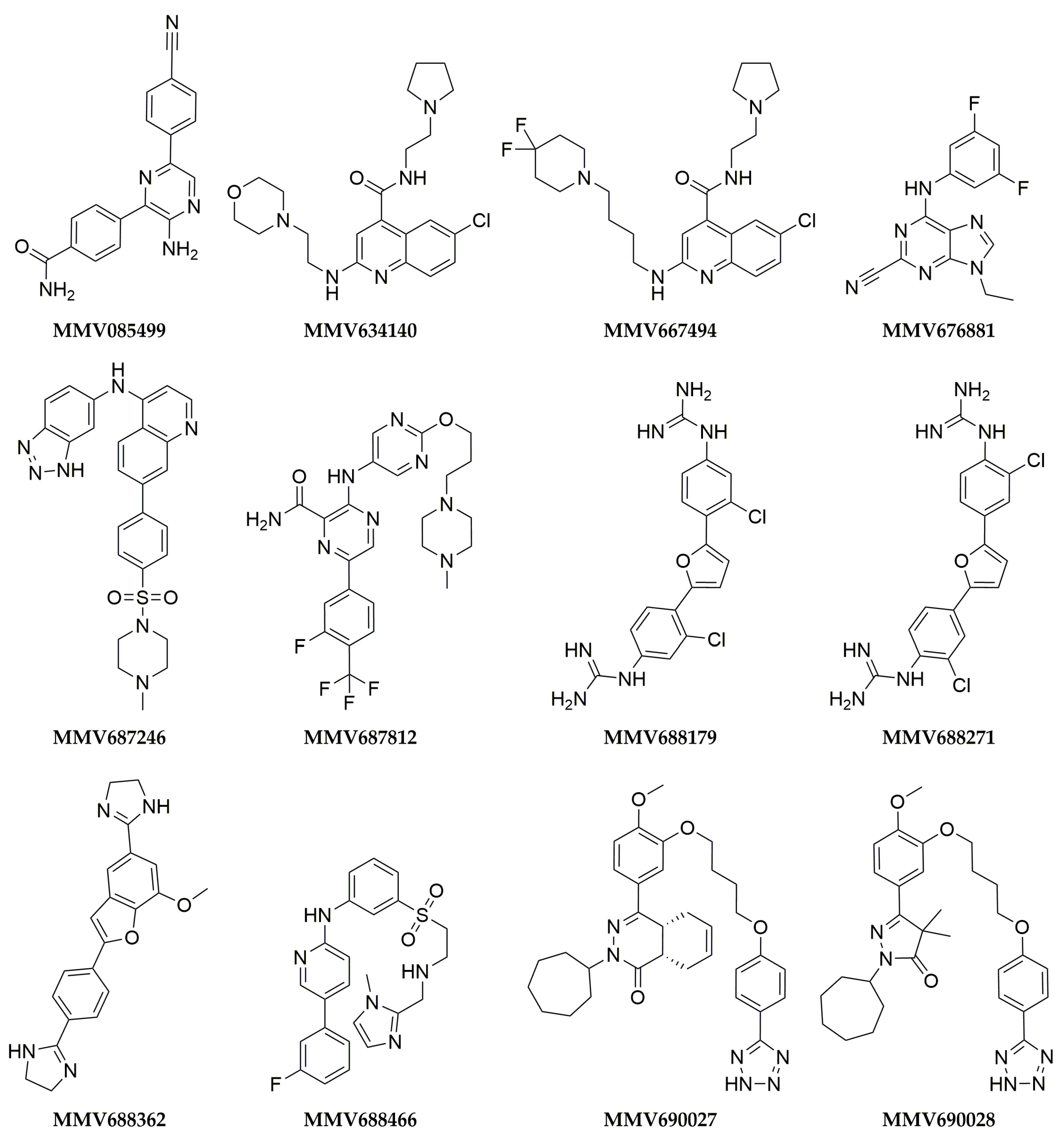
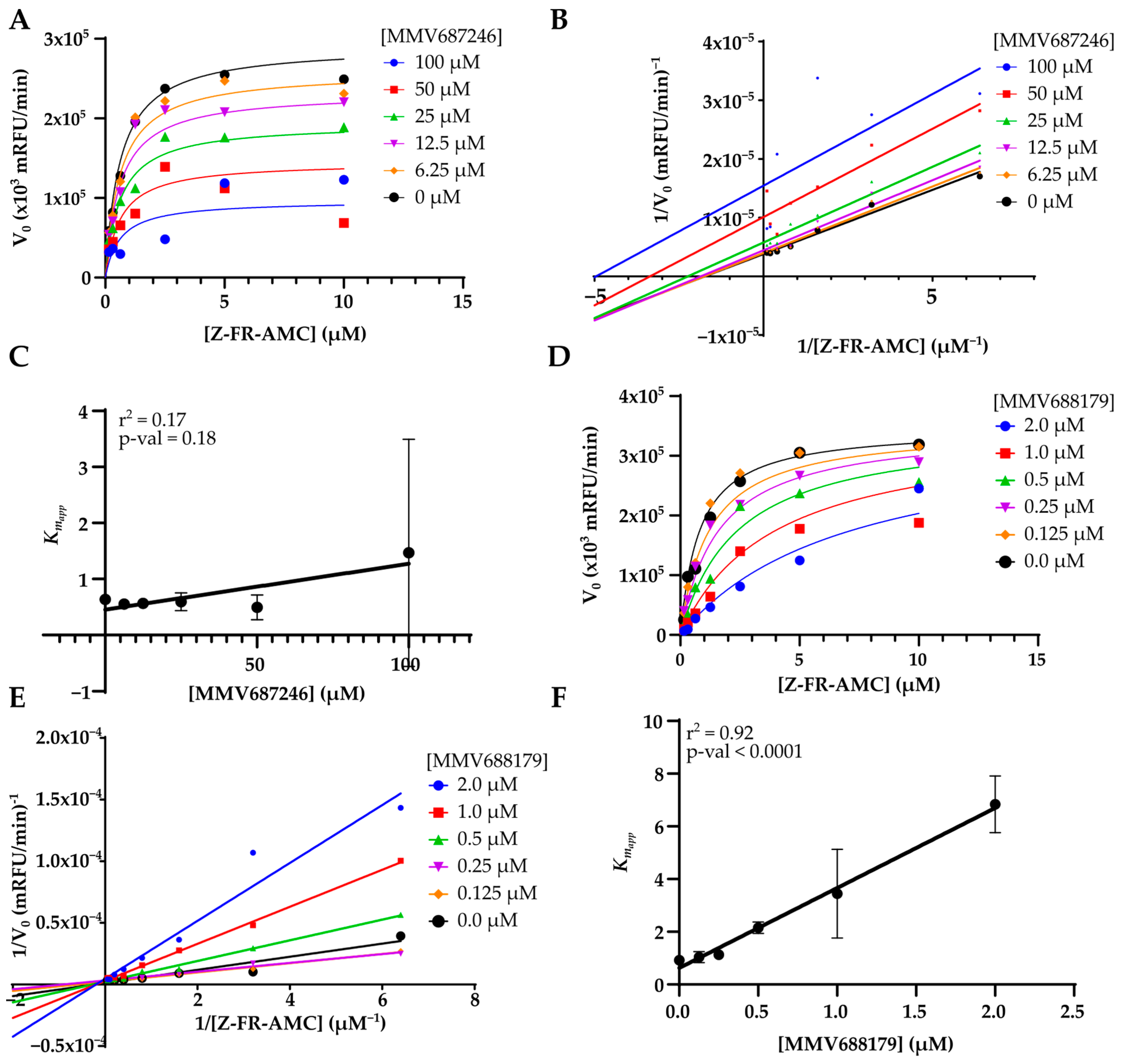
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Nussbaum, K.; Honek, J.; Cadmus, C.; Efferth, T. Trypanosomatid Parasites Causing Neglected Diseases. Curr. Med. Chem. 2010, 17, 1594–1617. [Google Scholar] [CrossRef] [PubMed]
- Stanaway, J.D.; Gregory, R. The Global Burden of Disease: Generating Evidence, Guiding Policy: European Union and European Free Trade Association Regional Edition; Institute for Health Metrics and Evaluation: Seattle, WA, USA, 2013; ISBN 978-0-9840910-6-5. [Google Scholar]
- Pinheiro, E.; Brum-Soares, L.; Reis, R.; Cubides, J.-C. Chagas Disease: Review of Needs, Neglect, and Obstacles to Treatment Access in Latin America. Rev. Soc. Bras. Med. Trop. 2017, 50, 296–300. [Google Scholar] [CrossRef]
- Aksoy, S.; Buscher, P.; Lehane, M.; Solano, P.; Van Den Abbeele, J. Human African Trypanosomiasis Control: Achievements and Challenges. PLoS Negl. Trop. Dis. 2017, 11, e0005454. [Google Scholar] [CrossRef] [PubMed]
- WHO Fact Sheet 259-Trypanosomiasis. Human African (Sleeping Sickness). Available online: http://www.who.int/mediacentre/factsheets/fs259/en/ (accessed on 8 December 2015).
- Medone, P.; Ceccarelli, S.; Parham, P.E.; Figuera, A.; Rabinovich, J.E. The Impact of Climate Change on the Geographical Distribution of Two Vectors of Chagas Disease: Implications for the Force of Infection. Philos. Trans. R. Soc. B Biol. Sci. 2015, 370, 20130560. [Google Scholar] [CrossRef]
- Molina, I.; Salvador, F.; Sánchez-Montalvá, A. Actualización en enfermedad de Chagas. Enferm. Infecc. Microbiol. Clínica 2016, 34, 132–138. [Google Scholar] [CrossRef]
- Patrick, G.L. An Introduction to Medicinal Chemistry, 5th ed.; Oxford University Press: Oxford, UK, 2013; ISBN 978-0-19-969739-7. [Google Scholar]
- Lenk, E.J.; Redekop, W.K.; Luyendijk, M.; Fitzpatrick, C.; Niessen, L.; Stolk, W.A.; Tediosi, F.; Rijnsburger, A.J.; Bakker, R.; Hontelez, J.A.C.; et al. Socioeconomic Benefit to Individuals of Achieving 2020 Targets for Four Neglected Tropical Diseases Controlled/Eliminated by Innovative and Intensified Disease Management: Human African Trypanosomiasis, Leprosy, Visceral Leishmaniasis, Chagas Disease. PLoS Negl. Trop. Dis. 2018, 12, e0006250. [Google Scholar] [CrossRef]
- Bermudez, J.; Davies, C.; Simonazzi, A.; Pablo Real, J.; Palma, S. Current Drug Therapy and Pharmaceutical Challenges for Chagas Disease. Acta Trop. 2016, 156, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, L.G.; Andricopulo, A.D. Targeting Cysteine Proteases in Trypanosomatid Disease Drug Discovery. Pharmacol. Ther. 2017, 180, 49–61. [Google Scholar] [CrossRef] [PubMed]
- Rocha, D.A.; Silva, E.B.; Fortes, I.S.; Lopes, M.S.; Ferreira, R.S.; Andrade, S.F. Synthesis and Structure-Activity Relationship Studies of Cruzain and Rhodesain Inhibitors. Eur. J. Med. Chem. 2018, 157, 1426–1459. [Google Scholar] [CrossRef]
- Caffrey, C.R.; Steverding, D.; Ferreira, R.S.; Oliveira, R.B.; O’Donoghue, A.J.; Monti, L.; Ballatore, C.; Bachovchin, K.A.; Ferrins, L.; Pollastri, M.P.; et al. Drug Discovery and Development for Kinetoplastid Diseases. In Burger’s Medicinal Chemistry and Drug Discovery; Wiley: Hoboken, NJ, USA, 2021; pp. 1–79. ISBN 978-0-470-27815-4. [Google Scholar]
- Morillo, C.A.; Marin-Neto, J.A.; Avezum, A.; Sosa-Estani, S.; Rassi, A.; Rosas, F.; Villena, E.; Quiroz, R.; Bonilla, R.; Britto, C.; et al. Randomized Trial of Benznidazole for Chronic Chagas’ Cardiomyopathy. N. Engl. J. Med. 2015, 373, 1295–1306. [Google Scholar] [CrossRef]
- Brun, R.; Blum, J.; Chappuis, F.; Burri, C. Human African Trypanosomiasis. Lancet 2010, 375, 148–159. [Google Scholar] [CrossRef] [PubMed]
- Büscher, P.; Cecchi, G.; Jamonneau, V.; Priotto, G. Human African Trypanosomiasis. Lancet 2017, 390, 2397–2409. [Google Scholar] [CrossRef]
- Fairlamb, A.H.; Horn, D. Melarsoprol Resistance in African Trypanosomiasis. Trends Parasitol. 2018, 34, 481–492. [Google Scholar] [CrossRef] [PubMed]
- Pépin, J.; Khonde, N.; Maiso, F.; Doua, F.; Jaffar, S.; Ngampo, S.; Mpia, B.; Mbulamberi, D.; Kuzoe, F. Short-Course Eflornithine in Gambian Trypanosomiasis: A Multicentre Randomized Controlled Trial. Bull. World Health Organ. 2000, 78, 1284–1295. [Google Scholar] [PubMed]
- Priotto, G.; Pinoges, L.; Fursa, I.B.; Burke, B.; Nicolay, N.; Grillet, G.; Hewison, C.; Balasegaram, M. Safety and Effectiveness of First Line Eflornithine for Trypanosoma Brucei Gambiense Sleeping Sickness in Sudan: Cohort Study. BMJ 2008, 336, 705–708. [Google Scholar] [CrossRef]
- Priotto, G.; Kasparian, S.; Mutombo, W.; Ngouama, D.; Ghorashian, S.; Arnold, U.; Ghabri, S.; Baudin, E.; Buard, V.; Kazadi-Kyanza, S.; et al. Nifurtimox-Eflornithine Combination Therapy for Second-Stage African Trypanosoma Brucei Gambiense Trypanosomiasis: A Multicentre, Randomised, Phase III, Non-Inferiority Trial. Lancet 2009, 374, 56–64. [Google Scholar] [CrossRef]
- Kuemmerle, A.; Schmid, C.; Bernhard, S.; Kande, V.; Mutombo, W.; Ilunga, M.; Lumpungu, I.; Mutanda, S.; Nganzobo, P.; Tete, D.N.; et al. Effectiveness of Nifurtimox Eflornithine Combination Therapy (NECT) in T. b. Gambiense Second Stage Sleeping Sickness Patients in the Democratic Republic of Congo: Report from a Field Study. PLoS Negl. Trop. Dis. 2021, 15, e0009903. [Google Scholar] [CrossRef]
- Kansiime, F.; Adibaku, S.; Wamboga, C.; Idi, F.; Kato, C.D.; Yamuah, L.; Vaillant, M.; Kioy, D.; Olliaro, P.; Matovu, E. A Multicentre, Randomised, Non-Inferiority Clinical Trial Comparing a Nifurtimox-Eflornithine Combination to Standard Eflornithine Monotherapy for Late Stage Trypanosoma Brucei Gambiense Human African Trypanosomiasis in Uganda. Parasites Vectors 2018, 11, 105. [Google Scholar] [CrossRef]
- Pollastri, M.P. Fexinidazole: A New Drug for African Sleeping Sickness on the Horizon. Trends Parasitol. 2018, 34, 178–179. [Google Scholar] [CrossRef]
- Kande Betu Kumesu, V.; Mutombo Kalonji, W.; Bardonneau, C.; Valverde Mordt, O.; Ngolo Tete, D.; Blesson, S.; Simon, F.; Delhomme, S.; Bernhard, S.; Nganzobo Ngima, P.; et al. Safety and Efficacy of Oral Fexinidazole in Children with Gambiense Human African Trypanosomiasis: A Multicentre, Single-Arm, Open-Label, Phase 2–3 Trial. Lancet Glob. Health 2022, 10, e1665–e1674. [Google Scholar] [CrossRef]
- Lindner, A.K.; Lejon, V.; Chappuis, F.; Seixas, J.; Kazumba, L.; Barrett, M.P.; Mwamba, E.; Erphas, O.; Akl, E.A.; Villanueva, G.; et al. New WHO Guidelines for Treatment of Gambiense Human African Trypanosomiasis Including Fexinidazole: Substantial Changes for Clinical Practice. Lancet Infect. Dis. 2020, 20, e38–e46. [Google Scholar] [CrossRef] [PubMed]
- Efficacy and Safety of Fexinidazole in Patients with Human African Trypanosomiasis (HAT) Due to Trypanosoma Brucei Rhodesiense. Available online: https://beta.clinicaltrials.gov/study/NCT03974178 (accessed on 20 December 2022).
- Chen, Y.T.; Brinen, L.S.; Kerr, I.D.; Hansell, E.; Doyle, P.S.; McKerrow, J.H.; Roush, W.R. In Vitro and In Vivo Studies of the Trypanocidal Properties of WRR-483 against Trypanosoma Cruzi. PLoS Negl. Trop. Dis. 2010, 4, e825. [Google Scholar] [CrossRef] [PubMed]
- Barr, S.C.; Warner, K.L.; Kornreic, B.G.; Piscitelli, J.; Wolfe, A.; Benet, L.; McKerrow, J.H. A Cysteine Protease Inhibitor Protects Dogs from Cardiac Damage during Infection by Trypanosoma Cruzi. Antimicrob. Agents Chemother. 2005, 49, 5160–5161. [Google Scholar] [CrossRef] [PubMed]
- da Silva, E.B.; do Nascimento Pereira, G.A.; Ferreira, R.S. Trypanosomal Cysteine Peptidases: Target Validation and Drug Design Strategies. In Comprehensive Analysis of Parasite Biology: From Metabolism to Drug Discovery; Müller, S., Cerdan, R., Radulescu, O., Eds.; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2016; pp. 121–145. ISBN 978-3-527-33904-4. [Google Scholar]
- dos Santos Nascimento, I.J.; de Aquino, T.M.; da Silva-Júnior, E.F. Cruzain and Rhodesain Inhibitors: Last Decade of Advances in Seeking for New Compounds Against American and African Trypanosomiases. Curr. Top. Med. Chem. 2021, 21, 1871–1899. [Google Scholar] [CrossRef] [PubMed]
- Santos, V.C.; Oliveira, A.E.R.; Campos, A.C.B.; Reis-Cunha, J.L.; Bartholomeu, D.C.; Teixeira, S.M.R.; Lima, A.P.C.A.; Ferreira, R.S. The Gene Repertoire of the Main Cysteine Protease of Trypanosoma Cruzi, Cruzipain, Reveals Four Sub-Types with Distinct Active Sites. Sci. Rep. 2021, 11, 18231. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, V.E.; Iribarren, P.A.; Niemirowicz, G.T.; Cazzulo, J.J. Update on Relevant Trypanosome Peptidases: Validated Targets and Future Challenges. Biochim. Biophys. Acta BBA-Proteins Proteom. 2021, 1869, 140577. [Google Scholar] [CrossRef]
- Giroud, M.; Dietzel, U.; Anselm, L.; Banner, D.; Kuglstatter, A.; Benz, J.; Blanc, J.-B.; Gaufreteau, D.; Liu, H.; Lin, X.; et al. Repurposing a Library of Human Cathepsin L Ligands: Identification of Macrocyclic Lactams as Potent Rhodesain and Trypanosoma Brucei Inhibitors. J. Med. Chem. 2018, 61, 3350–3369. [Google Scholar] [CrossRef]
- Eakin, A.E.; Mills, A.A.; Harth, G.; McKerrow, J.H.; Craik, C.S. The Sequence, Organization, and Expression of the Major Cysteine Protease (Cruzain) from Trypanosoma Cruzi. J. Biol. Chem. 1992, 267, 7411–7420. [Google Scholar] [CrossRef]
- Harth, G.; Andrews, N.; Mills, A.A.; Engel, J.C.; Smith, R.; McKerrow, J.H. Peptide-Fluoromethyl Ketones Arrest Intracellular Replication and Intercellular Transmission of Trypanosoma Cruzi. Mol. Biochem. Parasitol. 1993, 58, 17–24. [Google Scholar] [CrossRef]
- Meirelles, M.N.; Juliano, L.; Carmona, E.; Silva, S.G.; Costa, E.M.; Murta, A.C.; Scharfstein, J. Inhibitors of the Major Cysteinyl Proteinase (GP57/51) Impair Host Cell Invasion and Arrest the Intracellular Development of Trypanosoma Cruzi in Vitro. Mol. Biochem. Parasitol. 1992, 52, 175–184. [Google Scholar] [CrossRef]
- Aparicio, I.M.; Scharfstein, J.; Lima, A.P.C.A. A New Cruzipain-Mediated Pathway of Human Cell Invasion by Trypanosoma Cruzi Requires Trypomastigote Membranes. Infect. Immun. 2004, 72, 5892–5902. [Google Scholar] [CrossRef]
- Doyle, P.S.; Zhou, Y.M.; Hsieh, I.; Greenbaum, D.C.; McKerrow, J.H.; Engel, J.C. The Trypanosoma Cruzi Protease Cruzain Mediates Immune Evasion. PLoS Pathog. 2011, 7, e1002139. [Google Scholar] [CrossRef] [PubMed]
- Lonsdale-Eccles, J.D.; Grab, D.J. Trypanosome Hydrolases and the Blood–Brain Barrier. Trends Parasitol. 2002, 18, 17–19. [Google Scholar] [CrossRef] [PubMed]
- Mogk, S.; Boßelmann, C.M.; Mudogo, C.N.; Stein, J.; Wolburg, H.; Duszenko, M. African Trypanosomes and Brain Infection—The Unsolved Question: Brain Infection in African Sleeping Sickness. Biol. Rev. 2017, 92, 1675–1687. [Google Scholar] [CrossRef] [PubMed]
- Du, X.; Guo, C.; Hansell, E.; Doyle, P.S.; Caffrey, C.R.; Holler, T.P.; McKerrow, J.H.; Cohen, F.E. Synthesis and Structure−Activity Relationship Study of Potent Trypanocidal Thio Semicarbazone Inhibitors of the Trypanosomal Cysteine Protease Cruzain. J. Med. Chem. 2002, 45, 2695–2707. [Google Scholar] [CrossRef]
- Fonseca, N.C.; da Cruz, L.F.; da Silva Villela, F.; do Nascimento Pereira, G.A.; de Siqueira-Neto, J.L.; Kellar, D.; Suzuki, B.M.; Ray, D.; de Souza, T.B.; Alves, R.J.; et al. Synthesis of a Sugar-Based Thiosemicarbazone Series and Structure-Activity Relationship versus the Parasite Cysteine Proteases Rhodesain, Cruzain, and Schistosoma Mansoni Cathepsin B1. Antimicrob. Agents Chemother. 2015, 59, 2666–2677. [Google Scholar] [CrossRef] [PubMed]
- Mott, B.T.; Ferreira, R.S.; Simeonov, A.; Jadhav, A.; Ang, K.K.-H.; Leister, W.; Shen, M.; Silveira, J.T.; Doyle, P.S.; Arkin, M.R.; et al. Identification and Optimization of Inhibitors of Trypanosomal Cysteine Proteases: Cruzain, Rhodesain, and TbCatB. J. Med. Chem. 2010, 53, 52–60. [Google Scholar] [CrossRef]
- Alves, L.; Santos, D.A.; Cendron, R.; Rocho, F.R.; Matos, T.K.B.; Leitão, A.; Montanari, C.A. Nitrile-Based Peptoids as Cysteine Protease Inhibitors. Bioorg. Med. Chem. 2021, 41, 116211. [Google Scholar] [CrossRef]
- Braga, S.F.P.; Martins, L.C.; da Silva, E.B.; Sales Júnior, P.A.; Murta, S.M.F.; Romanha, A.J.; Soh, W.T.; Brandstetter, H.; Ferreira, R.S.; de Oliveira, R.B. Synthesis and Biological Evaluation of Potential Inhibitors of the Cysteine Proteases Cruzain and Rhodesain Designed by Molecular Simplification. Bioorg. Med. Chem. 2017, 25, 1889–1900. [Google Scholar] [CrossRef]
- Ferreira, R.S.; Dessoy, M.A.; Pauli, I.; Souza, M.L.; Krogh, R.; Sales, A.I.L.; Oliva, G.; Dias, L.C.; Andricopulo, A.D. Synthesis, Biological Evaluation, and Structure–Activity Relationships of Potent Noncovalent and Nonpeptidic Cruzain Inhibitors as Anti- Trypanosoma Cruzi Agents. J. Med. Chem. 2014, 57, 2380–2392. [Google Scholar] [CrossRef] [PubMed]
- Pauli, I.; Rezende, C.D.O., Jr.; Slafer, B.W.; Dessoy, M.A.; de Souza, M.L.; Ferreira, L.L.G.; Adjanohun, A.L.M.; Ferreira, R.S.; Magalhães, L.G.; Krogh, R.; et al. Multiparameter Optimization of Trypanocidal Cruzain Inhibitors With In Vivo Activity and Favorable Pharmacokinetics. Front. Pharmacol. 2022, 12, 3570. [Google Scholar] [CrossRef] [PubMed]
- Bryant, C.; Kerr, I.D.; Debnath, M.; Ang, K.K.H.; Ratnam, J.; Ferreira, R.S.; Jaishankar, P.; Zhao, D.; Arkin, M.R.; McKerrow, J.H.; et al. Novel Non-Peptidic Vinylsulfones Targeting the S2 and S3 Subsites of Parasite Cysteine Proteases. Bioorg. Med. Chem. Lett. 2009, 19, 6218–6221. [Google Scholar] [CrossRef]
- Doyle, P.S.; Zhou, Y.M.; Engel, J.C.; McKerrow, J.H. A Cysteine Protease Inhibitor Cures Chagas’ Disease in an Immunodeficient-Mouse Model of Infection. Antimicrob. Agents Chemother. 2007, 51, 3932–3939. [Google Scholar] [CrossRef] [PubMed]
- Barbosa Da Silva, E.; Sharma, V.; Hernandez-Alvarez, L.; Tang, A.H.; Stoye, A.; O’Donoghue, A.J.; Gerwick, W.H.; Payne, R.J.; McKerrow, J.H.; Podust, L.M. Intramolecular Interactions Enhance the Potency of Gallinamide A Analogues against Trypanosoma Cruzi. J. Med. Chem. 2022, 65, 4255–4269. [Google Scholar] [CrossRef] [PubMed]
- Barbosa da Silva, E.; Rocha, D.A.; Fortes, I.S.; Yang, W.; Monti, L.; Siqueira-Neto, J.L.; Caffrey, C.R.; McKerrow, J.; Andrade, S.F.; Ferreira, R.S. Structure-Based Optimization of Quinazolines as Cruzain and Tbr CATL Inhibitors. J. Med. Chem. 2021, 64, 13054–13071. [Google Scholar] [CrossRef]
- de Souza, M.L.; de Oliveira Rezende Junior, C.; Ferreira, R.S.; Espinoza Chávez, R.M.; Ferreira, L.L.G.; Slafer, B.W.; Magalhães, L.G.; Krogh, R.; Oliva, G.; Cruz, F.C.; et al. Discovery of Potent, Reversible, and Competitive Cruzain Inhibitors with Trypanocidal Activity: A Structure-Based Drug Design Approach. J. Chem. Inf. Model. 2020, 60, 1028–1041. [Google Scholar] [CrossRef]
- Greenbaum, D.C.; Mackey, Z.; Hansell, E.; Doyle, P.; Gut, J.; Caffrey, C.R.; Lehrman, J.; Rosenthal, P.J.; McKerrow, J.H.; Chibale, K. Synthesis and Structure−Activity Relationships of Parasiticidal Thiosemicarbazone Cysteine Protease Inhibitors against Plasmodium Falciparum, Trypanosoma Brucei, and Trypanosoma Cruzi. J. Med. Chem. 2004, 47, 3212–3219. [Google Scholar] [CrossRef]
- Ettari, R.; Tamborini, L.; Angelo, I.C.; Grasso, S.; Schirmeister, T.; Lo Presti, L.; De Micheli, C.; Pinto, A.; Conti, P. Development of Rhodesain Inhibitors with a 3-Bromoisoxazoline Warhead. ChemMedChem 2013, 8, 2070–2076. [Google Scholar] [CrossRef]
- Yang, P.-Y.; Wang, M.; Li, L.; Wu, H.; He, C.Y.; Yao, S.Q. Design, Synthesis and Biological Evaluation of Potent Azadipeptide Nitrile Inhibitors and Activity-Based Probes as Promising Anti—Trypanosoma Brucei Agents. Chem.-Eur. J. 2012, 18, 6528–6541. [Google Scholar] [CrossRef]
- Mallari, J.P.; Shelat, A.A.; Obrien, T.; Caffrey, C.R.; Kosinski, A.; Connelly, M.; Harbut, M.; Greenbaum, D.; McKerrow, J.H.; Guy, R.K. Development of Potent Purine-Derived Nitrile Inhibitors of the Trypanosomal Protease TbcatB. J. Med. Chem. 2008, 51, 545–552. [Google Scholar] [CrossRef]
- Kryshchyshyn, A.; Kaminskyy, D.; Karpenko, O.; Gzella, A.; Grellier, P.; Lesyk, R. Thiazolidinone/Thiazole Based Hybrids–New Class of Antitrypanosomal Agents. Eur. J. Med. Chem. 2019, 174, 292–308. [Google Scholar] [CrossRef] [PubMed]
- Havrylyuk, D.; Zimenkovsky, B.; Vasylenko, O.; Day, C.W.; Smee, D.F.; Grellier, P.; Lesyk, R. Synthesis and Biological Activity Evaluation of 5-Pyrazoline Substituted 4-Thiazolidinones. Eur. J. Med. Chem. 2013, 66, 228–237. [Google Scholar] [CrossRef]
- Silva, F.T.; Franco, C.H.; Favaro, D.C.; Freitas-Junior, L.H.; Moraes, C.B.; Ferreira, E.I. Design, Synthesis and Antitrypanosomal Activity of Some Nitrofurazone 1,2,4-Triazolic Bioisosteric Analogues. Eur. J. Med. Chem. 2016, 121, 553–560. [Google Scholar] [CrossRef] [PubMed]
- Kerr, I.D.; Lee, J.H.; Farady, C.J.; Marion, R.; Rickert, M.; Sajid, M.; Pandey, K.C.; Caffrey, C.R.; Legac, J.; Hansell, E.; et al. Vinyl Sulfones as Antiparasitic Agents and a Structural Basis for Drug Design. J. Biol. Chem. 2009, 284, 25697–25703. [Google Scholar] [CrossRef]
- Ettari, R.; Previti, S.; Maiorana, S.; Amendola, G.; Wagner, A.; Cosconati, S.; Schirmeister, T.; Hellmich, U.A.; Zappalà, M. Optimization Strategy of Novel Peptide-Based Michael Acceptors for the Treatment of Human African Trypanosomiasis. J. Med. Chem. 2019, 62, 10617–10629. [Google Scholar] [CrossRef]
- Previti, S.; Ettari, R.; Di Chio, C.; Ravichandran, R.; Bogacz, M.; Hellmich, U.A.; Schirmeister, T.; Cosconati, S.; Zappalà, M. Development of Reduced Peptide Bond Pseudopeptide Michael Acceptors for the Treatment of Human African Trypanosomiasis. Molecules 2022, 27, 3765. [Google Scholar] [CrossRef] [PubMed]
- Pereira, G.A.N.; da Silva, E.B.; Braga, S.F.P.; Leite, P.G.; Martins, L.C.; Vieira, R.P.; Soh, W.T.; Villela, F.S.; Costa, F.M.R.; Ray, D.; et al. Discovery and Characterization of Trypanocidal Cysteine Protease Inhibitors from the ‘Malaria Box’. Eur. J. Med. Chem. 2019, 179, 765–778. [Google Scholar] [CrossRef]
- Silva, L.R.; Guimarães, A.S.; do Nascimento, J.; do Santos Nascimento, I.J.; da Silva, E.B.; McKerrow, J.H.; Cardoso, S.H.; da Silva-Júnior, E.F. Computer-Aided Design of 1,4-Naphthoquinone-Based Inhibitors Targeting Cruzain and Rhodesain Cysteine Proteases. Bioorg. Med. Chem. 2021, 41, 116213. [Google Scholar] [CrossRef]
- Van Voorhis, W.C.; Adams, J.H.; Adelfio, R.; Ahyong, V.; Akabas, M.H.; Alano, P.; Alday, A.; Alemán Resto, Y.; Alsibaee, A.; Alzualde, A.; et al. Open Source Drug Discovery with the Malaria Box Compound Collection for Neglected Diseases and Beyond. PLoS Pathog. 2016, 12, e1005763. [Google Scholar] [CrossRef]
- Duffy, S.; Sykes, M.L.; Jones, A.J.; Shelper, T.B.; Simpson, M.; Lang, R.; Poulsen, S.-A.; Sleebs, B.E.; Avery, V.M. Screening the Medicines for Malaria Venture Pathogen Box across Multiple Pathogens Reclassifies Starting Points for Open-Source Drug Discovery. Antimicrob. Agents Chemother. 2017, 61, e00379-17. [Google Scholar] [CrossRef]
- Veale, C.G.L.; Laming, D.; Swart, T.; Chibale, K.; Hoppe, H.C. Exploring the Antiplasmodial 2-Aminopyridines as Potential Antitrypanosomal Agents. ChemMedChem 2019, 14, 2034–2041. [Google Scholar] [CrossRef] [PubMed]
- Gaulton, A.; Hersey, A.; Nowotka, M.; Bento, A.P.; Chambers, J.; Mendez, D.; Mutowo, P.; Atkinson, F.; Bellis, L.J.; Cibrián-Uhalte, E.; et al. The ChEMBL Database in 2017. Nucleic Acids Res. 2017, 45, D945–D954. [Google Scholar] [CrossRef]
- Veríssimo, G.C.; dos Santos Júnior, V.S.; de Almeida, I.A.D.R.; Ruas, M.S.M.; Coutinho, L.G.; de Oliveira, R.B.; Alves, R.J.; Maltarollo, V.G. The Brazilian Compound Library (BraCoLi) Database: A Repository of Chemical and Biological Information for Drug Design. Mol. Divers. 2022, 26, 3387–3397. [Google Scholar] [CrossRef]
- Bemis, G.W.; Murcko, M.A. The Properties of Known Drugs. 1. Molecular Frameworks. J. Med. Chem. 1996, 39, 2887–2893. [Google Scholar] [CrossRef]
- de Oliveira, R.B.; Vaz, A.B.; Alves, R.O.; Liarte, D.B.; Donnici, C.L.; Romanha, A.J.; Zani, C.L. Arylfurans as Potential Trypanosoma Cruzi Trypanothione Reductase Inhibitors. Mem. Inst. Oswaldo Cruz 2006, 101, 169–173. [Google Scholar] [CrossRef]
- Oliveira, R.B.D.; Zani, C.L.; Ferreira, R.S.; Leite, R.S.; Alves, T.M.A.; da Silva, T.H.A.; Romanha, A.J. Síntese, Avaliação Biológica e Modelagem Molecular de Arilfuranos Como Inibidores Da Enzima Tripanotiona Redutase. Quím. Nova 2008, 31, 261–267. [Google Scholar] [CrossRef]
- Ferreira, R.S.; Simeonov, A.; Jadhav, A.; Eidam, O.; Mott, B.T.; Keiser, M.J.; McKerrow, J.H.; Maloney, D.J.; Irwin, J.J.; Shoichet, B.K. Complementarity Between a Docking and a High-Throughput Screen in Discovering New Cruzain Inhibitors. J. Med. Chem. 2010, 53, 4891–4905. [Google Scholar] [CrossRef]
- Dolinsky, T.J.; Nielsen, J.E.; McCammon, J.A.; Baker, N.A. PDB2PQR: An Automated Pipeline for the Setup of Poisson-Boltzmann Electrostatics Calculations. Nucleic Acids Res. 2004, 32, W665–W667. [Google Scholar] [CrossRef]
- Turney, J.M.; Simmonett, A.C.; Parrish, R.M.; Hohenstein, E.G.; Evangelista, F.A.; Fermann, J.T.; Mintz, B.J.; Burns, L.A.; Wilke, J.J.; Abrams, M.L.; et al. Psi4: An Open-Source Ab Initio Electronic Structure Program: Psi4: An Electronic Structure Program. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2012, 2, 556–565. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the Speed and Accuracy of Docking with a New Scoring Function, Efficient Optimization and Multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Veale, C.G.L. Unpacking the Pathogen Box—An Open Source Tool for Fighting Neglected Tropical Disease. ChemMedChem 2019, 14, 386–453. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez, F.; Rozas, I.; Kaiser, M.; Brun, R.; Nguyen, B.; Wilson, W.D.; García, R.N.; Dardonville, C. New Bis(2-Aminoimidazoline) and Bisguanidine DNA Minor Groove Binders with Potent in Vivo Antitrypanosomal and Antiplasmodial Activity. J. Med. Chem. 2008, 51, 909–923. [Google Scholar] [CrossRef] [PubMed]
- Jadhav, A.; Ferreira, R.S.; Klumpp, C.; Mott, B.T.; Austin, C.P.; Inglese, J.; Thomas, C.J.; Maloney, D.J.; Shoichet, B.K.; Simeonov, A. Quantitative Analyses of Aggregation, Autofluorescence, and Reactivity Artifacts in a Screen for Inhibitors of a Thiol Protease. J. Med. Chem. 2010, 53, 37–51. [Google Scholar] [CrossRef] [PubMed]
- McGovern, S.L.; Caselli, E.; Grigorieff, N.; Shoichet, B.K. A Common Mechanism Underlying Promiscuous Inhibitors from Virtual and High-Throughput Screening. J. Med. Chem. 2002, 45, 1712–1722. [Google Scholar] [CrossRef] [PubMed]
- Coan, K.E.D.; Shoichet, B.K. Stability and Equilibria of Promiscuous Aggregates in High Protein Milieus. Mol. Biosyst. 2007, 3, 208–213. [Google Scholar] [CrossRef]
- Aulner, N.; Danckaert, A.; Ihm, J.; Shum, D.; Shorte, S.L. Next-Generation Phenotypic Screening in Early Drug Discovery for Infectious Diseases. Trends Parasitol. 2019, 35, 559–570. [Google Scholar] [CrossRef]
- Wall, R.J.; Carvalho, S.; Milne, R.; Bueren-Calabuig, J.A.; Moniz, S.; Cantizani-Perez, J.; MacLean, L.; Kessler, A.; Cotillo, I.; Sastry, L.; et al. The Q i Site of Cytochrome b Is a Promiscuous Drug Target in Trypanosoma Cruzi and Leishmania Donovani. ACS Infect. Dis. 2020, 6, 515–528. [Google Scholar] [CrossRef]
- Stephens, C.E.; Brun, R.; Salem, M.M.; Werbovetz, K.A.; Tanious, F.; Wilson, W.D.; Boykin, D.W. The Activity of Diguanidino and ‘Reversed’ Diamidino 2,5-Diarylfurans versus Trypanosoma Cruzi and Leishmania Donovani. Bioorg. Med. Chem. Lett. 2003, 13, 2065–2069. [Google Scholar] [CrossRef]
- Nagle, P.S.; Rodriguez, F.; Nguyen, B.; Wilson, W.D.; Rozas, I. High DNA Affinity of a Series of Peptide Linked Diaromatic Guanidinium-like Derivatives. J. Med. Chem. 2012, 55, 4397–4406. [Google Scholar] [CrossRef]
- Prati, F.; Uliassi, E.; Bolognesi, M.L. Two Diseases, One Approach: Multitarget Drug Discovery in Alzheimer’s and Neglected Tropical Diseases. MedChemComm 2014, 5, 853–861. [Google Scholar] [CrossRef]
- Previti, S.; Di Chio, C.; Ettari, R.; Zappalà, M. Dual Inhibition of Parasitic Targets: A Valuable Strategy to TreatMalaria and Neglected Tropical Diseases. Curr. Med. Chem. 2022, 29, 2952–2978. [Google Scholar] [CrossRef] [PubMed]
- Breidenbach, J.; Lemke, C.; Pillaiyar, T.; Schäkel, L.; Al Hamwi, G.; Diett, M.; Gedschold, R.; Geiger, N.; Lopez, V.; Mirza, S.; et al. Targeting the Main Protease of SARS-CoV-2: From the Establishment of High Throughput Screening to the Design of Tailored Inhibitors. Angew. Chem. Int. Ed. 2021, 60, 10423–10429. [Google Scholar] [CrossRef] [PubMed]
- Ehmann, A. The Australian National University Dept of Immunology Pathogen Box Compounds Screened. Available online: https://www.ebi.ac.uk/chembl/doc/inspect/CHEMBL3987221 (accessed on 6 January 2023).
- Motlekar, N.; Diamond, S.L.; Napper, A.D. Evaluation of an Orthogonal Pooling Strategy for Rapid High-Throughput Screening of Proteases. ASSAY Drug Dev. Technol. 2008, 6, 395–405. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Inhibition at 5 µM (Mean ± SEM %) a | Inhibition at 100 µM (Mean ± SEM %) b | IC50 (µM ± SEM %) c | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| TbrCatL | Cruzain | TbrCatL | Cruzain | TbrCatL | Cruzain | |||||||
| 0′ | 10′ inc | 0′ | 10′ inc | 0′ | 10′ inc | 0′ | 10′ inc | 0′ | 10′ inc | 0′ | 10′ inc | |
| MMV085499 | 12 ± 8 | 46 ± 5 | 2 ± 3 | 25 ± 4 | 26 ± 18 | 0 ± 0 | 0 ± 0 | 0 ± 0 | ND | ND | ND | ND |
| MMV634140 | 11 ± 1 | 42 ± 3 | 24 ± 1 | 31 ± 3 | ND | ND | ND | ND | ND | ND | ND | ND |
| MMV667494 | 31 ± 5 | 73 ± 1 | 40 ± 1 | 53 ± 1 | 0 ± 0 | 11 ± 8 | 0 ± 0 | 70 ± 3 | ND | ND | ND | ND |
| MMV676881 | 100 ± 0 | 100 ± 2 | 100 ± 2 | 100 ± 1 | ND | ND | ND | ND | ND | ND | ND | ND |
| MMV687246 | 29 ± 1 | 49 ± 4 | 24 ± 3 | 39 ± 3 | 92 ± 6 | 99 ± 1 | 72 ± 1 | 92 ± 3 | 9 ± 3 | 4.2 ± 0.6 | 14 ± 5 | 10 ± 2 |
| MMV687812 | 11 ± 3 | 5 ± 1 | 41 ± 2 | 27 ± 0 | 90 ± 2 | 93 ± 0 | 100 ± 0 | 95 ± 0 | 3.45 ± 0.05 | 3.6 ± 0.3 | 2.2 ± 0.3 | 2.6 ± 0.03 |
| MMV688179 | 50 ± 3 | 58 ± 25 | 98 ± 2 | 100 ± 0 | 100 ± 0 | 100 ± 0 | 100 ± 0 | 100 ± 0 | 4 ± 1 | 5 ± 2 | 0.46 ± 0.03 | 0.53 ± 0.03 |
| MMV688271 | 53 ± 5 | 43 ± 1 | 83 ± 10 | 50 ± 4 | ND | ND | ND | ND | ND | ND | ND | ND |
| MMV688362 | 9 ± 1 | 43 ± 4 | 19 ± 1 | 27 ± 3 | 100 ± 0 | 100 ± 0 | 100 ± 0 | 100 ± 0 | 2.9 ± 0.8 | 2.3 ± 0.1 | 4.2 ± 0.6 | 2.35 ± 0.02 |
| MMV688466 | 29 ± 2 | 54 ± 8 | 41 ± 2 | 49 ± 5 | 96 ± 3 | 100 ± 0 | 58 ± 27 | 98 ± 1 | 4 ± 1 | 11 ± 4 | 9 ± 1 | 25 ± 6 |
| MMV690027 | 6 ± 2 | 0 ± 6 | 50 ± 5 | 33 ± 2 | ND | ND | ND | ND | ND | ND | ND | ND |
| MMV690028 | 49 ± 4 | 23 ± 4 | 34 ± 8 | 27 ± 4 | 100 ± 0 | 88 ± 3 | 100 ± 0 | 100 ± 0 | 18 ± 7 | 27 ± 3 | 10 ± 3 | 13 ± 5 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
do Valle Moreira, T.; Martins, L.C.; Diniz, L.A.; Bernardes, T.C.D.; de Oliveira, R.B.; Ferreira, R.S. Screening the Pathogen Box to Discover and Characterize New Cruzain and TbrCatL Inhibitors. Pathogens 2023, 12, 251. https://doi.org/10.3390/pathogens12020251
do Valle Moreira T, Martins LC, Diniz LA, Bernardes TCD, de Oliveira RB, Ferreira RS. Screening the Pathogen Box to Discover and Characterize New Cruzain and TbrCatL Inhibitors. Pathogens. 2023; 12(2):251. https://doi.org/10.3390/pathogens12020251
Chicago/Turabian Styledo Valle Moreira, Thales, Luan Carvalho Martins, Lucas Abreu Diniz, Talita Cristina Diniz Bernardes, Renata Barbosa de Oliveira, and Rafaela Salgado Ferreira. 2023. "Screening the Pathogen Box to Discover and Characterize New Cruzain and TbrCatL Inhibitors" Pathogens 12, no. 2: 251. https://doi.org/10.3390/pathogens12020251
APA Styledo Valle Moreira, T., Martins, L. C., Diniz, L. A., Bernardes, T. C. D., de Oliveira, R. B., & Ferreira, R. S. (2023). Screening the Pathogen Box to Discover and Characterize New Cruzain and TbrCatL Inhibitors. Pathogens, 12(2), 251. https://doi.org/10.3390/pathogens12020251