
Exposure to Veterinary Antibiotics via Food Chain Disrupts Gut Microbiota and Drives Increased Escherichia coli Virulence and Drug Resistance in Young Adults

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Antibiotic Analysis of Urine Samples
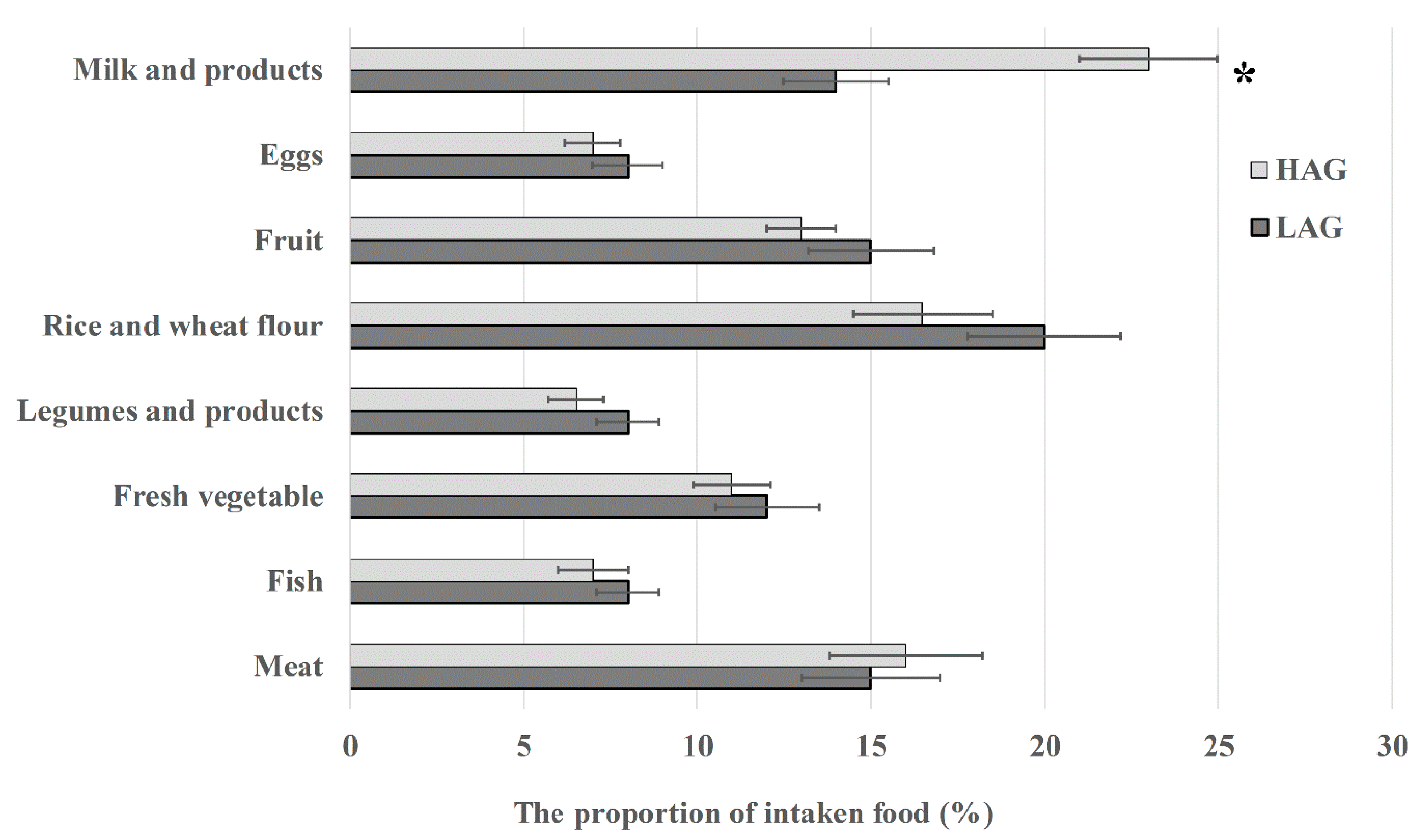
2.2. Dietary Investigation
2.3. Collection of Stool Samples and Extraction of Bacterial Genomic DNA for High-Throughout Sequencing
2.4. E. coli Quantification in Stool Samples
2.5. Isolation and Identification of E. coli from Stool Samples
2.6. The Virulence-Related Assays of the Isolates
2.7. Detection of Drug-Resistant Genes in the Isolates by PCR Assay
2.8. RNA Sequencing and Data Analysis
2.9. Validation of Gene Expression by Quantitative Real-Time PCR (Qpcr)
2.10. Statistical Analysis
3. Results
3.1. High Intake of Milk Is Positively Correlated to the Level of Antibiotic Residues in Urine Samples
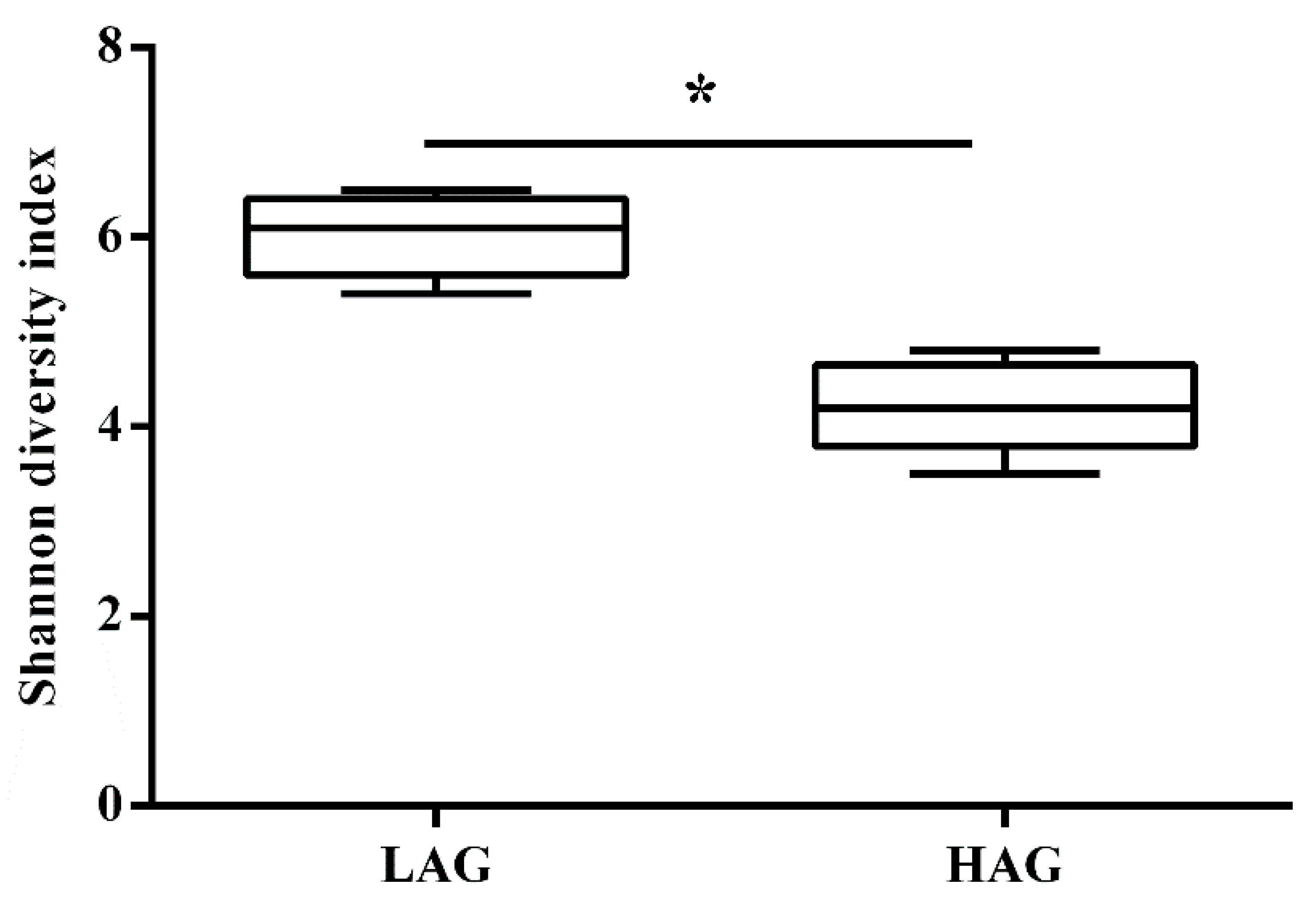
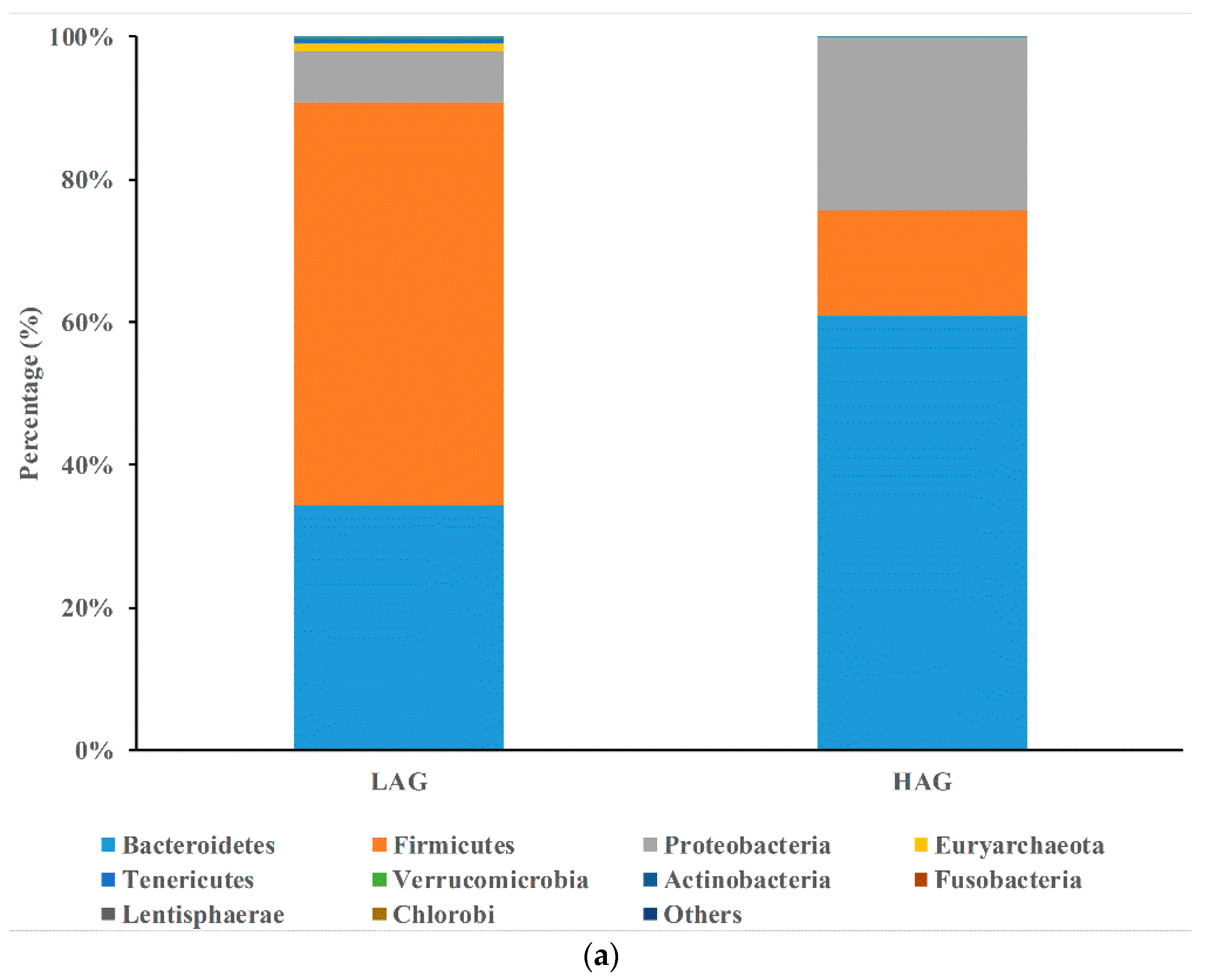
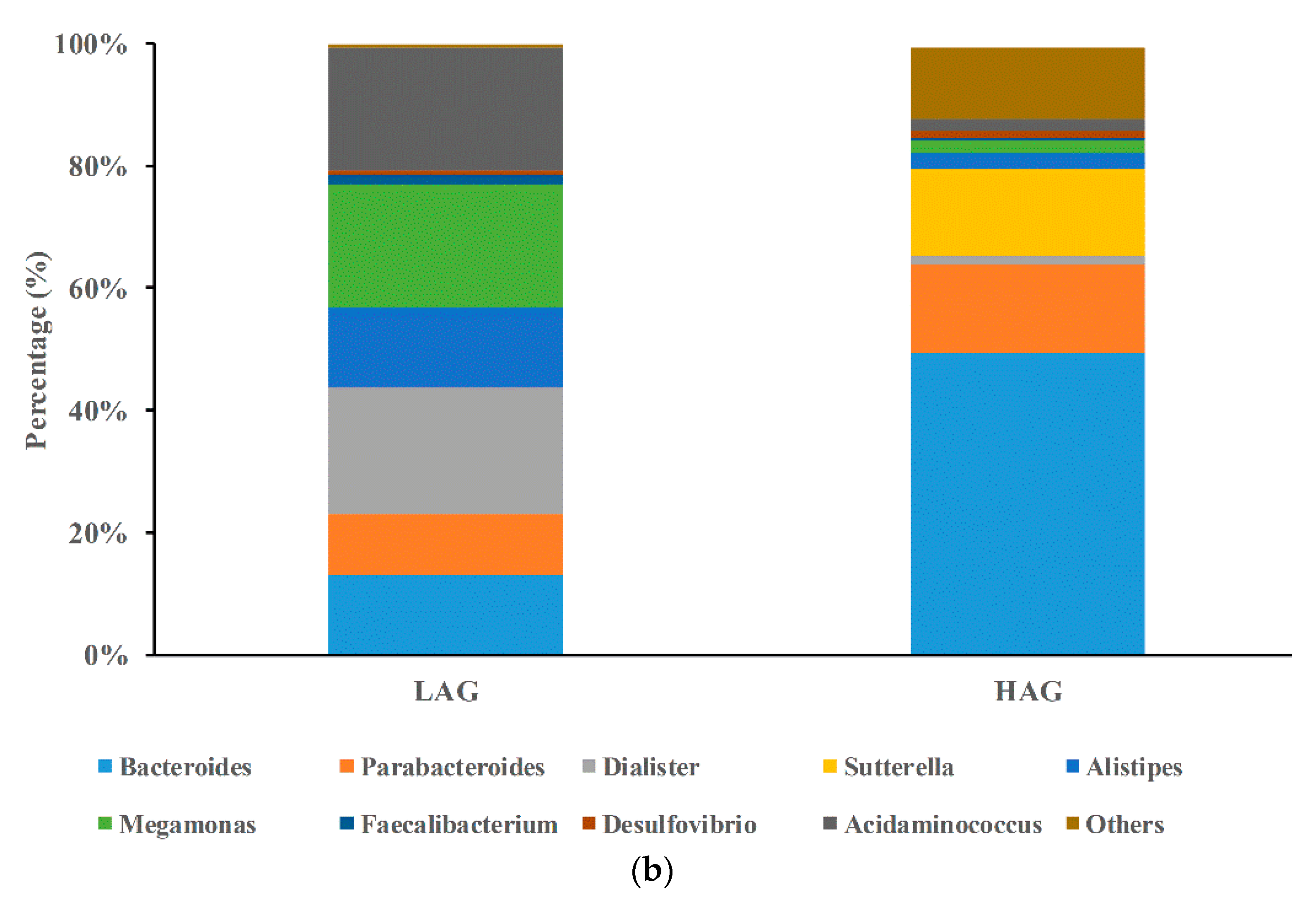
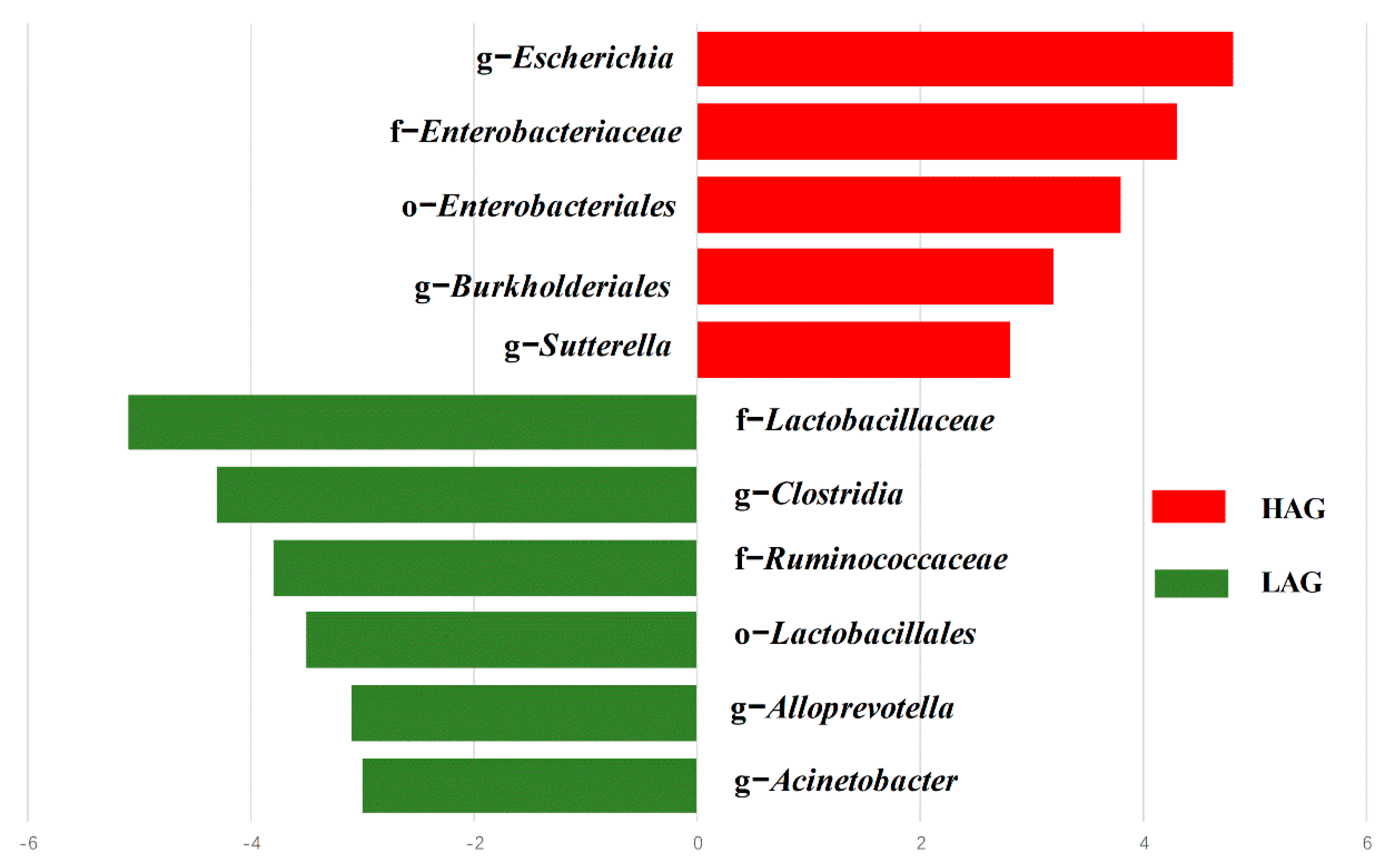
3.2. The Structure of Gut Microbiota in HAG Is Totally Different from LAG
3.3. HAG Has High abundance of E. coli
3.4. E. coli Isolated from HAG Harbors More Resistant Genes
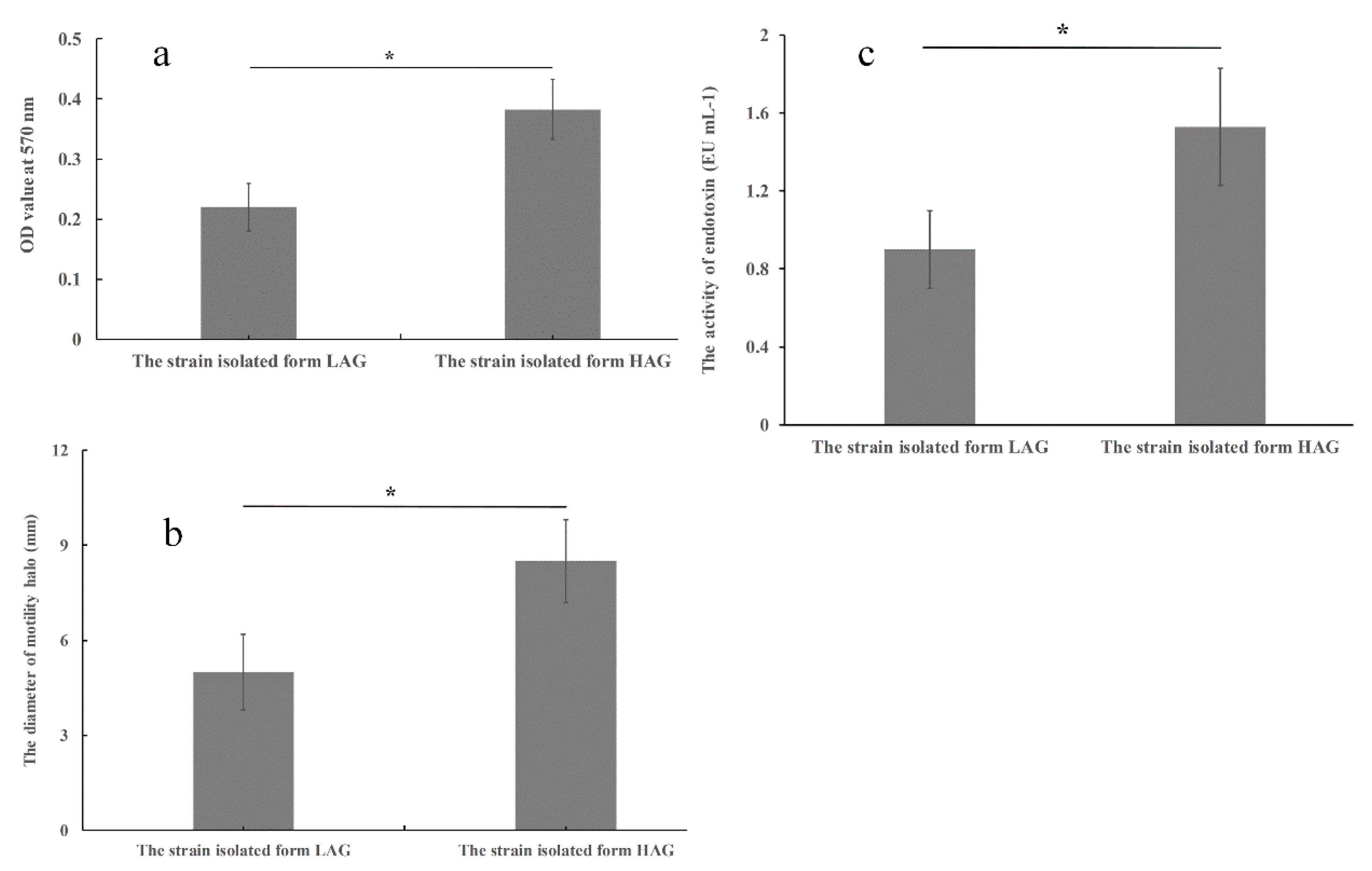
3.5. Virulence-Related Abilities of the Isolate from HAG Are Stronger Than the Isolate from LAG
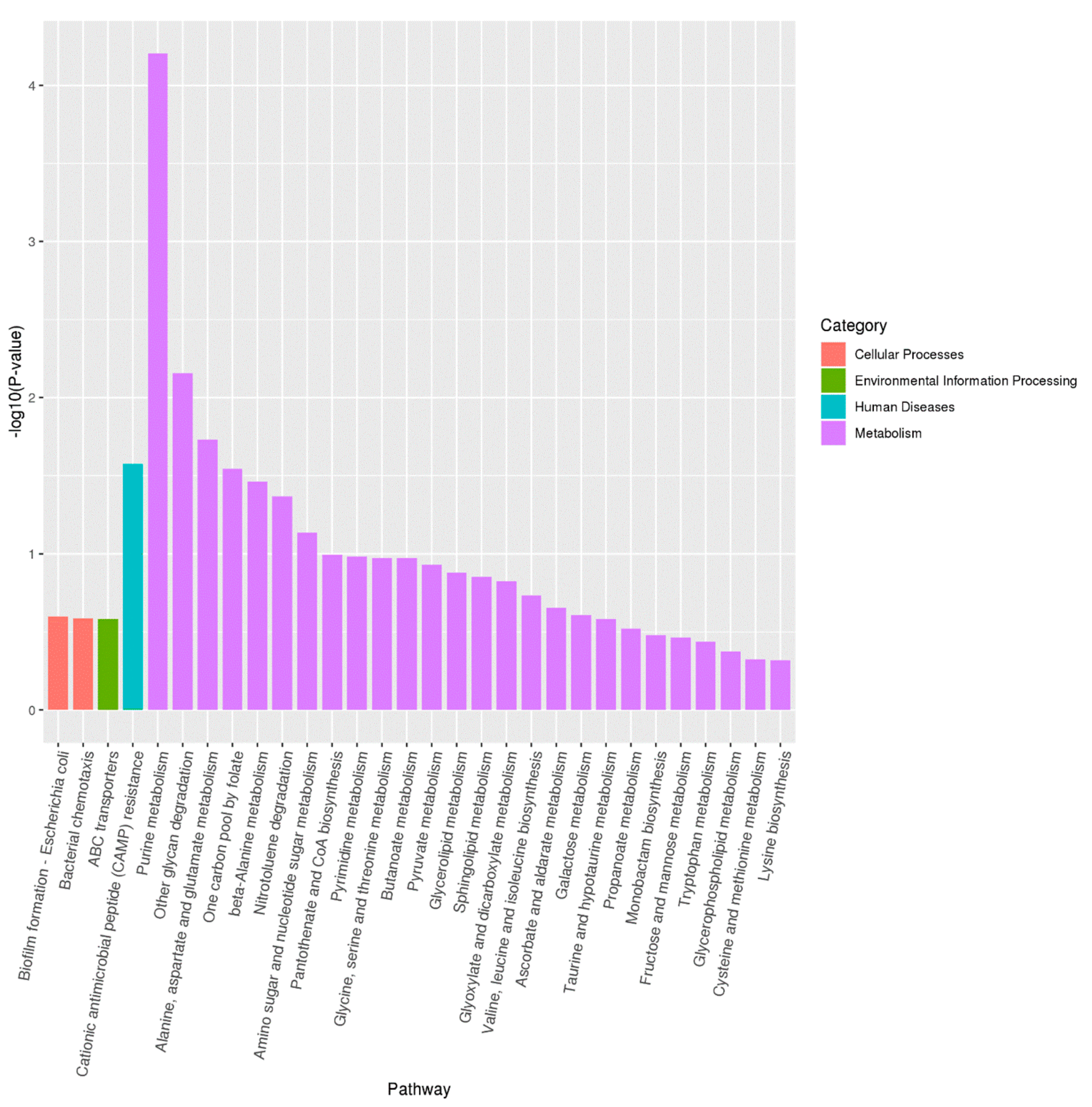
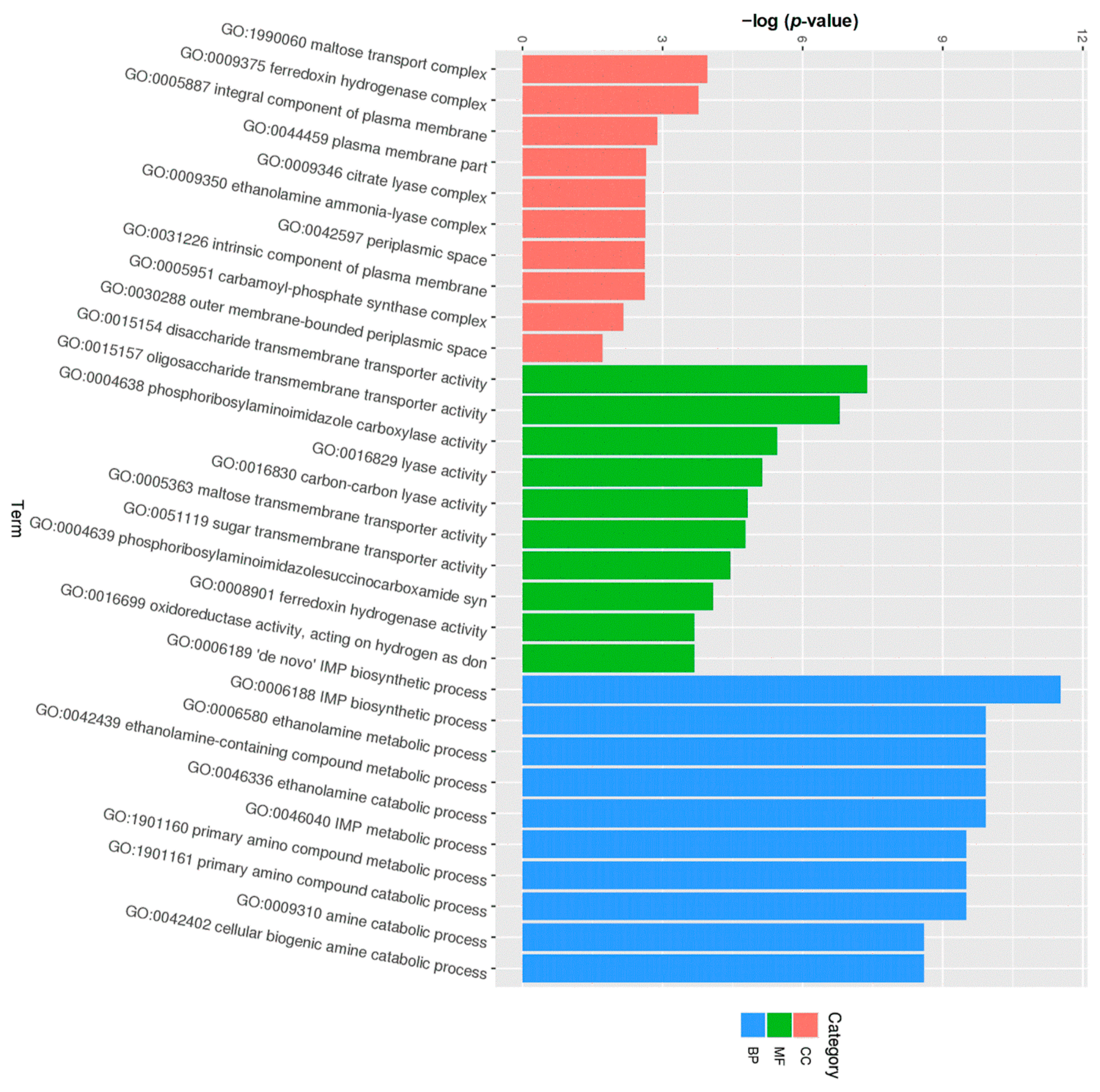
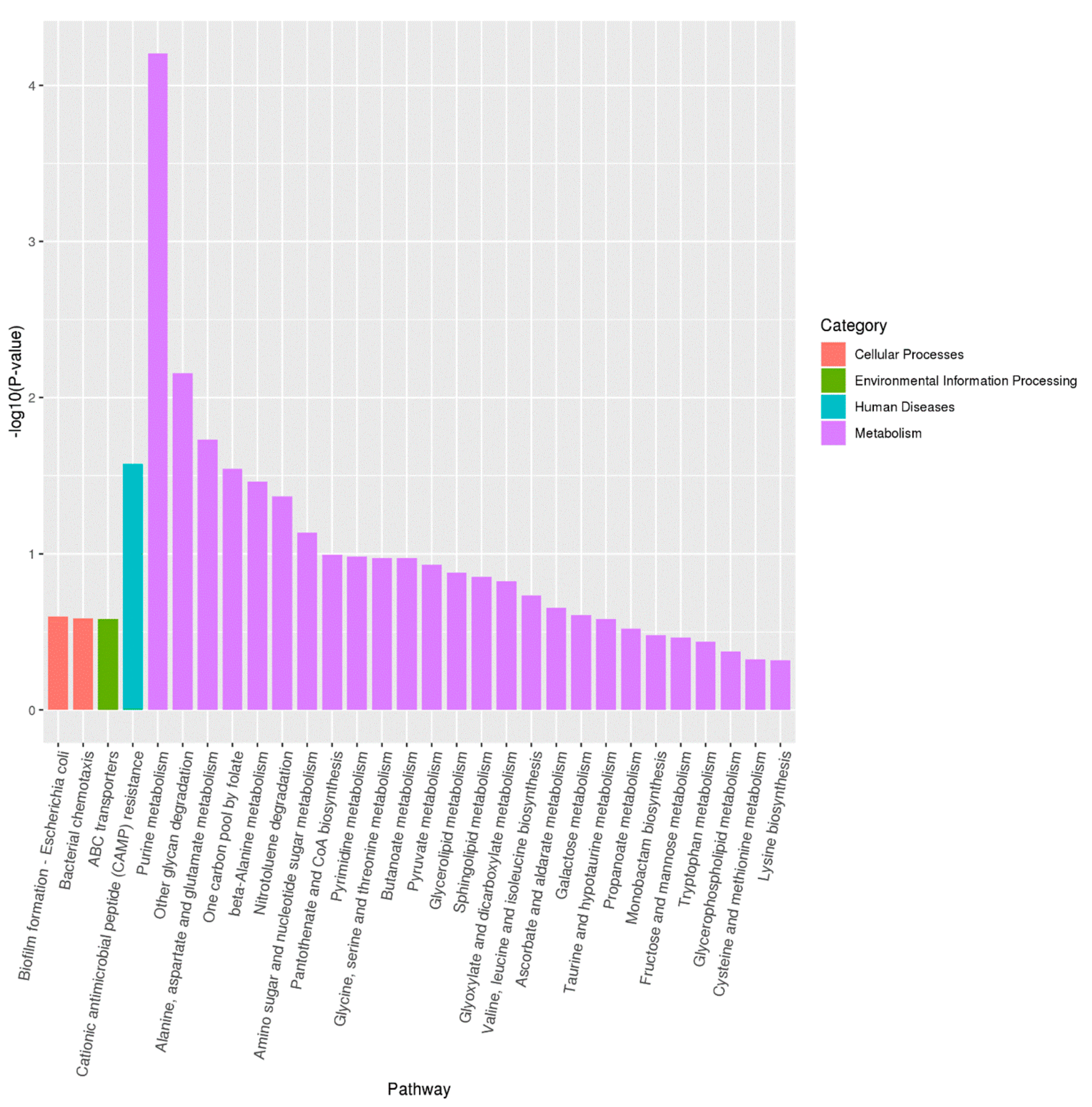
3.6. Transcriptome Analysis
3.7. Validation of the Results of Transcriptome Analysis via qPCR
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Adak, A.; Khan, M.R. An insight into gut microbiota and its functionalities. Cell Mol. Life Sci. 2019, 76, 473–493. [Google Scholar] [CrossRef] [PubMed]
- Gill, P.A.; Inniss, S.; Kumagai, T.; Rahman, F.Z.; Smith, A.M. The role of diet and gut microbiota in regulating gastrointestinal and inflammatory disease. Front. Immunol. 2022, 13, 866059. [Google Scholar] [CrossRef] [PubMed]
- Lamberte, L.E.; van Schaik, W. Antibiotic resistance in the commensal human gut microbiota. Curr. Opin. Microbiol. 2022, 68, 102150. [Google Scholar] [CrossRef] [PubMed]
- Pennycook, J.H.; Scanlan, P.D. Ecological and evolutionary responses to antibiotic treatment in the human gut microbiota. FEMS Microbiol. Rev. 2021, 45, fuab018. [Google Scholar] [CrossRef]
- Davinelli, S.; Scapagnini, G. Interactions between dietary polyphenols and aging gut microbiota: A review. BioFactors 2022, 48, 274–284. [Google Scholar] [CrossRef]
- Cook, T.M.; Mansuy-Aubert, V. Communication between the gut microbiota and peripheral nervous system in health and chronic disease. Gut microbes 2022, 14, 2068365. [Google Scholar] [CrossRef]
- Liu, L.; Xu, W.; Kong, P.; Dou, Y. The relationships among gut microbiota, hypoxia-inducible factor and anaemia with chronic kidney disease. Nephrology 2022. online ahead of print. [Google Scholar] [CrossRef]
- Hernández-Ceballos, W.; Cordova-Gallardo, J.; Mendez-Sanchez, N. Gut microbiota in metabolic-associated fatty liver disease and in other chronic metabolic diseases. J. Clin. Transl. Hepatol. 2021, 9, 227–238. [Google Scholar] [CrossRef]
- Ghimpețeanu, O.M.; Pogurschi, E.N.; Popa, D.C.; Dragomir, N.; Drăgotoiu, T.; Mihai, O.D.; Petcu, C.D. Antibiotic use in livestock and residues in food-A public health threat: A review. Foods 2022, 11, 1430. [Google Scholar] [CrossRef]
- Hossain, A.; Habibullah-Al-Mamun, M.; Nagano, I.; Masunaga, S.; Kitazawa, D.; Matsuda, H. Antibiotics, antibiotic-resistant bacteria, and resistance genes in aquaculture: Risks, current concern, and future thinking. Environ. Sci. Pollut. Res. Int. 2022, 29, 11054–11075. [Google Scholar] [CrossRef]
- Nelson, A.M.; Walk, S.T.; Taube, S.; Taniuchi, M.; Houpt, E.R.; Wobus, C.E.; Young, V.B. Disruption of the human gut microbiota following Norovirus infection. PLoS ONE 2012, 7, e48224. [Google Scholar] [CrossRef] [PubMed]
- Gallardo, P.; Izquierdo, M.; Vidal, R.M.; Soto, F.; Ossa, J.C.; Farfan, M.J. Gut microbiota-metabolome changes in children with diarrhea by diarrheagenic E. coli. Front. Cell Infect. Microbiol. 2020, 10, 485. [Google Scholar] [CrossRef] [PubMed]
- Luna-Guevara, J.J.; Arenas-Hernandez, M.M.P.; Martínez de la Peña, C.; Silva, J.L.; Luna-Guevara, M.L. The role of pathogenic E. coli in fresh vegetables: Behavior, contamination factors, and preventive measures. Int. J. Microbiol. 2019, 2019, 2894328. [Google Scholar] [CrossRef] [PubMed]
- Saxena, T.; Kaushik, P.; Krishna Mohan, M. Prevalence of E. coli O157:H7 in water sources: An overview on associated diseases, outbreaks and detection methods. Diagn. Microbiol. Infect. Dis. 2015, 82, 249–264. [Google Scholar] [CrossRef]
- Sapkota, S.; Adhikari, S.; Pandey, A.; Khadka, S.; Adhikari, M.; Kandel, H.; Pathak, S.; Pandey, A. Multi-drug resistant extended-spectrum beta-lactamase producing E. coli and Salmonella on raw vegetable salads served at hotels and restaurants in Bharatpur, Nepal. BMC Res. Notes 2019, 12, 516. [Google Scholar] [CrossRef]
- Liu, K.Y.; Zhang, J.J.; Geng, M.L.; Zhu, Y.T.; Liu, X.J.; Ding, P.; Wang, B.L.; Liu, W.W.; Liu, Y.H.; Tao, F.B. A Stable isotope dilution assay for multi-class antibiotics in pregnant urines by LC–MS/MS. Chromatographia 2020, 83, 507–521. [Google Scholar] [CrossRef]
- Zhao, W.; Hasegawa, K.; Chen, J. The use of food-frequency questionnaires for various purposes in China. Public Health Nutr. 2002, 5, 829–833. [Google Scholar] [CrossRef]
- Fazelahi, M.; Kia, V.; Kaghazian, H.; Paryan, M. Quantitative real-time PCR technique for the identification of E. coli residual DNA in streptokinase recombinant product. Prep. Biochem. Biotechnol. 2017, 47, 986–989. [Google Scholar] [CrossRef]
- Peeters, E.; Nelis, H.J.; Coenye, T. Comparison of multiple methods for quantification of microbial biofilms grown in microtiter plates. J. Microbiol. Methods 2008, 72, 157–165. [Google Scholar] [CrossRef]
- Wang, Q.; Li, Q.; Lin, Y.; Hou, Y.; Deng, Z.; Liu, W.; Wang, H.; Xia, Z. Biochemical and genetic basis of cadmium biosorption by Enterobacter ludwigii LY6, isolated from industrial contaminated soil. Environ. Pollut. 2020, 264, 114637. [Google Scholar] [CrossRef]
- Blair, J.M.; Webber, M.A.; Baylay, A.J.; Ogbolu, D.O.; Piddock, L.J. Molecular mechanisms of antibiotic resistance. Nat. Rev. Microbiol. 2015, 13, 42–51. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Stanford, K.; McAllister, T.A.; Johnson, R.P.; Chen, J.; Hou, H.; Zhang, G.; Niu, Y.D. Biofilm formation, virulence gene profiles, and antimicrobial resistance of nine serogroups of non-O157 Shiga toxin-producing Escherichia coli. Foodborne Pathog. Dis. 2016, 13, 316–324. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; He, R.; Zhang, R.; Yuan, J. Swarming motility without flagellar motor switching by reversal of swimming direction in E. coli. Front. Microbiol. 2020, 11, 1042. [Google Scholar] [CrossRef]
- Holzheimer, R.G. Antibiotic induced endotoxin release and clinical sepsis: A review. J. Chemother. 2001, 13, 159–172. [Google Scholar] [CrossRef]
- Chen, H.; Liu, S.; Xu, X.R.; Diao, Z.H.; Sun, K.F.; Hao, Q.W.; Liu, S.S.; Ying, G.G. Tissue distribution, bioaccumulation characteristics and health risk of antibiotics in cultured fish from a typical aquaculture area. J. Hazard Mater. 2018, 343, 140–148. [Google Scholar] [CrossRef]
- Klein, E.Y.; Van Boeckel, T.P.; Martinez, E.M.; Pant, S.; Gandra, S.; Levin, S.A.; Goossens, H.; Laxminarayan, R. Global increase and geographic convergence in antibiotic consumption between 2000 and 2015. Proc. Natl. Acad. Sci. USA 2018, 115, e3463–e3470. [Google Scholar] [CrossRef] [PubMed]
- Ter Kuile, B.H.; Kraupner, N.; Brul, S. The risk of low concentrations of antibiotics in agriculture for resistance in human health care. FEMS Microbiol. Lett. 2016, 363, fnw210. [Google Scholar] [CrossRef]
- Zainab, S.M.; Junaid, M.; Xu, N.; Malik, R.N. Antibiotics and antibiotic resistant genes (ARGs) in groundwater: A global review on dissemination, sources, interactions, environmental and human health risks. Water Res. 2020, 187, 116455. [Google Scholar] [CrossRef]
- Mo, W.Y.; Chen, Z.; Leung, H.M.; Leung, A.O. Application of veterinary antibiotics in China′s aquaculture industry and their potential human health risks. Environ. Sci. Pollut. Res. Int. 2017, 24, 8978–8989. [Google Scholar] [CrossRef]
- Zheng, N.; Wang, J.; Han, R.; Xu, X.; Zhen, Y.; Qu, X.; Sun, P.; Li, S.; Yu, Z. Occurrence of several main antibiotic residues in raw milk in 10 provinces of China. Food Addit. Contam. Part. B Surveill. 2013, 6, 84–89. [Google Scholar] [CrossRef]
- Sachi, S.; Ferdous, J.; Sikder, M.H.; Azizul Karim Hussani, S.M. Antibiotic residues in milk: Past, present, and future. J. Adv. Vet. Anim. Res. 2019, 6, 315–332. [Google Scholar] [CrossRef] [PubMed]
- Lebeaux, R.M.; Madan, J.C.; Nguyen, Q.P.; Coker, M.O.; Dade, E.F.; Moroishi, Y.; Palys, T.J.; Ross, B.D.; Pettigrew, M.M.; Morrison, H.G.; et al. Impact of antibiotics on off-target infant gut microbiota and resistance genes in cohort studies. Pediatr. Res. online ahead of print. 2022. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Cheng, X.; Zhong, S.; Liu, Z.; Liu, F.; Lin, X.; Zhao, Y.; Guan, M.; Xiao, T.; Jolkkonen, J.; et al. Long-term modification of gut microbiota by broad-spectrum antibiotics improves stroke outcome in rats. Stroke Vasc. Neurol. 2022, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Ramirez, J.; Guarner, F.; Bustos Fernandez, L.; Maruy, A.; Sdepanian, V.L.; Cohen, H. Antibiotics as major disruptors of gut microbiota. Front. Cell Infect. Microbiol. 2020, 10, 572912. [Google Scholar] [CrossRef]
- Schoch, S.F.; Castro-Mejía, J.L.; Krych, L.; Leng, B.; Kot, W.; Kohler, M.; Huber, R.; Rogler, G.; Biedermann, L.; Walser, J.C.; et al. From alpha diversity to zzz: Interactions among sleep, the brain, and gut microbiota in the first year of life. Prog. Neurobiol. 2022, 209, 102208. [Google Scholar] [CrossRef]
- Niestępski, S.; Harnisz, M.; Ciesielski, S.; Korzeniewska, E.; Osińska, A. Environmental fate of Bacteroidetes, with particular emphasis on Bacteroides fragilis group bacteria and their specific antibiotic resistance genes, in activated sludge wastewater treatment plants. J. Hazard. Mater. 2020, 394, 122544. [Google Scholar] [CrossRef]
- Kamal, S.M.; Simpson, D.J.; Wang, Z.; Gänzle, M.; Römling, U. Horizontal transmission of stress resistance genes shape the ecology of Beta- and Gamma-Proteobacteria. Front. Microbiol. 2021, 12, 696522. [Google Scholar] [CrossRef]
- Dela, H.; Egyir, B.; Majekodunmi, A.O.; Behene, E.; Yeboah, C.; Ackah, D.; Bongo, R.N.A.; Bonfoh, B.; Zinsstag, J.; Bimi, L.; et al. Diarrhoeagenic E. coli occurrence and antimicrobial resistance of extended spectrum Beta-Lactamases isolated from diarrhoea patients attending health facilities in Accra, Ghana. PLoS ONE 2022, 17, e0268991. [Google Scholar] [CrossRef]
- Thänert, R.; Sawhney, S.S.; Schwartz, D.J.; Dantas, G. The resistance within: Antibiotic disruption of the gut microbiome and resistome dynamics in infancy. Cell Host Microbe 2022, 30, 675–683. [Google Scholar] [CrossRef]
- Brubaker, J.; Zhang, X.; Bourgeois, A.L.; Harro, C.; Sack, D.A.; Chakraborty, S. Intestinal and systemic inflammation induced by symptomatic and asymptomatic enterotoxigenic E. coli infection and impact on intestinal colonization and ETEC specific immune responses in an experimental human challenge model. Gut Microbes 2021, 13, 1891852. [Google Scholar] [CrossRef]
- Musa, L.; Proietti, P.C.; Marenzoni, M.L.; Stefanetti, V.; Kika, T.S.; Blasi, F.; Magistrali, C.F.; Toppi, V.; Ranucci, D.; Branciari, R.; et al. Susceptibility of commensal E. coli isolated from conventional, antibiotic-free, and organic meat chickens on farms and at slaughter toward antimicrobials with public health relevance. Antibiotics 2021, 10, 1321. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Zhao, G.; Ji, X.; Liu, J.; Zhao, T.; Gao, Y.; Gao, S.; Hao, Y.; Gao, Y.; Wang, L.; et al. Metabolome and transcriptome analysis reveals the transcriptional regulatory mechanism of triterpenoid saponin biosynthesis in soapberry (Sapindus mukorossi Gaertn.). J. Agric. Food Chem. 2022, 70, 7095–7109. [Google Scholar] [CrossRef] [PubMed]
- Terabayashi, T.; Germino, G.G.; Menezes, L.F. Pathway identification through transcriptome analysis. Cell. Signal. 2020, 74, 109701. [Google Scholar] [CrossRef]
- Thorrold, C.A.; Letsoalo, M.E.; Dusé, A.G.; Marais, E. Efflux pump activity in fluoroquinolone and tetracycline resistant Salmonella and E. coli implicated in reduced susceptibility to household antimicrobial cleaning agents. Int. J. Food Microbiol. 2007, 113, 315–320. [Google Scholar] [CrossRef] [PubMed]
- Andersson, D.I.; Hughes, D. Antibiotic resistance and its cost: Is it possible to reverse resistance? Nat. Rev. Microbiol. 2010, 8, 260–271. [Google Scholar] [CrossRef] [PubMed]
- Cepas, V.; Soto, S.M. Relationship between virulence and resistance among Gram-negative bacteria. Antibiotics 2020, 9, 71. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antibiotic Type | Antibiotics | Healthy Group (ng/mL) | Patient Group (ng/mL) | p-Value |
|---|---|---|---|---|
| HAs | Levofloxacin | 6.22 | 6.35 | NS |
| Clarithromycin | 0.97 | 0.94 | NS | |
| VAs | Enrofloxacin | 2.79 | 2.66 | NS |
| Sulfachloropyridazine | 51.34 | 61.56 | * | |
| Sulphaquinoxaline | 1.24 | 2.67 | * | |
| Sulfaclozine sodium monohydrate | 13.44 | 51.54 | * | |
| N4-acetylsulfamonomethoxine | 4.35 | 4.55 | NS | |
| Cefquinome sulfate | 59.57 | 92.94 | * | |
| Ceftiofur | 0.00 | 1.55 | * | |
| Cyadox | 315.38 | 322.81 | NS | |
| PVAs | Lomefloxacin hydrochloride | 5.53 | 8.96 | * |
| Ofloxacin | 0.00 | 10.17 | * | |
| Ciprofloxacin | 13.48 | 14.22 | NS | |
| Norfloxacin | 2.43 | 5.26 | * | |
| Trimethoprim | 4.90 | 4.33 | NS | |
| Sulfametoxydiazine | 12.65 | 24.37 | * | |
| Sulfamethoxazole | 4.42 | 7.37 | * | |
| Sulfadiazine | 75.74 | 111.23 | * | |
| Erythromycin | 54.43 | 73.63 | * | |
| Lincomycin hydrochloride | 11.24 | 26.29 | * | |
| Doxycycline hydrochloride | 84.39 | 110.01 | * | |
| Tetracycline hydrochloride | 4.34 | 3.89 | NS | |
| Cefotaxime sodium | 22.19 | 30.23 | * | |
| Amoxicillin trihydrate | 62.12 | 229.51 | * | |
| Penicillin-G sodium salt | 133.06 | 421.33 | * | |
| Penicillin V | 0.00 | 4.23 | * |
| Isolate’s Name | Antibiotic-Resistant Genes Detected in the Isolate |
|---|---|
| E. coli strain isolated from LAG | cmlA, tetA, mdtF, mdtb, ctx-M1, qnr-A |
| E. coli strain isolated from HAG | cmlA, tetA, OXY, mdtl, mdtG, mdtF, mdtb, ctx-M1, qnr-s, qnr-B, qnr-A |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, Y.; Wu, Y.; Wu, J.; Li, X.; Yu, L.; Xie, K.; Zhang, M.; Ren, L.; Ji, Y.; Li, Y. Exposure to Veterinary Antibiotics via Food Chain Disrupts Gut Microbiota and Drives Increased Escherichia coli Virulence and Drug Resistance in Young Adults. Pathogens 2022, 11, 1062. https://doi.org/10.3390/pathogens11091062
Liu Y, Wu Y, Wu J, Li X, Yu L, Xie K, Zhang M, Ren L, Ji Y, Li Y. Exposure to Veterinary Antibiotics via Food Chain Disrupts Gut Microbiota and Drives Increased Escherichia coli Virulence and Drug Resistance in Young Adults. Pathogens. 2022; 11(9):1062. https://doi.org/10.3390/pathogens11091062
Chicago/Turabian StyleLiu, Yehao, Yifan Wu, Jie Wu, Xin Li, Lingling Yu, Ke Xie, Mingyi Zhang, Lingling Ren, Yanli Ji, and Yuhui Li. 2022. "Exposure to Veterinary Antibiotics via Food Chain Disrupts Gut Microbiota and Drives Increased Escherichia coli Virulence and Drug Resistance in Young Adults" Pathogens 11, no. 9: 1062. https://doi.org/10.3390/pathogens11091062
APA StyleLiu, Y., Wu, Y., Wu, J., Li, X., Yu, L., Xie, K., Zhang, M., Ren, L., Ji, Y., & Li, Y. (2022). Exposure to Veterinary Antibiotics via Food Chain Disrupts Gut Microbiota and Drives Increased Escherichia coli Virulence and Drug Resistance in Young Adults. Pathogens, 11(9), 1062. https://doi.org/10.3390/pathogens11091062