
Do or Die: HPV E5, E6 and E7 in Cell Death Evasion




{kind=link}
{kind=link}
Abstract
1. Introduction
2. HPV Genome Organization and Life Cycle
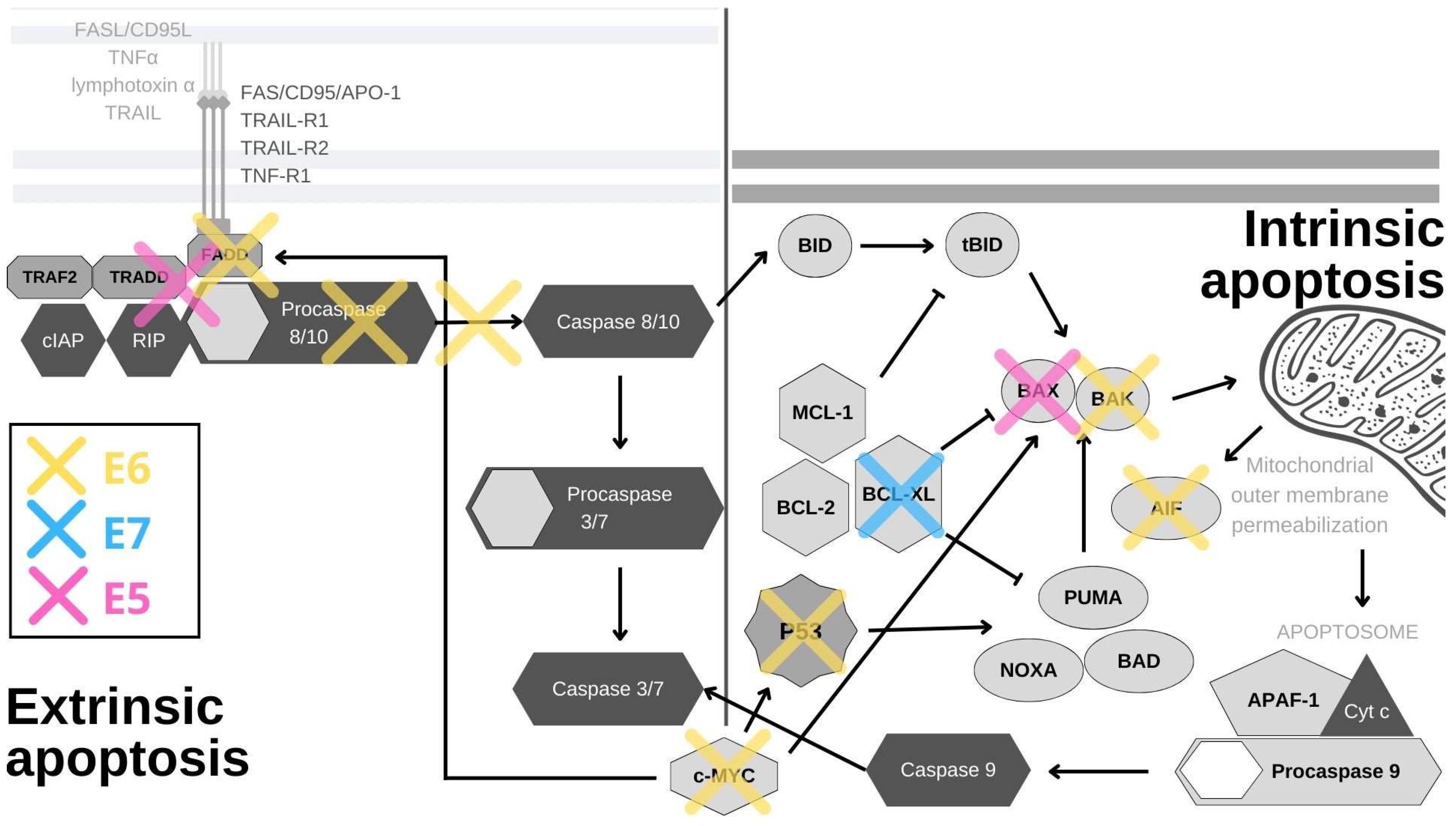
3. Apoptosis
4. Autophagy
5. Anoikis and Pyroptosis
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bernard, H.-U.; Burk, R.D.; Chen, Z.; van Doorslaer, K.; zur Hausen, H.; de Villiers, E.-M. Classification of Papillomaviruses (PVs) Based on 189 PV Types and Proposal of Taxonomic Amendments. Virology 2010, 401, 70–79. [Google Scholar] [CrossRef] [PubMed]
- Bzhalava, D.; Eklund, C.; Dillner, J. International Standardization and Classification of Human Papillomavirus Types. Virology 2015, 476, 341–344. [Google Scholar] [CrossRef]
- Tomaić, V. Functional Roles of E6 and E7 Oncoproteins in HPV-Induced Malignancies at Diverse Anatomical Sites. Cancers 2016, 8, 95. [Google Scholar] [CrossRef] [PubMed]
- Đukić, A.; Lulić, L.; Thomas, M.; Skelin, J.; Saidu, N.E.B.; Grce, M.; Banks, L.; Tomaić, V. HPV Oncoproteins and the Ubiquitin Proteasome System: A Signature of Malignancy? Pathogens 2020, 9, 133. [Google Scholar] [CrossRef] [PubMed]
- World Health Organisation International Agency for Research on Cancer Iarc Monographs on the Evaluation of Carcinogenic Risks to Humans Volume 90 Human Papillomaviruses; International Agency for Research on Cancer: Lyon Cedex, France, 2007; Volume 90, ISBN 9789283212904.
- Bouvard, V.; Baan, R.; Straif, K.; Grosse, Y.; Secretan, B.; El Ghissassi, F.; Benbrahim-Tallaa, L.; Guha, N.; Freeman, C.; Galichet, L.; et al. A Review of Human Carcinogens--Part B: Biological Agents. Lancet Oncol. 2009, 10, 321–322. [Google Scholar] [CrossRef]
- Lont, A.P.; Kroon, B.K.; Horenblas, S.; Gallee, M.P.W.; Berkhof, J.; Meijer, C.J.L.M.; Snijders, P.J.F. Presence of High-Risk Human Papillomavirus DNA in Penile Carcinoma Predicts Favorable Outcome in Survival. Int. J. Cancer 2006, 119, 1078–1081. [Google Scholar] [CrossRef] [PubMed]
- Madsen, B.S.; Jensen, H.L.; Van Den Brule, A.J.C.; Wohlfahrt, J.; Frisch, M. Risk Factors for Invasive Squamous Cell Carcinoma of the Vulva and Vagina-Population-Based Case-Control Study in Denmark. Int. J. Cancer 2008, 122, 2827–2834. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Franceschi, S.; Howell-Jones, R.; Snijders, P.J.F.; Clifford, G.M. Human Papillomavirus Type Distribution in 30,848 Invasive Cervical Cancers Worldwide: Variation by Geographical Region, Histological Type and Year of Publication. Int. J. Cancer 2011, 128, 927–935. [Google Scholar] [CrossRef] [PubMed]
- Doorbar, J.; Quint, W.; Banks, L.; Bravo, I.G.; Stoler, M.; Broker, T.R.; Stanley, M.A. The Biology and Life-Cycle of Human Papillomaviruses. Vaccine 2012, 30 (Suppl. 5), F55–F70. [Google Scholar] [CrossRef]
- Graham, S.V.; Faizo, A.A.A. Control of Human Papillomavirus Gene Expression by Alternative Splicing. Virus Res. 2017, 231, 83–95. [Google Scholar] [CrossRef]
- Doorbar, J. Model Systems of Human Papillomavirus-Associated Disease. J. Pathol. 2016, 238, 166–179. [Google Scholar] [CrossRef]
- Maglennon, G.A.; McIntosh, P.; Doorbar, J. Persistence of Viral DNA in the Epithelial Basal Layer Suggests a Model for Papillomavirus Latency Following Immune Regression. Virology 2011, 414, 153–163. [Google Scholar] [CrossRef] [PubMed]
- McBride, A.A. Mechanisms and Strategies of Papillomavirus Replication. Biol. Chem. 2017, 398, 919–927. [Google Scholar] [CrossRef]
- Gutierrez-Xicotencatl, L.; Pedroza-Saavedra, A.; Chihu-Amparan, L.; Salazar-Piña, A.; Maldonado-Gama, M.; Esquivel-Guadarrama, F. Cellular Functions of HPV16 E5 Oncoprotein during Oncogenic Transformation. Mol. Cancer Res. 2021, 19, 167–179. [Google Scholar] [CrossRef]
- Nakahara, T.; Peh, W.L.; Doorbar, J.; Lee, D.; Lambert, P.F. Human Papillomavirus Type 16 E1circumflexE4 Contributes to Multiple Facets of the Papillomavirus Life Cycle. J. Virol. 2005, 79, 13150–13165. [Google Scholar] [CrossRef]
- Scarth, J.A.; Patterson, M.R.; Morgan, E.L.; Macdonald, A. The Human Papillomavirus Oncoproteins: A Review of the Host Pathways Targeted on the Road to Transformation. J. Gen. Virol. 2021, 102, 001540. [Google Scholar] [CrossRef] [PubMed]
- McBride, A.A.; Warburton, A. The Role of Integration in Oncogenic Progression of HPV-Associated Cancers. PLoS Pathog. 2017, 13, e1006211. [Google Scholar] [CrossRef]
- Cosper, P.F.; Bradley, S.; Luo, L.; Kimple, R.J. Biology of HPV Mediated Carcinogenesis and Tumor Progression. Semin. Radiat. Oncol. 2021, 31, 265–273. [Google Scholar] [CrossRef]
- Lorenzon, L.; Mazzetta, F.; Venuti, A.; Frega, A.; Torrisi, M.R.; French, D. In Vivo HPV 16 E5 MRNA: Expression Pattern in Patients with Squamous Intra-Epithelial Lesions of the Cervix. J. Clin. Virol. 2011, 52, 79–83. [Google Scholar] [CrossRef]
- Barbaresi, S.; Cortese, M.S.; Quinn, J.; Ashrafi, G.H.; Graham, S.V.; Campo, M.S. Effects of Human Papillomavirus Type 16 E5 Deletion Mutants on Epithelial Morphology: Functional Characterization of Each Transmembrane Domain. J. Gen. Virol. 2010, 91, 521–530. [Google Scholar] [CrossRef]
- Stoler, M.H.; Rhodes, C.R.; Whitbeck, A.; Wolinsky, S.M.; Chow, L.T.; Broker, T.R. Human Papillomavirus Type 16 and 18 Gene Expression in Cervical Neoplasias. Hum. Pathol. 1992, 23, 117–128. [Google Scholar] [CrossRef]
- D’Arcy, M.S. Cell Death: A Review of the Major Forms of Apoptosis, Necrosis and Autophagy. Cell Biol. Int. 2019, 43, 582–592. [Google Scholar] [CrossRef] [PubMed]
- Xiong, S.; Mu, T.; Wang, G.; Jiang, X. Mitochondria-Mediated Apoptosis in Mammals. Protein Cell 2014, 5, 737–749. [Google Scholar] [CrossRef] [PubMed]
- Dorstyn, L.; Akey, C.W.; Kumar, S. New Insights into Apoptosome Structure and Function. Cell Death Differ. 2018, 25, 1194–1208. [Google Scholar] [CrossRef]
- Walczak, H. Death Receptor-Ligand Systems in Cancer, Cell Death, and Inflammation. Cold Spring Harb. Perspect. Biol. 2013, 5, a008698. [Google Scholar] [CrossRef]
- Gough, P.; Myles, I.A. Tumor Necrosis Factor Receptors: Pleiotropic Signaling Complexes and Their Differential Effects. Front. Immunol. 2020, 11, 585880. [Google Scholar] [CrossRef]
- Sessler, T.; Healy, S.; Samali, A.; Szegezdi, E. Structural Determinants of DISC Function: New Insights into Death Receptor-Mediated Apoptosis Signalling. Pharmacol. Ther. 2013, 140, 186–199. [Google Scholar] [CrossRef]
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W.; et al. Molecular Mechanisms of Cell Death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018, 25, 486–541. [Google Scholar] [CrossRef]
- Roufayel, R.; Younes, K.; Al-Sabi, A.; Murshid, N. BH3-Only Proteins Noxa and Puma Are Key Regulators of Induced Apoptosis. Life 2022, 12, 256. [Google Scholar] [CrossRef]
- Hafner, A.; Bulyk, M.L.; Jambhekar, A.; Lahav, G. The Multiple Mechanisms That Regulate P53 Activity and Cell Fate. Nat. Rev. Mol. Cell Biol. 2019, 20, 199–210. [Google Scholar] [CrossRef]
- Huibregtse, J.M.; Scheffner, M.; Howley, P.M. Localization of the E6-AP Regions That Direct Human Papillomavirus E6 Binding, Association with P53, and Ubiquitination of Associated Proteins. Mol. Cell. Biol. 1993, 13, 4918–4927. [Google Scholar] [CrossRef]
- Scheffner, M.; Huibregtse, J.M.; Vierstra, R.D.; Howley, P.M. The HPV-16 E6 and E6-AP Complex Functions as a Ubiquitin-Protein Ligase in the Ubiquitination of P53. Cell 1993, 75, 495–505. [Google Scholar] [CrossRef]
- Tomaić, V.; Pim, D.; Banks, L. The Stability of the Human Papillomavirus E6 Oncoprotein Is E6AP Dependent. Virology 2009, 393, 7–10. [Google Scholar] [CrossRef] [PubMed]
- Kranjec, C.; Tomaić, V.; Massimi, P.; Nicolaides, L.; Doorbar, J.; Banks, L. The High-Risk HPV E6 Target Scribble (HScrib) Is Required for HPV E6 Expression in Cervical Tumour-Derived Cell Lines. Papillomavirus Res. 2016, 2, 70–77. [Google Scholar] [CrossRef]
- Massimi, P.; Shai, A.; Lambert, P.; Banks, L. HPV E6 Degradation of P53 and PDZ Containing Substrates in an E6AP Null Background. Oncogene 2008, 27, 1800–1804. [Google Scholar] [CrossRef] [PubMed]
- Lechner, M.S.; Laimins, L.A. Inhibition of P53 DNA Binding by Human Papillomavirus E6 Proteins. J. Virol. 1994, 68, 4262–4273. [Google Scholar] [CrossRef]
- Zimmermann, H.; Degenkolbe, R.; Bernard, H.U.; O’Connor, M.J. The Human Papillomavirus Type 16 E6 Oncoprotein Can Down-Regulate P53 Activity by Targeting the Transcriptional Coactivator CBP/P300. J. Virol. 1999, 73, 6209–6219. [Google Scholar] [CrossRef]
- Sekaric, P.; Shamanin, V.A.; Luo, J.; Androphy, E.J. HAda3 Regulates P14ARF-Induced P53 Acetylation and Senescence. Oncogene 2007, 26, 6261–6268. [Google Scholar] [CrossRef]
- Jha, S.; Vande Pol, S.; Banerjee, N.S.; Dutta, A.B.; Chow, L.T.; Dutta, A. Destabilization of TIP60 by Human Papillomavirus E6 Results in Attenuation of TIP60-Dependent Transcriptional Regulation and Apoptotic Pathway. Mol. Cell 2010, 38, 700–711. [Google Scholar] [CrossRef]
- Thomas, M.C.; Chiang, C.-M. E6 Oncoprotein Represses P53-Dependent Gene Activation via Inhibition of Protein Acetylation Independently of Inducing P53 Degradation. Mol. Cell 2005, 17, 251–264. [Google Scholar] [CrossRef]
- Ajay, A.K.; Meena, A.S.; Bhat, M.K. Human Papillomavirus 18 E6 Inhibits Phosphorylation of P53 Expressed in HeLa Cells. Cell Biosci. 2012, 2, 2. [Google Scholar] [CrossRef] [PubMed]
- Ji, L.; Gong, C.; Ge, L.; Song, L.; Chen, F.; Jin, C.; Zhu, H.; Zhou, G. Orphan Nuclear Receptor Nurr1 as a Potential Novel Marker for Progression in Human Pancreatic Ductal Adenocarcinoma. Exp. Ther. Med. 2017, 13, 551–559. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Basheeth, N.; Patil, N. Biomarkers in Head and Neck Cancer an Update. Indian J. Otolaryngol. Head neck Surg. Off. Publ. Assoc. Otolaryngol. India 2019, 71, 1002–1011. [Google Scholar] [CrossRef] [PubMed]
- He, G.; Wang, Q.; Zhou, Y.; Wu, X.; Wang, L.; Duru, N.; Kong, X.; Zhang, P.; Wan, B.; Sui, L.; et al. YY1 Is a Novel Potential Therapeutic Target for the Treatment of HPV Infection-Induced Cervical Cancer by Arsenic Trioxide. Int. J. Gynecol. cancer Off. J. Int. Gynecol. Cancer Soc. 2011, 21, 1097–1104. [Google Scholar] [CrossRef] [PubMed]
- Warowicka, A.; Broniarczyk, J.; Węglewska, M.; Kwaśniewski, W.; Goździcka-Józefiak, A. Dual Role of YY1 in HPV Life Cycle and Cervical Cancer Development. Int. J. Mol. Sci. 2022, 23, 3453. [Google Scholar] [CrossRef]
- Borbély, Á.A.; Murvai, M.; Kónya, J.; Beck, Z.; Gergely, L.; Li, F.; Veress, G. Effects of Human Papillomavirus Type 16 Oncoproteins on Survivin Gene Expression. J. Gen. Virol. 2006, 87, 287–294. [Google Scholar] [CrossRef]
- Branca, M.; Giorgi, C.; Santini, D.; Di Bonito, L.; Ciotti, M.; Costa, S.; Benedetto, A.; Casolati, E.A.; Favalli, C.; Paba, P.; et al. Survivin as a Marker of Cervical Intraepithelial Neoplasia and High-Risk Human Papillomavirus and a Predictor of Virus Clearance and Prognosis in Cervical Cancer. Am. J. Clin. Pathol. 2005, 124, 113–121. [Google Scholar] [CrossRef]
- Liu, X.; Roberts, J.; Dakic, A.; Zhang, Y.; Schlegel, R. HPV E7 Contributes to the Telomerase Activity of Immortalized and Tumorigenic Cells and Augments E6-Induced HTERT Promoter Function. Virology 2008, 375, 611–623. [Google Scholar] [CrossRef] [PubMed]
- Gross-Mesilaty, S.; Reinstein, E.; Bercovich, B.; Tobias, K.E.; Schwartz, A.L.; Kahana, C.; Ciechanover, A. Basal and Human Papillomavirus E6 Oncoprotein-Induced Degradation of Myc Proteins by the Ubiquitin Pathway. Proc. Natl. Acad. Sci. USA 1998, 95, 8058–8063. [Google Scholar] [CrossRef]
- Thomas, M.; Banks, L. Inhibition of Bak-Induced Apoptosis by HPV-18 E6. Oncogene 1998, 17, 2943–2954. [Google Scholar] [CrossRef]
- Kübler, K.; Heinenberg, S.; Rudlowski, C.; Keyver-Paik, M.-D.; Abramian, A.; Merkelbach-Bruse, S.; Büttner, R.; Kuhn, W.; Schildhaus, H.-U. C-Myc Copy Number Gain Is a Powerful Prognosticator of Disease Outcome in Cervical Dysplasia. Oncotarget 2015, 6, 825–835. [Google Scholar] [CrossRef] [PubMed]
- Ji, W.; Lou, W.; Hong, Z.; Qiu, L.; Di, W. Genomic Amplification of HPV, H-TERC and C-MYC in Liquid-based Cytological Specimens for Screening of Cervical Intraepithelial Neoplasia and Cancer. Oncol Lett 2019, 17, 2099–2106. [Google Scholar] [CrossRef] [PubMed]
- Shimada, M.; Yamashita, A.; Saito, M.; Ichino, M.; Kinjo, T.; Mizuki, N.; Klinman, D.M.; Okuda, K. The Human Papillomavirus E6 Protein Targets Apoptosis-Inducing Factor (AIF) for Degradation. Sci. Rep. 2020, 10, 14195. [Google Scholar] [CrossRef] [PubMed]
- Cabeça, T.K.; de Mello Abreu, A.; Andrette, R.; de Souza Lino, V.; Morale, M.G.; Aguayo, F.; Termini, L.; Villa, L.L.; Lepique, A.P.; Boccardo, E. HPV-Mediated Resistance to TNF and TRAIL Is Characterized by Global Alterations in Apoptosis Regulatory Factors, Dysregulation of Death Receptors, and Induction of ROS/RNS. Int. J. Mol. Sci. 2019, 20, 198. [Google Scholar] [CrossRef]
- Filippova, M.; Song, H.; Connolly, J.L.; Dermody, T.S.; Duerksen-Hughes, P.J. The Human Papillomavirus 16 E6 Protein Binds to Tumor Necrosis Factor (TNF) R1 and Protects Cells from TNF-Induced Apoptosis. J. Biol. Chem. 2002, 277, 21730–21739. [Google Scholar] [CrossRef]
- Filippova, M.; Parkhurst, L.; Duerksen-Hughes, P.J. The Human Papillomavirus 16 E6 Protein Binds to Fas-Associated Death Domain and Protects Cells from Fas-Triggered Apoptosis. J. Biol. Chem. 2004, 279, 25729–25744. [Google Scholar] [CrossRef]
- Garnett, T.O.; Filippova, M.; Duerksen-Hughes, P.J. Accelerated Degradation of FADD and Procaspase 8 in Cells Expressing Human Papilloma Virus 16 E6 Impairs TRAIL-Mediated Apoptosis. Cell Death Differ. 2006, 13, 1915–1926. [Google Scholar] [CrossRef]
- Tang, S.; Ding, S.; Yu, L.; Shen, H.; Wan, Y.; Wu, Y. Effects of HPV16 E6 Protein on Daxx-Induced Apoptosis in C33A Cells. Cell. Mol. Biol. Lett. 2020, 25, 38. [Google Scholar] [CrossRef]
- Gaykalova, D.A.; Manola, J.B.; Ozawa, H.; Zizkova, V.; Morton, K.; Bishop, J.A.; Sharma, R.; Zhang, C.; Michailidi, C.; Considine, M.; et al. NF-ΚB and Stat3 Transcription Factor Signatures Differentiate HPV-Positive and HPV-Negative Head and Neck Squamous Cell Carcinoma. Int. J. Cancer 2015, 137, 1879–1889. [Google Scholar] [CrossRef]
- Fan, Y.; Shen, Z. The Clinical Value of HPV E6/E7 and STAT3 MRNA Detection in Cervical Cancer Screening. Pathol.-Res. Pract. 2018, 214, 767–775. [Google Scholar] [CrossRef] [PubMed]
- Morgan, E.L.; Macdonald, A. Autocrine STAT3 Activation in HPV Positive Cervical Cancer through a Virus-Driven Rac1—NFκB—IL-6 Signalling Axis. PLoS Pathog. 2019, 15, e1007835. [Google Scholar] [CrossRef] [PubMed]
- Morgan, E.L.; Wasson, C.W.; Hanson, L.; Kealy, D.; Pentland, I.; McGuire, V.; Scarpini, C.; Coleman, N.; Arthur, J.S.C.; Parish, J.L.; et al. STAT3 Activation by E6 Is Essential for the Differentiation-Dependent HPV18 Life Cycle. PLoS Pathog. 2018, 14, e1006975. [Google Scholar] [CrossRef] [PubMed]
- Thomas, M.; Narayan, N.; Pim, D.; Tomaić, V.; Massimi, P.; Nagasaka, K.; Kranjec, C.; Gammoh, N.; Banks, L.; Thomas, M.; et al. Human Papillomaviruses, Cervical Cancer and Cell Polarity. Oncogene 2008, 27, 7018–7030. [Google Scholar] [CrossRef] [PubMed]
- Pim, D.; Bergant, M.; Boon, S.S.; Ganti, K.; Kranjec, C.; Massimi, P.; Subbaiah, V.K.; Thomas, M.; Tomaić, V.; Banks, L. Human Papillomaviruses and the Specificity of PDZ Domain Targeting. FEBS J. 2012, 279, 3530–3537. [Google Scholar] [CrossRef]
- Ganti, K.; Broniarczyk, J.; Manoubi, W.; Massimi, P.; Mittal, S.; Pim, D.; Szalmas, A.; Thatte, J.; Thomas, M.; Tomaić, V.; et al. The Human Papillomavirus E6 PDZ Binding Motif: From Life Cycle to Malignancy. Viruses 2015, 7, 3530–3551. [Google Scholar] [CrossRef]
- Kranjec, C.; Massimi, P.; Banks, L. Restoration of MAGI-1 Expression in Human Papillomavirus-Positive Tumor Cells Induces Cell Growth Arrest and Apoptosis. J. Virol. 2014, 88, 7155–7169. [Google Scholar] [CrossRef]
- James, M.A.; Lee, J.H.; Klingelhutz, A.J. Human Papillomavirus Type 16 E6 Activates NF-KappaB, Induces CIAP-2 Expression, and Protects against Apoptosis in a PDZ Binding Motif-Dependent Manner. J. Virol. 2006, 80, 5301–5307. [Google Scholar] [CrossRef]
- Singh, P.; Ravanan, P.; Talwar, P. Death Associated Protein Kinase 1 (DAPK1): A Regulator of Apoptosis and Autophagy. Front. Mol. Neurosci. 2016, 9, 46. [Google Scholar] [CrossRef]
- Banzai, C.; Nishino, K.; Quan, J.; Yoshihara, K.; Sekine, M.; Yahata, T.; Tanaka, K. Promoter Methylation of DAPK1, FHIT, MGMT, and CDKN2A Genes in Cervical Carcinoma. Int. J. Clin. Oncol. 2014, 19, 127–132. [Google Scholar] [CrossRef]
- Yanatatsaneejit, P.; Chalertpet, K.; Sukbhattee, J.; Nuchcharoen, I.; Phumcharoen, P.; Mutirangura, A. Promoter Methylation of Tumor Suppressor Genes Induced by Human Papillomavirus in Cervical Cancer. Oncol. Lett. 2020, 20, 955–961. [Google Scholar] [CrossRef]
- Ekanayake Weeramange, C.; Tang, K.D.; Vasani, S.; Langton-Lockton, J.; Kenny, L.; Punyadeera, C. DNA Methylation Changes in Human Papillomavirus-Driven Head and Neck Cancers. Cells 2020, 9, 1359. [Google Scholar] [CrossRef] [PubMed]
- Frazzi, R.; Cusenza Ylenia, V.; Pistoni, M.; Canovi, L.; Cascione, L.; Bertoni, F.; Merli, F. KLF4, DAPK1 and SPG20 Promoter Methylation Is Not Affected by DNMT1 Silencing and Hypomethylating Drugs in Lymphoma Cells. Oncol Rep 2022, 47, 10. [Google Scholar] [CrossRef] [PubMed]
- Feng, Q.; Balasubramanian, A.; Hawes, S.E.; Toure, P.; Sow, P.S.; Dem, A.; Dembele, B.; Critchlow, C.W.; Xi, L.; Lu, H.; et al. Detection of Hypermethylated Genes in Women with and without Cervical Neoplasia. J. Natl. Cancer Inst. 2005, 97, 273–282. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Tan, W.; Yang, H.; Zhang, S.; Dai, Y. Detection of Host Cell Gene/HPV DNA Methylation Markers: A Promising Triage Approach for Cervical Cancer. Front. Oncol. 2022, 12, 831949. [Google Scholar] [CrossRef]
- Lee, H.J.; Kim, M.J.; Kim, Y.S.; Choi, M.Y.; Cho, G.J.; Choi, W.S. UHRF1 Silences Gelsolin to Inhibit Cell Death in Early Stage Cervical Cancer. Biochem. Biophys. Res. Commun. 2020, 526, 1061–1068. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Qiao, L.; Wang, X.; Ding, C.; Chen, J.J. UHRF1 Epigenetically Down-Regulates UbcH8 to Inhibit Apoptosis in Cervical Cancer Cells. Cell Cycle 2018, 17, 300–308. [Google Scholar] [CrossRef]
- Huh, K.; Zhou, X.; Hayakawa, H.; Cho, J.-Y.; Libermann, T.A.; Jin, J.; Harper, J.W.; Munger, K. Human Papillomavirus Type 16 E7 Oncoprotein Associates with the Cullin 2 Ubiquitin Ligase Complex, Which Contributes to Degradation of the Retinoblastoma Tumor Suppressor. J. Virol. 2007, 81, 9737–9747. [Google Scholar] [CrossRef]
- Alunni-Fabbroni, M.; Littlewood, T.; Deleu, L.; Caldeira, S.; Giarrè, M.; Dell’ Orco, M.; Tommasino, M. Induction of S Phase and Apoptosis by the Human Papillomavirus Type 16 E7 Protein Are Separable Events in Immortalized Rodent Fibroblasts. Oncogene 2000, 19, 2277–2285. [Google Scholar] [CrossRef]
- Aguilar-Lemarroy, A.; Gariglio, P.; Whitaker, N.J.; Eichhorst, S.T.; Hausen, H.z.; Krammer, P.H.; Rösl, F. Restoration of P53 Expression Sensitizes Human Papillomavirus Type 16 Immortalized Human Keratinocytes to CD95-Mediated Apoptosis. Oncogene 2002, 21, 165–175. [Google Scholar] [CrossRef]
- Basile, J.R.; Zacny, V.; Münger, K. The Cytokines Tumor Necrosis Factor-α (TNF-α) and TNF-Related Apoptosis-Inducing Ligand Differentially Modulate Proliferation and Apoptotic Pathways in Human Keratinocytes Expressing the Human Papillomavirus-16 E7 Oncoprotein. J. Biol. Chem. 2001, 276, 22522–22528. [Google Scholar] [CrossRef]
- Thompson, D.A.; Zacny, V.; Belinsky, G.S.; Classon, M.; Jones, D.L.; Schlegel, R.; Münger, K. The HPV E7 Oncoprotein Inhibits Tumor Necrosis Factor α-Mediated Apoptosis in Normal Human Fibroblasts. Oncogene 2001, 20, 3629–3640. [Google Scholar] [CrossRef]
- Santer, F.R.; Moser, B.; Spoden, G.A.; Jansen-Dürr, P.; Zwerschke, W. Human Papillomavirus Type 16 E7 Oncoprotein Inhibits Apoptosis Mediated by Nuclear Insulin-like Growth Factor-Binding Protein-3 by Enhancing Its Ubiquitin/Proteasome-Dependent Degradation. Carcinogenesis 2007, 28, 2511–2520. [Google Scholar] [CrossRef][Green Version]
- Shim, J.-H.; Cho, K.-J.; Lee, K.-A.; Kim, S.-H.; Myung, P.-K.; Choe, Y.-K.; Yoon, D.-Y. E7-Expressing HaCaT Keratinocyte Cells Are Resistant to Oxidative Stress-Induced Cell Death via the Induction of Catalase. Proteomics 2005, 5, 2112–2122. [Google Scholar] [CrossRef]
- Zhang, W.; Chen, H.; Chen, Y.; Liu, J.; Wang, X.; Yu, X.; Chen, J.J.; Zhao, W. Cancerous Inhibitor of Protein Phosphatase 2A Contributes to Human Papillomavirus Oncoprotein E7-Induced Cell Proliferation via E2F1. Oncotarget 2015, 6, 5253–5262. [Google Scholar] [CrossRef]
- Severino, A.; Abbruzzese, C.; Manente, L.; Valderas, Á.A.; Mattarocci, S.; Federico, A.; Starace, G.; Chersi, A.; Mileo, A.M.; Paggi, M.G. Human Papillomavirus-16 E7 Interacts with Siva-1 and Modulates Apoptosis in HaCaT Human Immortalized Keratinocytes. J. Cell. Physiol. 2007, 212, 118–125. [Google Scholar] [CrossRef]
- Longworth, M.S.; Laimins, L.A. The Binding of Histone Deacetylases and the Integrity of Zinc Finger-like Motifs of the E7 Protein Are Essential for the Life Cycle of Human Papillomavirus Type 31. J. Virol. 2004, 78, 3533–3541. [Google Scholar] [CrossRef]
- Finzer, P.; Krueger, A.; Stöhr, M.; Brenner, D.; Soto, U.; Kuntzen, C.; Krammer, P.H.; Rösl, F. HDAC Inhibitors Trigger Apoptosis in HPV-Positive Cells by Inducing the E2F-P73 Pathway. Oncogene 2004, 23, 4807–4817. [Google Scholar] [CrossRef]
- Darvas, K.; Rosenberger, S.; Brenner, D.; Fritsch, C.; Gmelin, N.; Krammer, P.H.; Rösl, F. Histone Deacetylase Inhibitor-Induced Sensitization to TNFalpha/TRAIL-Mediated Apoptosis in Cervical Carcinoma Cells Is Dependent on HPV Oncogene Expression. Int. J. Cancer 2010, 127, 1384–1392. [Google Scholar] [CrossRef]
- Oh, J.-M.; Kim, S.-H.; Cho, E.-A.; Song, Y.-S.; Kim, W.-H.; Juhnn, Y.-S. Human Papillomavirus Type 16 E5 Protein Inhibits Hydrogen-Peroxide-Induced Apoptosis by Stimulating Ubiquitin-Proteasome-Mediated Degradation of Bax in Human Cervical Cancer Cells. Carcinogenesis 2010, 31, 402–410. [Google Scholar] [CrossRef]
- Zhang, B.; Spandau, D.F.; Roman, A. E5 Protein of Human Papillomavirus Type 16 Protects Human Foreskin Keratinocytes from UV B-Irradiation-Induced Apoptosis. J. Virol. 2002, 76, 220–231. [Google Scholar] [CrossRef]
- Kabsch, K.; Mossadegh, N.; Kohl, A.; Komposch, G.; Schenkel, J.; Alonso, A.; Tomakidi, P. The HPV-16 E5 Protein Inhibits TRAIL- and FasL-Mediated Apoptosis in Human Keratinocyte Raft Cultures. Intervirology 2004, 47, 48–56. [Google Scholar] [CrossRef] [PubMed]
- Sudarshan, S.R.; Schlegel, R.; Liu, X. The HPV-16 E5 Protein Represses Expression of Stress Pathway Genes XBP-1 and COX-2 in Genital Keratinocytes. Biochem. Biophys. Res. Commun. 2010, 399, 617–622. [Google Scholar] [CrossRef]
- Chang, J.L.; Tsao, Y.P.; Liu, D.W.; Huang, S.J.; Lee, W.H.; Chen, S.L. The Expression of HPV-16 E5 Protein in Squamous Neoplastic Changes in the Uterine Cervix. J. Biomed. Sci. 2001, 8, 206–213. [Google Scholar] [CrossRef] [PubMed]
- Um, S.H.; Mundi, N.; Yoo, J.; Palma, D.A.; Fung, K.; MacNeil, D.; Wehrli, B.; Mymryk, J.S.; Barrett, J.W.; Nichols, A.C. Variable Expression of the Forgotten Oncogene E5 in HPV-Positive Oropharyngeal Cancer. J. Clin. Virol. 2014, 61, 94–100. [Google Scholar] [CrossRef]
- Khandia, R.; Dadar, M.; Munjal, A.; Dhama, K.; Karthik, K.; Tiwari, R.; Yatoo, M.I.; Iqbal, H.M.N.; Singh, K.P.; Joshi, S.K.; et al. A Comprehensive Review of Autophagy and Its Various Roles in Infectious, Non-Infectious, and Lifestyle Diseases: Current Knowledge and Prospects for Disease Prevention, Novel Drug Design, and Therapy. Cells 2019, 8, 674. [Google Scholar] [CrossRef]
- Kaushik, S.; Cuervo, A.M. The Coming of Age of Chaperone-Mediated Autophagy. Nat. Rev. Mol. Cell Biol. 2018, 19, 365–381. [Google Scholar] [CrossRef]
- Li, W.; Li, J.; Bao, J. Microautophagy: Lesser-Known Self-Eating. Cell. Mol. Life Sci. 2012, 69, 1125–1136. [Google Scholar] [CrossRef]
- Vescovo, T.; Pagni, B.; Piacentini, M.; Fimia, G.M.; Antonioli, M. Regulation of Autophagy in Cells Infected With Oncogenic Human Viruses and Its Impact on Cancer Development. Front. Cell Dev. Biol. 2020, 8, 47. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, L.; Mostowy, S.; Sancho-Shimizu, V. Autophagy-Virus Interplay: From Cell Biology to Human Disease. Front. Cell Dev. Biol. 2018, 6, 155. [Google Scholar] [CrossRef]
- Surviladze, Z.; Sterk, R.T.; DeHaro, S.A.; Ozbun, M.A. Cellular Entry of Human Papillomavirus Type 16 Involves Activation of the Phosphatidylinositol 3-Kinase/Akt/MTOR Pathway and Inhibition of Autophagy. J. Virol. 2013, 87, 2508–2517. [Google Scholar] [CrossRef]
- Griffin, L.M.; Cicchini, L.; Pyeon, D. Human Papillomavirus Infection Is Inhibited by Host Autophagy in Primary Human Keratinocytes. Virology 2013, 437, 12–19. [Google Scholar] [CrossRef]
- Hanning, J.E.; Saini, H.K.; Murray, M.J.; Caffarel, M.M.; van Dongen, S.; Ward, D.; Barker, E.M.; Scarpini, C.G.; Groves, I.J.; Stanley, M.A.; et al. Depletion of HPV16 Early Genes Induces Autophagy and Senescence in a Cervical Carcinogenesis Model, Regardless of Viral Physical State. J. Pathol. 2013, 231, 354–366. [Google Scholar] [CrossRef] [PubMed]
- Mattoscio, D.; Medda, A.; Chiocca, S. Human Papilloma Virus and Autophagy. Int. J. Mol. Sci. 2018, 19, 1775. [Google Scholar] [CrossRef]
- Thomas, M.; Tomaić, V.; Pim, D.; Myers, M.P.; Tommasino, M.; Banks, L. Interactions between E6AP and E6 Proteins from Alpha and Beta HPV Types. Virology 2013, 435, 357–362. [Google Scholar] [CrossRef]
- Antonioli, M.; Pagni, B.; Vescovo, T.; Ellis, R.; Cosway, B.; Rollo, F.; Bordoni, V.; Agrati, C.; Labus, M.; Covello, R.; et al. HPV Sensitizes OPSCC Cells to Cisplatin-Induced Apoptosis by Inhibiting Autophagy through E7-Mediated Degradation of AMBRA1. Autophagy 2021, 17, 2842–2855. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Münger, K. Expression of the Human Papillomavirus Type 16 E7 Oncoprotein Induces an Autophagy-Related Process and Sensitizes Normal Human Keratinocytes to Cell Death in Response to Growth Factor Deprivation. Virology 2009, 385, 192–197. [Google Scholar] [CrossRef] [PubMed]
- Pim, D.; Massimi, P.; Dilworth, S.M.; Banks, L. Activation of the Protein Kinase B Pathway by the HPV-16 E7 Oncoprotein Occurs through a Mechanism Involving Interaction with PP2A. Oncogene 2005, 24, 7830–7838. [Google Scholar] [CrossRef] [PubMed]
- Menges, C.W.; Baglia, L.A.; Lapoint, R.; McCance, D.J. Human Papillomavirus Type 16 E7 Up-Regulates AKT Activity through the Retinoblastoma Protein. Cancer Res. 2006, 66, 5555–5559. [Google Scholar] [CrossRef]
- Molinolo, A.A.; Marsh, C.; El Dinali, M.; Gangane, N.; Jennison, K.; Hewitt, S.; Patel, V.; Seiwert, T.Y.; Gutkind, J.S. MTOR as a Molecular Target in HPV-Associated Oral and Cervical Squamous Carcinomas. Clin. Cancer Res. 2012, 18, 2558–2568. [Google Scholar] [CrossRef]
- Zhang, L.; Wu, J.; Ling, M.T.; Zhao, L.; Zhao, K.-N. The Role of the PI3K/Akt/MTOR Signalling Pathway in Human Cancers Induced by Infection with Human Papillomaviruses. Mol. Cancer 2015, 14, 87. [Google Scholar] [CrossRef]
- Rezazadeh, D.; Norooznezhad, A.H.; Mansouri, K.; Jahani, M.; Mostafaie, A.; Mohammadi, M.H.; Modarressi, M.H. Rapamycin Reduces Cervical Cancer Cells Viability in Hypoxic Condition: Investigation of the Role of Autophagy and Apoptosis. Onco. Targets. Ther. 2020, 13, 4239–4247. [Google Scholar] [CrossRef] [PubMed]
- Thomas, R.J.; Oleinik, N.; Panneer Selvam, S.; Vaena, S.G.; Dany, M.; Nganga, R.N.; Depalma, R.; Baron, K.D.; Kim, J.; Szulc, Z.M.; et al. HPV/E7 Induces Chemotherapy-Mediated Tumor Suppression by Ceramide-Dependent Mitophagy. EMBO Mol. Med. 2017, 9, 1030–1051. [Google Scholar] [CrossRef]
- Belleudi, F.; Nanni, M.; Raffa, S.; Torrisi, M.R. HPV16 E5 Deregulates the Autophagic Process in Human Keratinocytes. Oncotarget 2015, 6, 9370. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.; Pan, X.; Li, F.; Zhang, Y.; Lu, X. Expression of Beclin 1 and LC3 in FIGO Stage I-II Cervical Squamous Cell Carcinoma and Relationship to Survival. Tumour Biol. J. Int. Soc. Oncodevelopmental Biol. Med. 2012, 33, 1653–1659. [Google Scholar] [CrossRef] [PubMed]
- Hu, D.; Zhou, J.; Wang, F.; Shi, H.; Li, Y.; Li, B. HPV-16 E6/E7 Promotes Cell Migration and Invasion in Cervical Cancer via Regulating Cadherin Switch in Vitro and in Vivo. Arch. Gynecol. Obstet. 2015, 292, 1345–1354. [Google Scholar] [CrossRef]
- Carchman, E.H.; Matkowskyj, K.A.; Meske, L.; Lambert, P.F. Dysregulation of Autophagy Contributes to Anal Carcinogenesis. PLoS ONE 2016, 11, e0164273. [Google Scholar] [CrossRef] [PubMed]
- Paoli, P.; Giannoni, E.; Chiarugi, P. Anoikis Molecular Pathways and Its Role in Cancer Progression. Biochim. Biophys. Acta 2013, 1833, 3481–3498. [Google Scholar] [CrossRef]
- Rangarajan, A.; Syal, R.; Selvarajah, S.; Chakrabarti, O.; Sarin, A.; Krishna, S. Activated Notch1 Signaling Cooperates with Papillomavirus Oncogenes in Transformation and Generates Resistance to Apoptosis on Matrix Withdrawal through PKB/Akt. Virology 2001, 286, 23–30. [Google Scholar] [CrossRef]
- Wan, P.K.-T.; Leung, T.H.-Y.; Siu, M.K.-Y.; Mo, X.-T.; Tang, H.W.-M.; Chan, K.K.-L.; Cheung, A.N.-Y.; Ngan, H.Y.-S. HPV-Induced Nurr1 Promotes Cancer Aggressiveness, Self-Renewal, and Radioresistance via ERK and AKT Signaling in Cervical Cancer. Cancer Lett. 2021, 497, 14–27. [Google Scholar] [CrossRef]
- Henken, F.E.; Banerjee, N.S.; Snijders, P.J.F.; Meijer, C.J.L.M.; De-Castro Arce, J.; Rösl, F.; Broker, T.R.; Chow, L.T.; Steenbergen, R.D.M. PIK3CA-Mediated PI3-Kinase Signalling Is Essential for HPV-Induced Transformation in Vitro. Mol. Cancer 2011, 10, 71. [Google Scholar] [CrossRef]
- Beaty, B.T.; Moon, D.H.; Shen, C.J.; Amdur, R.J.; Weiss, J.; Grilley-Olson, J.; Patel, S.; Zanation, A.; Hackman, T.G.; Thorp, B.; et al. PIK3CA Mutation in HPV-Associated OPSCC Patients Receiving Deintensified Chemoradiation. J. Natl. Cancer Inst. 2020, 112, 855–858. [Google Scholar] [CrossRef]
- Litwin, T.R.; Clarke, M.A.; Dean, M.; Wentzensen, N. Somatic Host Cell Alterations in HPV Carcinogenesis. Viruses 2017, 9, 206. [Google Scholar] [CrossRef]
- Backsch, C.; Rudolph, B.; Steinbach, D.; Scheungraber, C.; Liesenfeld, M.; Häfner, N.; Hildner, M.; Habenicht, A.; Runnebaum, I.B.; Dürst, M. An Integrative Functional Genomic and Gene Expression Approach Revealed SORBS2 as a Putative Tumour Suppressor Gene Involved in Cervical Carcinogenesis. Carcinogenesis 2011, 32, 1100–1106. [Google Scholar] [CrossRef]
- Sun, Y.; Zhang, Q.; Yao, L.; Wang, S.; Zhang, Z. Comprehensive Analysis Reveals Novel Gene Signature in Head and Neck Squamous Cell Carcinoma: Predicting Is Associated with Poor Prognosis in Patients. Transl. Cancer Res. 2020, 9, 5882–5892. [Google Scholar] [CrossRef]
- Zhao, L.; Wang, W.; Huang, S.; Yang, Z.; Xu, L.; Yang, Q.; Zhou, X.; Wang, J.; Shen, Q.; Wang, C.; et al. The RNA Binding Protein SORBS2 Suppresses Metastatic Colonization of Ovarian Cancer by Stabilizing Tumor-Suppressive Immunomodulatory Transcripts. Genome Biol. 2018, 19, 35. [Google Scholar] [CrossRef]
- Yan, B.; Peng, Z.; Xing, C. SORBS2, Mediated by MEF2D, Suppresses the Metastasis of Human Hepatocellular Carcinoma by Inhibitiing the c-Abl-ERK Signaling Pathway. Am. J. Cancer Res. 2019, 9, 2706–2718. [Google Scholar]
- Jung, A.C.; Ray, A.-M.; Ramolu, L.; Macabre, C.; Simon, F.; Noulet, F.; Blandin, A.-F.; Renner, G.; Lehmann, M.; Choulier, L.; et al. Caveolin-1-Negative Head and Neck Squamous Cell Carcinoma Primary Tumors Display Increased Epithelial to Mesenchymal Transition and Prometastatic Properties. Oncotarget 2015, 6, 41884–41901. [Google Scholar] [CrossRef]
- Razani, B.; Altschuler, Y.; Zhu, L.; Pestell, R.G.; Mostov, K.E.; Lisanti, M.P. Caveolin-1 Expression Is down-Regulated in Cells Transformed by the Human Papilloma Virus in a P53-Dependent Manner. Replacement of Caveolin-1 Expression Suppresses HPV-Mediated Cell Transformation. Biochemistry 2000, 39, 13916–13924. [Google Scholar] [CrossRef]
- Suprynowicz, F.A.; Disbrow, G.L.; Krawczyk, E.; Simic, V.; Lantzky, K.; Schlegel, R. HPV-16 E5 Oncoprotein Upregulates Lipid Raft Components Caveolin-1 and Ganglioside GM1 at the Plasma Membrane of Cervical Cells. Oncogene 2008, 27, 1071–1078. [Google Scholar] [CrossRef] [PubMed]
- Spanos, W.C.; Hoover, A.; Harris, G.F.; Wu, S.; Strand, G.L.; Anderson, M.E.; Klingelhutz, A.J.; Hendriks, W.; Bossler, A.D.; Lee, J.H. The PDZ Binding Motif of Human Papillomavirus Type 16 E6 Induces PTPN13 Loss, Which Allows Anchorage-Independent Growth and Synergizes with Ras for Invasive Growth. J. Virol. 2008, 82, 2493–2500. [Google Scholar] [CrossRef]
- Szalmás, A.; Tomaić, V.; Basukala, O.; Massimi, P.; Mittal, S.; Kónya, J.; Banks, L. The PTPN14 Tumor Suppressor Is a Degradation Target of Human Papillomavirus E7. J. Virol. 2017, 91, e00057-17. [Google Scholar] [CrossRef] [PubMed]
- Hatterschide, J.; Bohidar, A.E.; Grace, M.; Nulton, T.J.; Kim, H.W.; Windle, B.; Morgan, I.M.; Munger, K.; White, E.A. PTPN14 Degradation by High-Risk Human Papillomavirus E7 Limits Keratinocyte Differentiation and Contributes to HPV-Mediated Oncogenesis. Proc. Natl. Acad. Sci. USA 2019, 116, 7033–7042. [Google Scholar] [CrossRef]
- Yu, P.; Zhang, X.; Liu, N.; Tang, L.; Peng, C.; Chen, X. Pyroptosis: Mechanisms and Diseases. Signal Transduct. Target. Ther. 2021, 6, 128. [Google Scholar] [CrossRef]
- So, D.; Shin, H.-W.; Kim, J.; Lee, M.; Myeong, J.; Chun, Y.-S.; Park, J.-W. Cervical Cancer Is Addicted to SIRT1 Disarming the AIM2 Antiviral Defense. Oncogene 2018, 37, 5191–5204. [Google Scholar] [CrossRef]
- Velez-Perez, A.; Wang, X.I.; Li, M.; Zhang, S. SIRT1 Overexpression in Cervical Squamous Intraepithelial Lesions and Invasive Squamous Cell Carcinoma. Hum. Pathol. 2017, 59, 102–107. [Google Scholar] [CrossRef]
- Song, Y.; Wu, X.; Xu, Y.; Zhu, J.; Li, J.; Zou, Z.; Chen, L.; Zhang, B.; Hua, C.; Rui, H.; et al. HPV E7 Inhibits Cell Pyroptosis by Promoting TRIM21-Mediated Degradation and Ubiquitination of the IFI16 Inflammasome. Int. J. Biol. Sci. 2020, 16, 2924–2937. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Skelin, J.; Sabol, I.; Tomaić, V. Do or Die: HPV E5, E6 and E7 in Cell Death Evasion. Pathogens 2022, 11, 1027. https://doi.org/10.3390/pathogens11091027
Skelin J, Sabol I, Tomaić V. Do or Die: HPV E5, E6 and E7 in Cell Death Evasion. Pathogens. 2022; 11(9):1027. https://doi.org/10.3390/pathogens11091027
Chicago/Turabian StyleSkelin, Josipa, Ivan Sabol, and Vjekoslav Tomaić. 2022. "Do or Die: HPV E5, E6 and E7 in Cell Death Evasion" Pathogens 11, no. 9: 1027. https://doi.org/10.3390/pathogens11091027
APA StyleSkelin, J., Sabol, I., & Tomaić, V. (2022). Do or Die: HPV E5, E6 and E7 in Cell Death Evasion. Pathogens, 11(9), 1027. https://doi.org/10.3390/pathogens11091027