
How GBS Got Its Hump: Genomic Analysis of Group B Streptococcus from Camels Identifies Host Restriction as well as Mobile Genetic Elements Shared across Hosts and Pathogens

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Methods
2.1. Dataset Selection
2.2. Sequencing, Assembly and Typing
2.3. Core Genome Analysis of Camel Isolates
2.4. Genome-Wide Association Studies (GWAS)
2.5. Analysis of Mobile Genetic Elements in Camel Isolates
3. Results
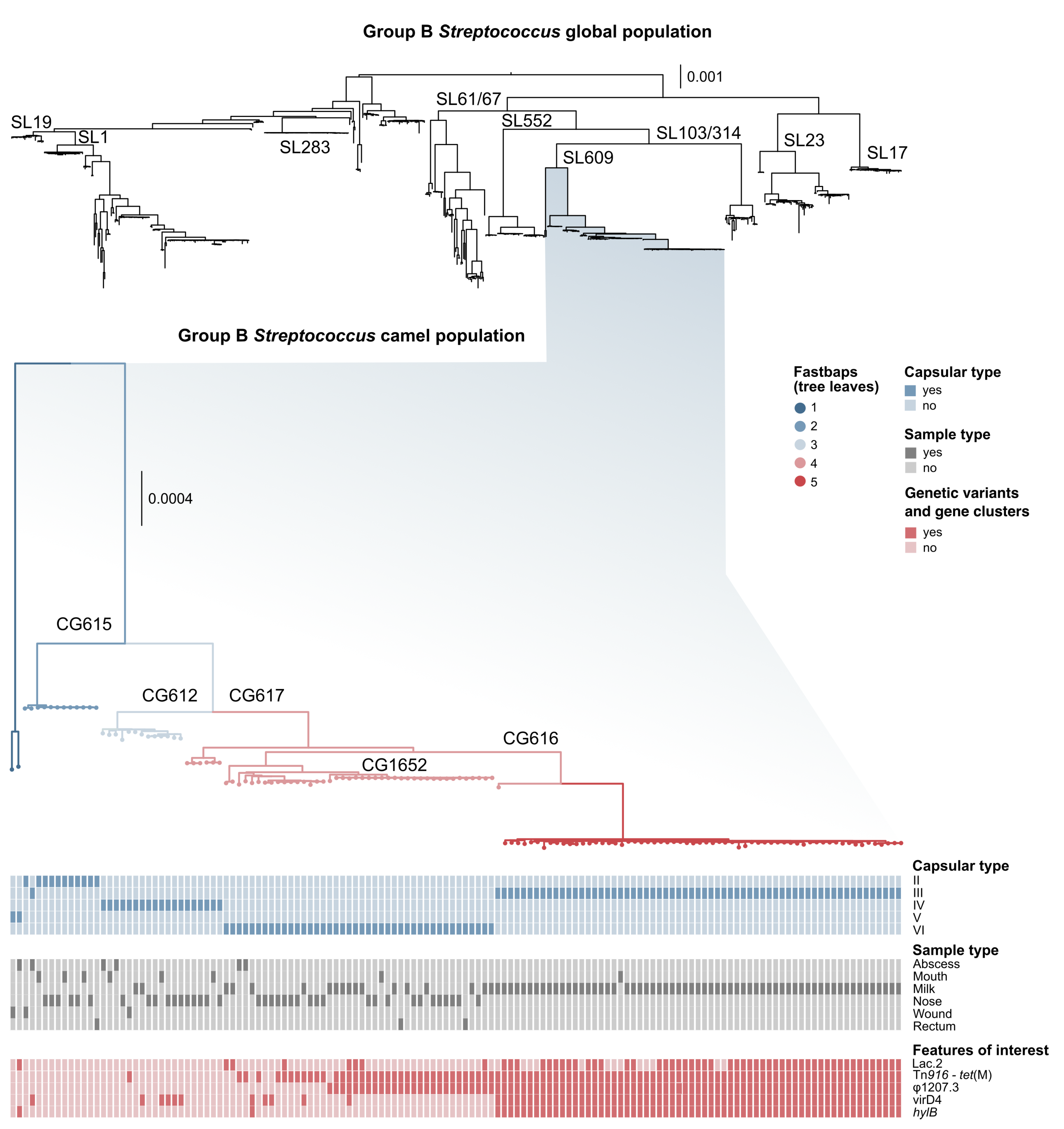
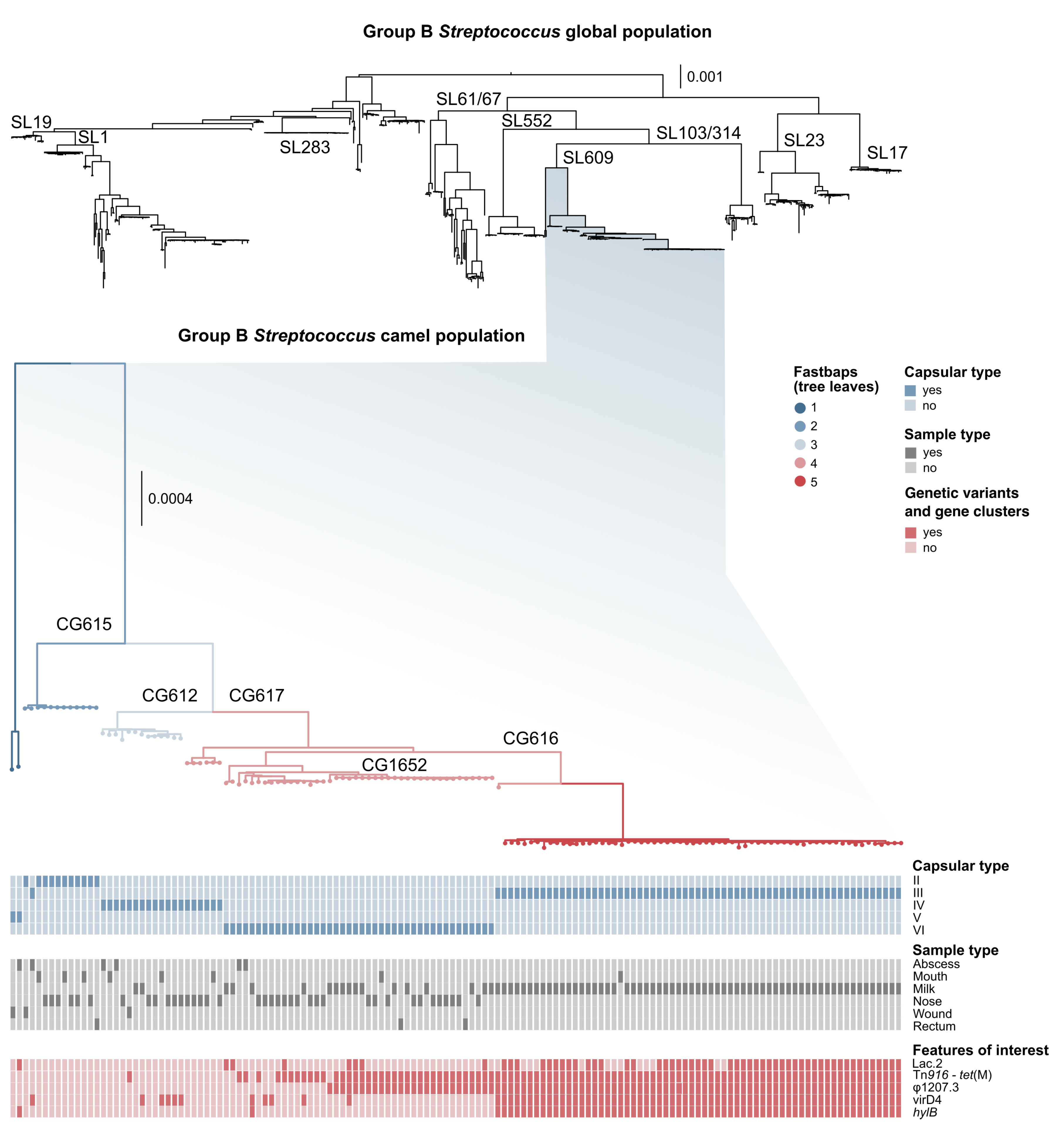
3.1. Genotyping and Serotyping
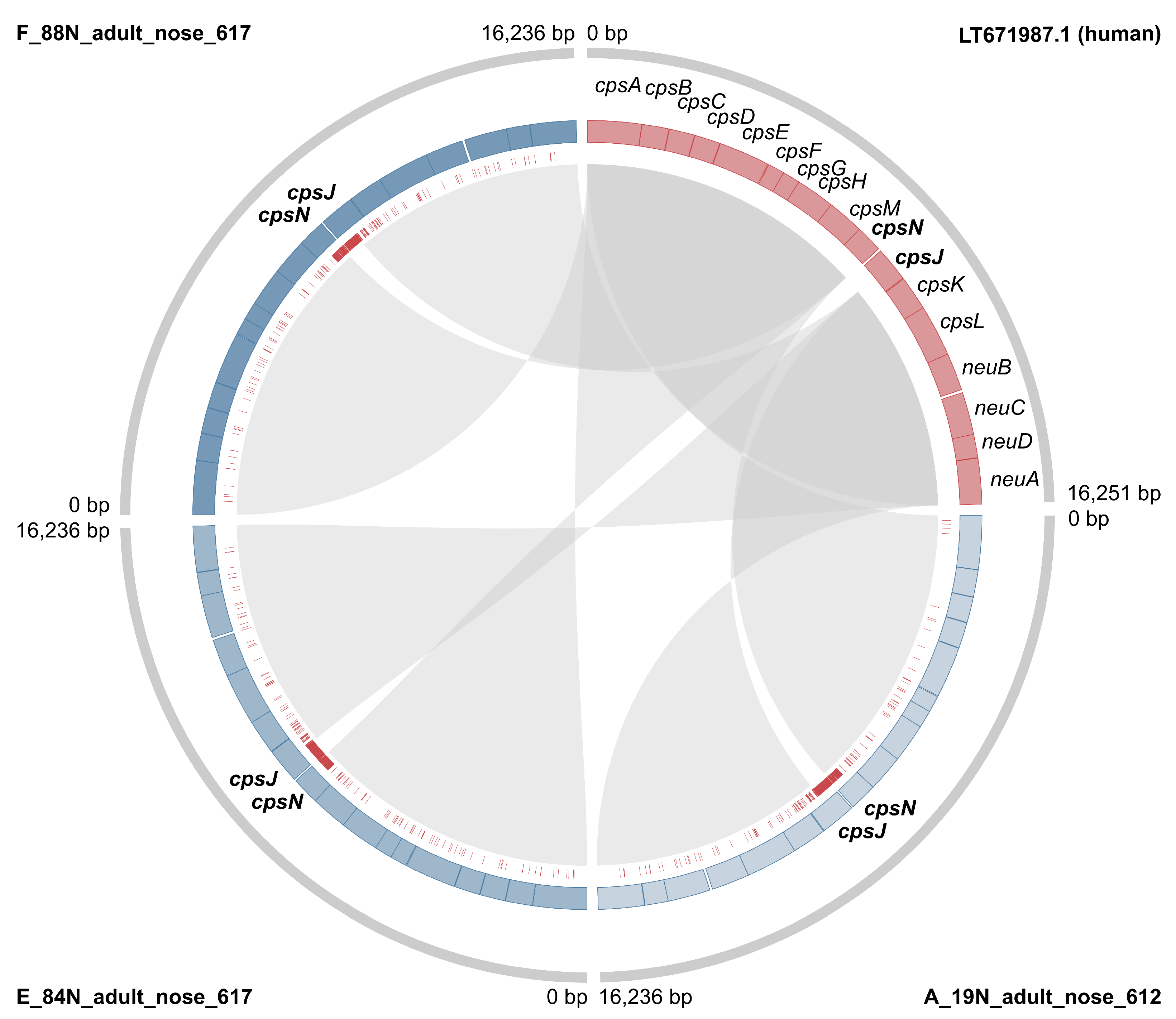
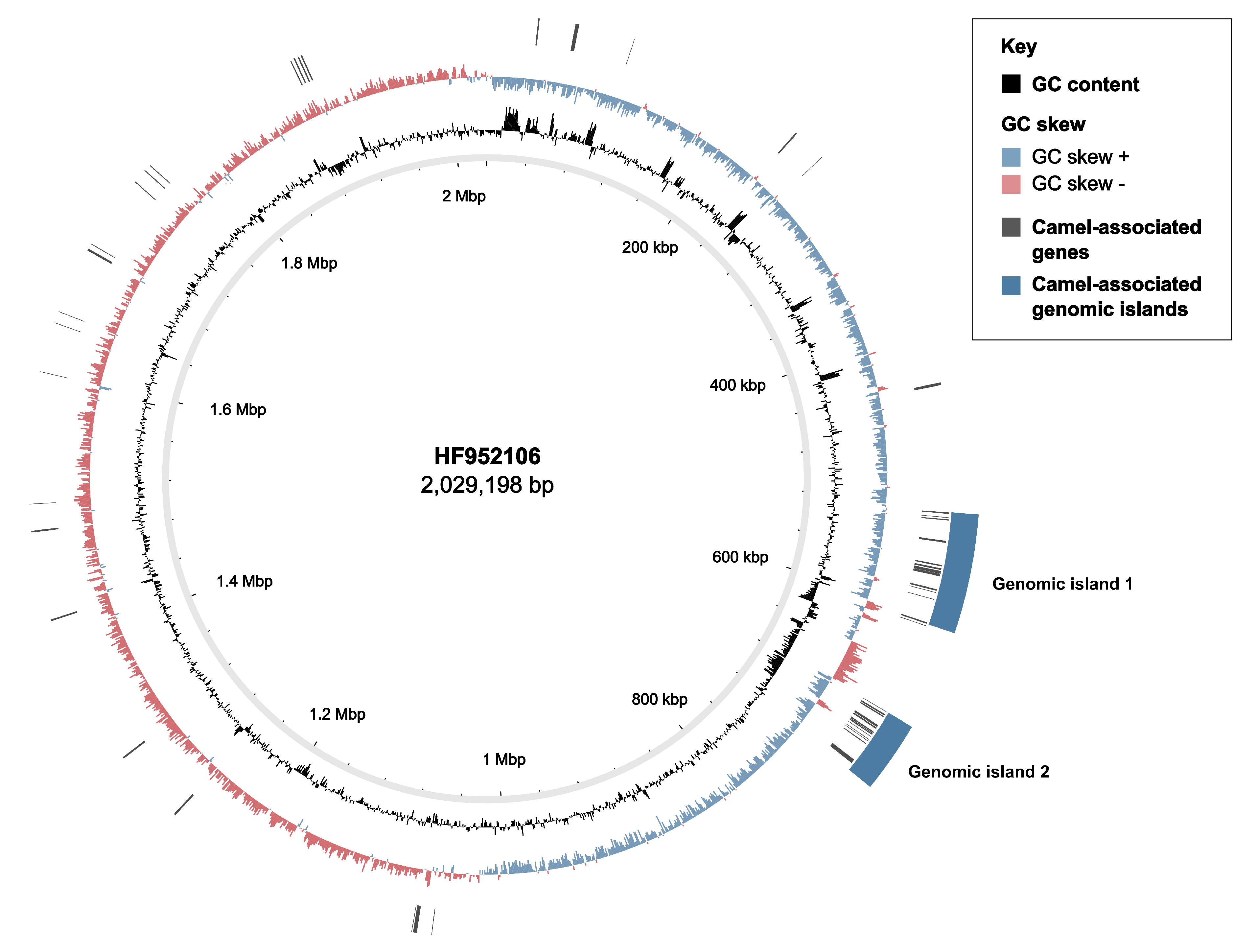
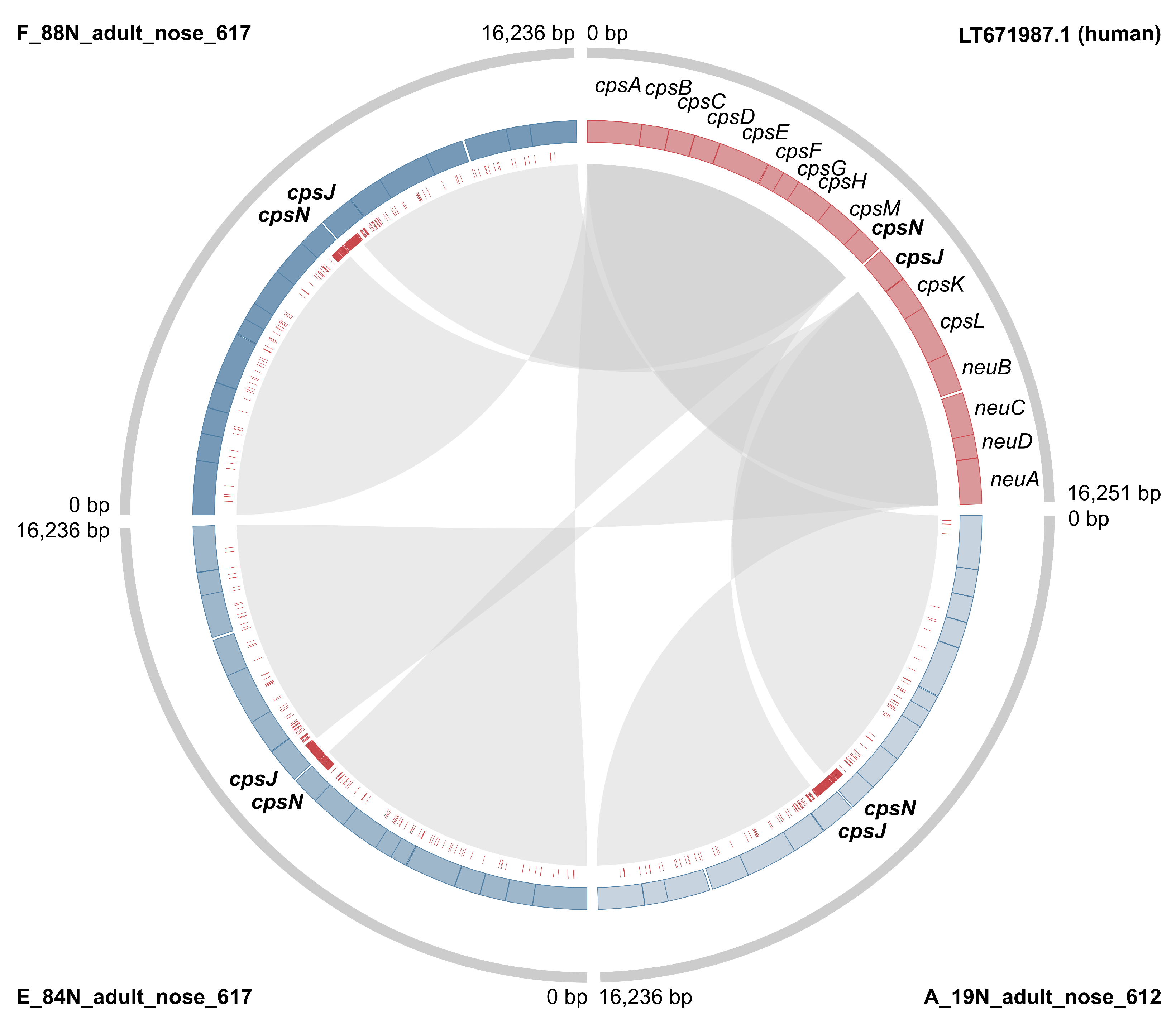
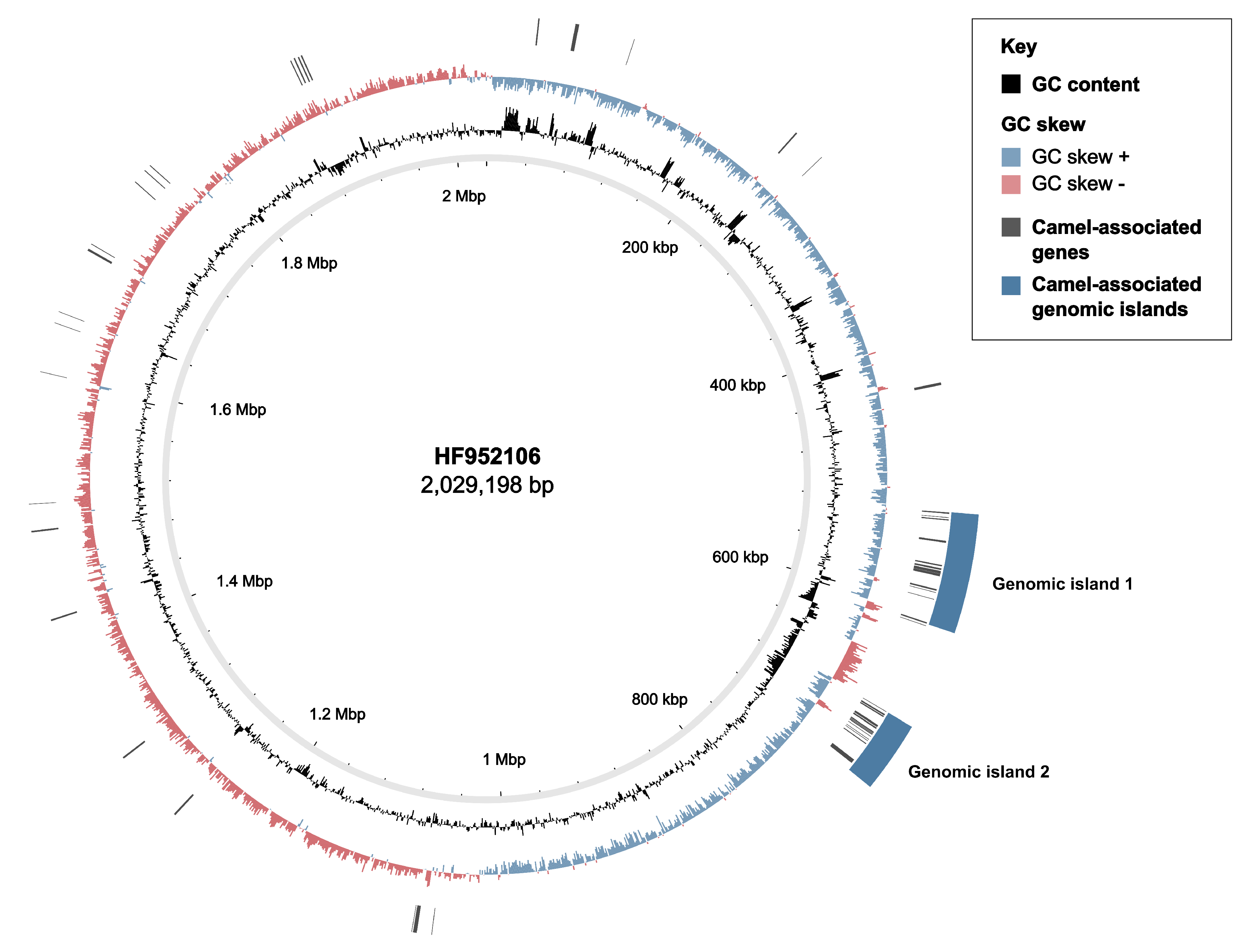
3.2. Markers of Camel-Association Within GBS
3.3. Markers of Mastitis-Association within Camel GBS
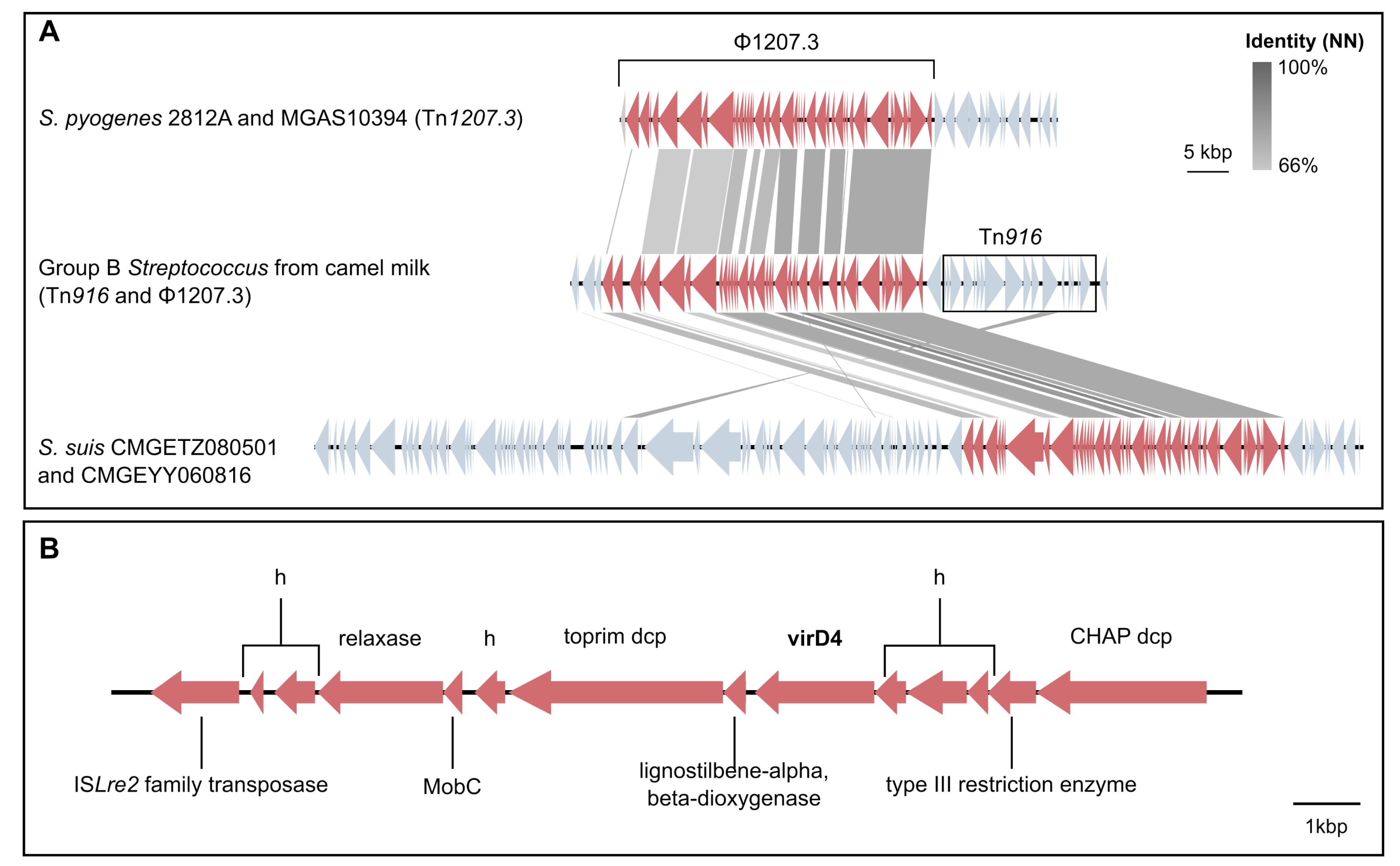
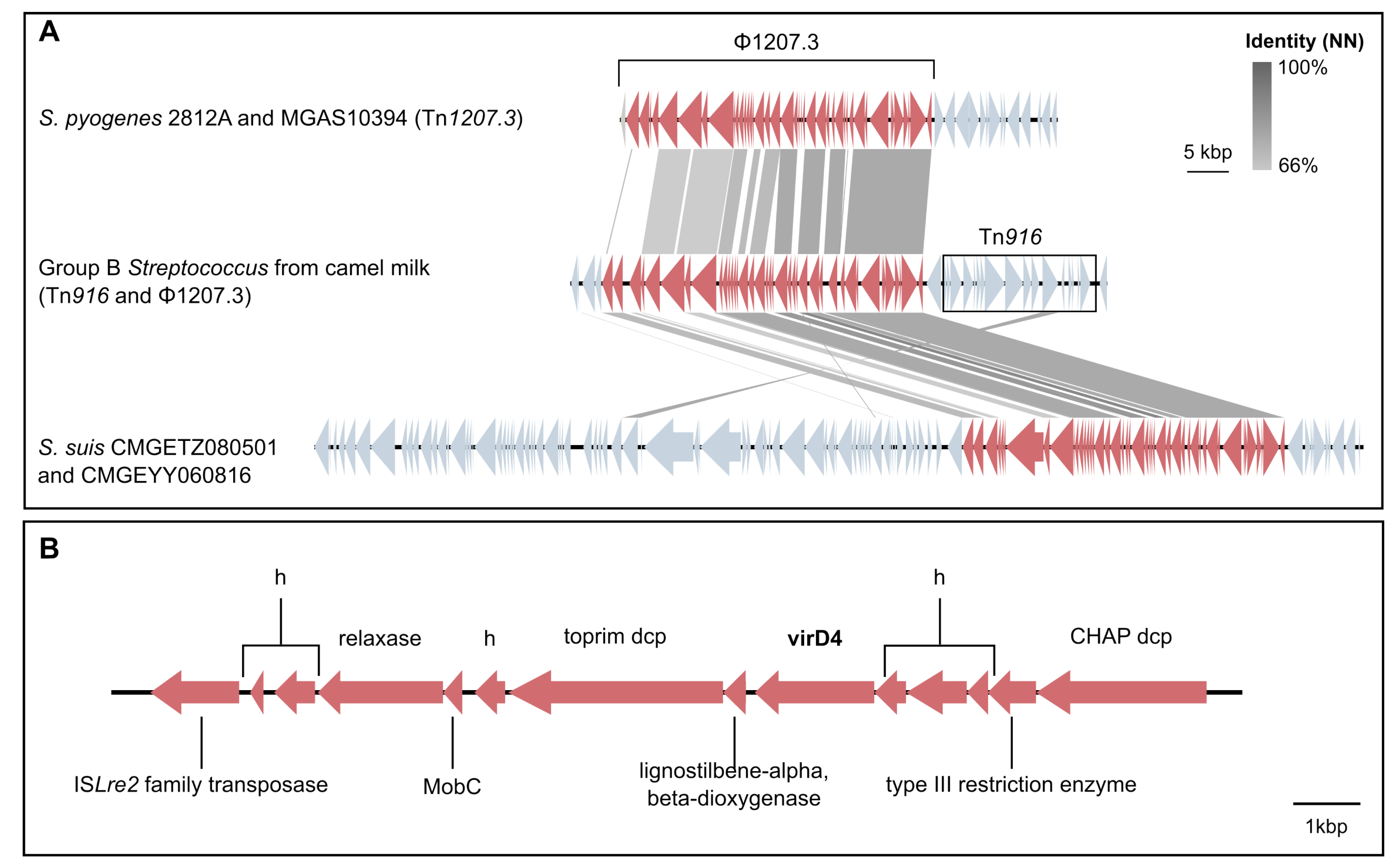
3.4. Mobile Genetic Elements in Camel GBS
4. Discussion
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| CC | clonal complex |
| CG | clonal group |
| GBS | Group B Streptococcus |
| GEI | genomic islands |
| GWAS | genome-wide association study |
| MGE | mobile genetic elements |
| ML | maximum likelihood |
| MLST | multilocus sequence-typing |
| NT | non-typeable |
| PICI | phage-inducible chromosomal islands |
| SL | sublineage |
| ST | sequence type |
| TcR | tetracycline resistance |
References
- Seale, A.C.; Bianchi-Jassir, F.; Russell, N.J.; Kohli-Lynch, M.; Tann, C.J.; Hall, J.; Madrid, L.; Blencowe, H.; Cousens, S.; Baker, C.J.; et al. Estimates of the burden of group B streptococcal disease worldwide for pregnant women, stillbirths, and children. Clin. Infect. Dis. 2017, 65, S200–S219. [Google Scholar] [CrossRef] [PubMed]
- Lyhs, U.; Kulkas, L.; Katholm, J.; Waller, K.P.; Saha, K.; Tomusk, R.J.; Zadoks, R.N. Streptococcus agalactiae Serotype IV Humans Cattle, Northern Europe. Emerg. Infect. Dis. 2016, 22, 2097. [Google Scholar] [CrossRef] [PubMed]
- Barkham, T.; Zadoks, R.N.; Azmai, M.N.A.; Baker, S.; Bich, V.T.N.; Chalker, V.; Chau, M.L.; Dance, D.; Deepak, R.N.; van Doorn, H.R.; et al. One hypervirulent clone, sequence type 283, accounts for a large proportion of invasive Streptococcus agalactiae isolated from humans and diseased tilapia in Southeast Asia. PLoS Negl. Trop. Dis. 2019, 13, e0007421. [Google Scholar] [CrossRef]
- Paveenkittiporn, W.; Ungcharoen, R.; Kerdsin, A. Streptococcus agalactiae infections and clinical relevance in adults, Thailand. Diagn. Microbiol. Infect. Dis. 2020, 97, 115005. [Google Scholar] [CrossRef]
- Brochet, M.; Couvé, E.; Zouine, M.; Vallaeys, T.; Rusniok, C.; Lamy, M.C.; Buchrieser, C.; Trieu-Cuot, P.; Kunst, F.; Poyart, C.; et al. Genomic diversity and evolution within the species Streptococcus agalactiae. Microbes Infect. 2006, 8, 1227–1243. [Google Scholar] [CrossRef] [PubMed]
- Delannoy, C.M.; Zadoks, R.N.; Crumlish, M.; Rodgers, D.; Lainson, F.A.; Ferguson, H.; Turnbull, J.; Fontaine, M.C. Genomic comparison of virulent and non-virulent Streptococcus agalactiae Fish. J. Fish Dis. 2016, 39, 13–29. [Google Scholar] [CrossRef] [PubMed]
- Richards, V.P.; Velsko, I.M.; Alam, T.; Zadoks, R.N.; Manning, S.D.; Pavinski Bitar, P.D.; Hasler, H.B.; Crestani, C.; Springer, G.H.; Probert, B.M.; et al. Population gene introgression and high genome plasticity for the zoonotic pathogen Streptococcus agalactiae. Mol. Biol. Evol. 2019, 36, 2572–2590. [Google Scholar] [CrossRef] [PubMed]
- Viana, D.; Comos, M.; McAdam, P.R.; Ward, M.J.; Selva, L.; Guinane, C.M.; González-Muñoz, B.M.; Tristan, A.; Foster, S.J.; Fitzgerald, J.R.; et al. A single natural nucleotide mutation alters bacterial pathogen host tropism. Nat. Genet. 2015, 47, 361–366. [Google Scholar] [CrossRef] [PubMed]
- Richardson, E.J.; Bacigalupe, R.; Harrison, E.M.; Weinert, L.A.; Lycett, S.; Vrieling, M.; Robb, K.; Hoskisson, P.A.; Holden, M.T.; Feil, E.J.; et al. Gene exchange drives the ecological success of a multi-host bacterial pathogen. Nat. Ecol. Evol. 2018, 2, 1468. [Google Scholar] [CrossRef] [PubMed]
- Rosinski-Chupin, I.; Sauvage, E.; Mairey, B.; Mangenot, S.; Ma, L.; Da Cunha, V.; Rusniok, C.; Bouchier, C.; Barbe, V.; Glaser, P. Reductive evolution in Streptococcus agalactiae and the emergence of a host adapted lineage. BMC Genom. 2013, 14, 252. [Google Scholar] [CrossRef] [Green Version]
- Penadés, J.R.; Christie, G.E. The phage-inducible chromosomal islands: A family of highly evolved molecular parasites. Annu. Rev. Virol. 2015, 2, 181–201. [Google Scholar] [CrossRef] [PubMed]
- Crestani, C.; Forde, T.L.; Zadoks, R.N. Development and application of a prophage integrase typing scheme for Group B Streptococcus. Front. Microbiol. 2020, 11, 1993. [Google Scholar] [CrossRef]
- Rouli, L.; Merhej, V.; Fournier, P.E.; Raoult, D. The bacterial pangenome as a new tool for analysing pathogenic bacteria. New Microbes New Infect. 2015, 7, 72–85. [Google Scholar] [CrossRef]
- Sheppard, S.K.; Guttman, D.S.; Fitzgerald, J.R. Population genomics of bacterial host adaptation. Nat. Rev. Genet. 2018, 19, 549–565. [Google Scholar] [CrossRef] [PubMed]
- Crestani, C.; Forde, T.L.; Lycett, S.J.; Holmes, M.A.; Fasth, C.; Persson-Waller, K.; Zadoks, R.N. The fall and rise of group B Streptococcus Dairy Cattle: Reintroduction due to human-to-cattle host jumps? Microb. Genom. 2021, 7, 000648. [Google Scholar] [CrossRef] [PubMed]
- Botelho, A.C.N.; Oliveira, J.G.; Damasco, A.P.; Santos, K.T.; Ferreira, A.F.M.; Rocha, G.T.; Marinho, P.S.; Bornia, R.B.; Pinto, T.C.; Américo, M.A.; et al. Streptococcus agalactiae Carriage Pregnant Women Living Rio De Janeiro, Brazil, Over a Period of Eight Years. PLoS ONE 2018, 13, e0196925. [Google Scholar] [CrossRef]
- Gori, A.; Harrison, O.B.; Mlia, E.; Nishihara, Y.; Chan, J.M.; Msefula, J.; Mallewa, M.; Dube, Q.; Swarthout, T.D.; Nobbs, A.H.; et al. Pan-GWAS of Streptococcus agalactiae Highlights Lineage-Specific Genes Associated with Virulence and Niche Adaptation. mBio 2020, 11, e00728-20. [Google Scholar] [CrossRef] [PubMed]
- Fischer, A.; Liljander, A.; Kaspar, H.; Muriuki, C.; Fuxelius, H.H.; Bongcam-Rudloff, E.; de Villiers, E.P.; Huber, C.A.; Frey, J.; Daubenberger, C.; et al. Camel Streptococcus agalactiae Populations are Associated with Specific Disease Complexes and Acquired the Tetracycline Resistance Gene tetM via a Tn916-Like Element. Vet. Res. 2013, 44, 86. [Google Scholar] [CrossRef] [PubMed]
- Zubair, S.; de Villiers, E.; Younan, M.; Andersson, G.; Tettelin, H.; Riley, D.; Jores, J.; Bongcam-Rudloff, E.; Bishop, R.P. Genome sequences of two pathogenic Streptococcus agalactiae isolates from the One-humped camel Camelus dromedarius. Genome Announc. 2013, 1, e00515-13. [Google Scholar] [CrossRef] [PubMed]
- Seligsohn, D.; Nyman, A.; Younan, M.; Sake, W.; Persson, Y.; Bornstein, S.; Maichomo, M.; de Verdier, K.; Morrell, J.; Chenais, E. Subclinical mastitis in pastoralist dairy camel herds in Isiolo, Kenya: Prevalence, risk factors, and antimicrobial susceptibility. J. Dairy Sci. 2020, 103, 4717–4731. [Google Scholar] [CrossRef] [PubMed]
- Iyer, A.P.; Albaik, M.; Baghallab, I. Mastitis in camels in African and Middle East countries. J. Bacteriol. Parasitol. 2014, 5, 1. [Google Scholar] [CrossRef]
- Obied, A.; Bagadi, H.; Mukhtar, M. Mastitis in Camelus dromedarius Somat. Cell Content Camels’ Milk. Res. Vet. Sci. 1996, 61, 55–58. [Google Scholar] [CrossRef]
- Bekele, T.; Molla, B. Mastitis in lactating camels (Camelus dromedarius) in Afar region, north-eastern Ethiopia. Berl. Und Münchener Tierärztliche Wochenschr. 2001, 114, 169–172. [Google Scholar]
- Younan, M.; Bornstein, S. Lancefield group B and C streptococci in East African camels (Camelus dromedarius). Vet. Rec. 2007, 160, 330–335. [Google Scholar] [CrossRef] [PubMed]
- Seligsohn, D.; Crestani, C.; Gitahi, N.; Flodin, E.L.; Chenais, E.; Zadoks, R.N. Investigation of extramammary sources of Group B Streptococcus reveals its unusual ecology and epidemiology in camels. PLoS ONE 2021, 16, e0252973. [Google Scholar] [CrossRef]
- Rothen, J.; Schindler, T.; Pothier, J.F.; Younan, M.; Certa, U.; Daubenberger, C.; Pflüger, V.; Jores, J. Draft genome sequences of seven Streptococcus agalactiae strains isolated from Camelus dromedarius at the horn of Africa. Genome Announc. 2017, 5, e00525-17. [Google Scholar] [CrossRef]
- Seligsohn, D.; Crestani, C.; Gitahi, N.; Flodin, E.L.; Chenais, E.; Zadoks, R.N. Genomic analysis of Group B Streptococcus from milk demonstrates the need for improved biosecurity: A cross-sectional study of pastoralist camels in Kenya. BMC Microbiol. 2021, 21, 217. [Google Scholar] [CrossRef]
- Brynildsrud, O.; Bohlin, J.; Scheffer, L.; Eldholm, V. Rapid scoring of genes in microbial pan-genome-wide association studies with Scoary. Genome Biol. 2016, 17, 238. [Google Scholar] [CrossRef]
- Gurevich, A.; Saveliev, V.; Vyahhi, N.; Tesler, G. QUAST: Quality assessment tool for genome assemblies. Bioinformatics 2013, 29, 1072–1075. [Google Scholar] [CrossRef] [PubMed]
- Hasman, H.; Saputra, D.; Sicheritz-Ponten, T.; Lund, O.; Svendsen, C.A.; Frimodt-Møller, N.; Aarestrup, F.M. Rapid whole-genome sequencing for detection and characterization of microorganisms directly from clinical samples. J. Clin. Microbiol. 2014, 52, 139–146. [Google Scholar] [CrossRef]
- Inouye, M.; Dashnow, H.; Raven, L.A.; Schultz, M.B.; Pope, B.J.; Tomita, T.; Zobel, J.; Holt, K.E. SRST2: Rapid genomic surveillance for public health and hospital microbiology labs. Genome Med. 2014, 6, 90. [Google Scholar] [CrossRef] [Green Version]
- Larsen, M.V.; Cosentino, S.; Rasmussen, S.; Friis, C.; Hasman, H.; Marvig, R.L.; Jelsbak, L.; Sicheritz-Pontén, T.; Ussery, D.W.; Aarestrup, F.M.; et al. Multilocus sequence typing of total-genome-sequenced bacteria. J. Clin. Microbiol. 2012, 50, 1355–1361. [Google Scholar] [CrossRef]
- Metcalf, B.; Chochua, S.; Gertz, R., Jr.; Hawkins, P.; Ricaldi, J.; Li, Z.; Walker, H.; Tran, T.; Rivers, J.; Mathis, S.; et al. Short-read whole genome sequencing for determination of antimicrobial resistance mechanisms and capsular serotypes of current invasive Streptococcus agalactiae recovered in the USA. Clin. Microbiol. Infect. 2017, 23, 574.e7–574.e14. [Google Scholar] [CrossRef]
- Imperi, M.; Pataracchia, M.; Alfarone, G.; Baldassarri, L.; Orefici, G.; Creti, R. A multiplex PCR assay for the direct identification of the capsular type (Ia to IX) of Streptococcus agalactiae. J. Microbiol. Methods 2010, 80, 212–214. [Google Scholar] [CrossRef] [PubMed]
- Tiruvayipati, S.; Ying, T.W.; Barkham, T.; Chen, S.L. GBS-SBG—GBS serotyping by genome sequencing. Microb. Genom. 2021, 7, 000688. [Google Scholar] [CrossRef] [PubMed]
- Seemann, T. Prokka: Rapid prokaryotic genome annotation. Bioinformatics 2014, 30, 2068–2069. [Google Scholar] [CrossRef]
- Kozlov, A.M.; Darriba, D.; Flouri, T.; Morel, B.; Stamatakis, A. RAxML-NG: A fast, scalable and user-friendly tool for maximum likelihood phylogenetic inference. Bioinformatics 2019, 35, 4453–4455. [Google Scholar] [CrossRef] [PubMed]
- Tonkin-Hill, G.; Lees, J.A.; Bentley, S.D.; Frost, S.D.; Corander, J. Fast hierarchical Bayesian analysis of population structure. Nucleic Acids Res. 2019, 47, 5539–5549. [Google Scholar] [CrossRef] [PubMed]
- RStudio Team. RStudio: Integrated Development Environment for R; RStudio, PBC: Boston, MA, USA, 2020; Available online: http://www.rstudio.com/ (accessed on 1 September 2022).
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2018; Available online: https://www.R-project.org/ (accessed on 1 September 2022).
- Page, A.J.; Cummins, C.A.; Hunt, M.; Wong, V.K.; Reuter, S.; Holden, M.T.; Fookes, M.; Falush, D.; Keane, J.A.; Parkhill, J. Roary: Rapid large-scale prokaryote pan genome analysis. Bioinformatics 2015, 31, 3691–3693. [Google Scholar] [CrossRef] [PubMed]
- Arndt, D.; Grant, J.R.; Marcu, A.; Sajed, T.; Pon, A.; Liang, Y.; Wishart, D.S. PHASTER: A better, faster version of the PHAST phage search tool. Nucleic Acids Res. 2016, 44, W16–W21. [Google Scholar] [CrossRef] [PubMed]
- Hadfield, J.; Croucher, N.J.; Goater, R.J.; Abudahab, K.; Aanensen, D.M.; Harris, S.R. Phandango: An interactive viewer for bacterial population genomics. Bioinformatics 2018, 34, 292–293. [Google Scholar] [CrossRef] [PubMed]
- Krzywinski, M.; Schein, J.; Birol, I.; Connors, J.; Gascoyne, R.; Horsman, D.; Jones, S.J.; Marra, M.A. Circos: An information aesthetic for comparative genomics. Genome Res. 2009, 19, 1639–1645. [Google Scholar] [CrossRef] [PubMed]
- Alikhan, N.F.; Petty, N.K.; Zakour, N.L.B.; Beatson, S.A. BLAST Ring Image Generator (BRIG): Simple prokaryote genome comparisons. BMC Genom. 2011, 12, 402. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, M.J.; Petty, N.K.; Beatson, S.A. Easyfig: A genome comparison visualizer. Bioinformatics 2011, 27, 1009–1010. [Google Scholar] [CrossRef] [PubMed]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef] [PubMed]
- Brenciani, A.; Tiberi, E.; Bacciaglia, A.; Petrelli, D.; Varaldo, P.E.; Giovanetti, E. Two distinct genetic elements are responsible for Erm(TR)-Mediat. Erythromycin Resist. Tetracycline-Susceptible Tetracycline-Resist. Strains Streptococcus pyogenes. Antimicrob. Agents Chemother. 2011, 55, 2106–2112. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Li, X.; Xie, Y.; Bi, D.; Sun, J.; Li, J.; Tai, C.; Deng, Z.; Ou, H.Y. ICEberg 2.0: An updated database of bacterial integrative and conjugative elements. Nucleic Acids Res. 2018, 47, D660–D665. [Google Scholar] [CrossRef] [PubMed]
- Santagati, M.; Iannelli, F.; Cascone, C.; Campanile, F.; Oggioni, M.R.; Stefani, S.; Pozzi, G. The novel conjugative transposon Tn1207.3 Carries Macrolide Efflux Gene mef(A) Streptococcus pyogenes. Microb. Drug Resist. 2003, 9, 243–247. [Google Scholar] [CrossRef] [PubMed]
- Iannelli, F.; Santagati, M.; Santoro, F.; Oggioni, M.R.; Stefani, S.; Pozzi, G. Nucleotide sequence of conjugative prophage Φ1207.3 (formerly Tn1207.3) carrying the mef(A)/msr(D) genes for efflux resistance to macrolides in Streptococcus pyogenes. Front. Microbiol. 2014, 5, 687. [Google Scholar] [CrossRef] [PubMed]
- Kawasaki, M.; Delamare-Deboutteville, J.; Bowater, R.O.; Walker, M.J.; Beatson, S.; Zakour, N.L.B.; Barnes, A.C. Microevolution of Streptococcus agalactiae ST-261 from Australia indicates dissemination via imported tilapia and ongoing adaptation to marine hosts or environment. Appl. Environ. Microbiol. 2018, 84, e00859-18. [Google Scholar] [CrossRef]
- Da Cunha, V.; Davies, M.R.; Douarre, P.E.; Rosinski-Chupin, I.; Margarit, I.; Spinali, S.; Perkins, T.; Lechat, P.; Dmytruk, N.; Sauvage, E.; et al. Streptococcus agalactiae clones infecting humans were selected and fixed through the extensive use of tetracycline. Nat. Commun. 2014, 5, 5544. [Google Scholar] [CrossRef]
- Richards, V.P.; Lang, P.; Bitar, P.D.P.; Lefébure, T.; Schukken, Y.H.; Zadoks, R.N.; Stanhope, M.J. Comparative genomics and the role of lateral gene transfer in the evolution of bovine adapted Streptococcus agalactiae. Infect. Genet. Evol. 2011, 11, 1263–1275. [Google Scholar] [CrossRef] [PubMed]
- Almeida, A.; Alves-Barroco, C.; Sauvage, E.; Bexiga, R.; Albuquerque, P.; Tavares, F.; Santos-Sanches, I.; Glaser, P. Persistence of a dominant bovine lineage of group B Streptococcus reveals genomic signatures of host adaptation. Environ. Microbiol. 2016, 18, 4216–4229. [Google Scholar] [CrossRef] [PubMed]
- Brueggemann, A.B.; Pai, R.; Crook, D.W.; Beall, B. Vaccine escape recombinants emerge after pneumococcal vaccination in the United States. PLoS Pathog. 2007, 3, e168. [Google Scholar] [CrossRef]
- Neemuchwala, A.; Teatero, S.; Athey, T.B.; McGeer, A.; Fittipaldi, N. Capsular switching and other large-scale recombination events in invasive sequence type 1 group B Streptococcus. Emerg. Infect. Dis. 2016, 22, 1941. [Google Scholar] [CrossRef] [PubMed]
- Bellais, S.; Six, A.; Fouet, A.; Longo, M.; Dmytruk, N.; Glaser, P.; Trieu-Cuot, P.; Poyart, C. Capsular switching in group B Streptococcus CC17 Hypervirulent Clone: A future challenge for polysaccharide vaccine development. J. Infect. Dis. 2012, 206, 1745–1752. [Google Scholar] [CrossRef] [PubMed]
- Martins, E.R.; Melo-Cristino, J.; Ramirez, M. Evidence for rare capsular switching in Streptococcus agalactiae. J. Bacteriol. 2010, 192, 1361–1369. [Google Scholar] [CrossRef]
- Maisey, H.C.; Doran, K.S.; Nizet, V. Recent advances in understanding the molecular basis of group B Streptococcus Virulence. Expert Rev. Mol. Med. 2008, 10, e27. [Google Scholar] [CrossRef] [PubMed]
- Herbert, M.A.; Beveridge, C.J.; Saunders, N.J. Bacterial virulence factors in neonatal sepsis: Group B Streptococcus. Curr. Opin. Infect. Dis. 2004, 17, 225–229. [Google Scholar] [CrossRef]
- Li, L.; Wang, R.; Huang, Y.; Huang, T.; Luo, F.; Huang, W.; Yang, X.; Lei, A.; Chen, M.; Gan, X. High incidence of pathogenic Streptococcus agalactiae ST485 Strain Pregnant/puerperal women and isolation of hyper-virulent human CC67 strain. Front. Microbiol. 2018, 9, 50. [Google Scholar] [CrossRef]
- Frost, L.S.; Leplae, R.; Summers, A.O.; Toussaint, A. Mobile genetic elements: The agents of open source evolution. Nat. Rev. Microbiol. 2005, 3, 722. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Quiles-Puchalt, N.; Chiang, Y.N.; Bacigalupe, R.; Fillol-Salom, A.; Chee, M.S.J.; Fitzgerald, J.R.; Penadés, J.R. Genome hypermobility by lateral transduction. Science 2018, 362, 207–212. [Google Scholar] [CrossRef]
- Humphrey, S.; Fillol-Salom, A.; Quiles-Puchalt, N.; Ibarra-Chávez, R.; Haag, A.F.; Chen, J.; Penadés, J.R. Bacterial chromosomal mobility via lateral transduction exceeds that of classical mobile genetic elements. Nat. Commun. 2021, 12, 6509. [Google Scholar] [CrossRef] [PubMed]
- Takai, S.; Hines, S.A.; Sekizaki, T.; Nicholson, V.M.; Alperin, D.A.; Osaki, M.; Takamatsu, D.; Nakamura, M.; Suzuki, K.; Ogino, N.; et al. DNA sequence and comparison of virulence plasmids from Rhodococcus equi ATCC 33701 and 103. Infect. Immun. 2000, 68, 6840–6847. [Google Scholar] [CrossRef] [PubMed]
- Ji, X.; Sun, Y.; Liu, J.; Zhu, L.; Guo, X.; Lang, X.; Feng, S. A novel virulence-associated protein, VapE, Streptococcus suis Serotype 2. Mol. Med. Rep. 2016, 13, 2871–2877. [Google Scholar] [CrossRef] [PubMed]
- Shaw, D. The association between satellite prophages and pneumococcal carriage and disease. In Proceedings of the Abstract for the 31st European Congress of Clinical Microbiology and Infectious Diseases, Online Event, 9–12 July 2021. [Google Scholar]
- Holt, K.E.; Wertheim, H.; Zadoks, R.N.; Baker, S.; Whitehouse, C.A.; Dance, D.; Jenney, A.; Connor, T.R.; Hsu, L.Y.; Severin, J.; et al. Genomic analysis of diversity, population structure, virulence, and antimicrobial resistance in Klebsiella pneumoniae, an urgent threat to public health. Proc. Natl. Acad. Sci. USA 2015, 112, E3574–E3581. [Google Scholar] [CrossRef]
- Blum, S.; Heller, E.; Krifucks, O.; Sela, S.; Hammer-Muntz, O.; Leitner, G. Identification of a bovine mastitis Escherichia coli subset. Vet. Microbiol. 2008, 132, 135–148. [Google Scholar] [CrossRef]
- Mortensen, B.L.; Skaar, E.P. The contribution of nutrient metal acquisition and metabolism to Acinetobacter baumannii survival within the host. Front. Cell. Infect. Microbiol. 2013, 3, 95. [Google Scholar] [CrossRef]
- Affandi, T.; McEvoy, M.M. Mechanism of metal ion-induced activation of a two-component sensor kinase. Biochem. J. 2019, 476, 115–135. [Google Scholar] [CrossRef]
- Hanczvikkel, A.; Füzi, M.; Ungvári, E.; Tóth, Á. Transmissible silver resistance readily evolves in high-risk clone isolates of Klebsiella pneumoniae. Acta Microbiol. Immunol. Hung. 2018, 65, 387–403. [Google Scholar] [CrossRef] [PubMed]
- Faye, B.; Bengoumi, M. Trace-element metabolism in camel. In Proceedings of the Third Annual Meeting for Animal Production Under Arid Conditions, Al-Ain, United Arab Emirates, 2–3 May 1998; Volume 1, pp. 9–35. [Google Scholar]
- Rolland, K.; Marois, C.; Siquier, V.; Cattier, B.; Quentin, R. Genetic features of Streptococcus agalactiae strains causing severe neonatal infections, as revealed by pulsed-field gel electrophoresis and hylB gene analysis. J. Clin. Microbiol. 1999, 37, 1892–1898. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sukhnanand, S.; Dogan, B.; Ayodele, M.O.; Zadoks, R.N.; Craver, M.P.J.; Dumas, N.B.; Schukken, Y.H.; Boor, K.J.; Wiedmann, M. Molecular subtyping and characterization of bovine and human Streptococcus agalactiae isolates. J. Clin. Microbiol. 2005, 43, 1177–1186. [Google Scholar] [CrossRef] [PubMed]
- Domelier, A.S.; van der Mee-Marquet, N.; Grandet, A.; Mereghetti, L.; Rosenau, A.; Quentin, R. Loss of catabolic function in Streptococcus agalactiae strains and its association with neonatal meningitis. J. Clin. Microbiol. 2006, 44, 3245–3250. [Google Scholar] [CrossRef] [PubMed]
- Behrooz, S.K.; Lida, L.; Ali, S.; Mehdi, M.; Rasoul, M.; Elnaz, O.; Farid, B.T.; Gholamreza, I. Study of MazEF, sam, and phd-doc putative toxin–antitoxin systems in Staphylococcus epidermidis. Acta Microbiol. Immunol. Hung. 2018, 65, 81–91. [Google Scholar] [CrossRef]
- Chan, W.T.; Yeo, C.C.; Sadowy, E.; Espinosa, M. Functional validation of putative toxin-antitoxin genes from the Gram-positive pathogen Streptococcus pneumoniae: Phd-Doc Is Fourth Bona-Fide Operon. Front. Microbiol. 2014, 5, 677. [Google Scholar] [CrossRef] [PubMed]
- Guérout, A.M.; Iqbal, N.; Mine, N.; Ducos-Galand, M.; Van Melderen, L.; Mazel, D. Characterization of the phd-doc and ccd toxin-antitoxin cassettes from Vibrio Superintegrons. J. Bacteriol. 2013, 195, 2270–2283. [Google Scholar] [CrossRef]
- Lehnherr, H.; Maguin, E.; Jafri, S.; Yarmolinsky, M.B. Plasmid addiction genes of bacteriophage P1: Doc, which causes cell death on curing of prophage, and phd, which prevents host death when prophage is retained. J. Mol. Biol. 1993, 233, 414–428. [Google Scholar] [CrossRef]
- Yang, Q.E.; Walsh, T.R. Toxin–antitoxin systems and their role in disseminating and maintaining antimicrobial resistance. FEMS Microbiol. Rev. 2017, 41, 343–353. [Google Scholar] [CrossRef]
- Schulein, R.; Guye, P.; Rhomberg, T.A.; Schmid, M.C.; Schröder, G.; Vergunst, A.C.; Carena, I.; Dehio, C. A bipartite signal mediates the transfer of type IV secretion substrates of Bartonella henselae human cells. Proc. Natl. Acad. Sci. USA 2005, 102, 856–861. [Google Scholar] [CrossRef]
- Jiang, X.; Yang, Y.; Zhou, J.; Zhu, L.; Gu, Y.; Zhang, X.; Li, X.; Fang, W. Roles of the putative type IV-like secretion system key component VirD4 and PrsA in pathogenesis of Streptococcus suis Type 2. Front. Cell. Infect. Microbiol. 2016, 6, 172. [Google Scholar] [CrossRef]
- Wallden, K.; Rivera-Calzada, A.; Waksman, G. Microreview: Type IV secretion systems: Versatility and diversity in function. Cell. Microbiol. 2010, 12, 1203–1212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alvarez-Martinez, C.E.; Christie, P.J. Biological diversity of prokaryotic type IV secretion systems. Microbiol. Mol. Biol. Rev. 2009, 73, 775–808. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Crestani, C.; Seligsohn, D.; Forde, T.L.; Zadoks, R.N. How GBS Got Its Hump: Genomic Analysis of Group B Streptococcus from Camels Identifies Host Restriction as well as Mobile Genetic Elements Shared across Hosts and Pathogens. Pathogens 2022, 11, 1025. https://doi.org/10.3390/pathogens11091025
Crestani C, Seligsohn D, Forde TL, Zadoks RN. How GBS Got Its Hump: Genomic Analysis of Group B Streptococcus from Camels Identifies Host Restriction as well as Mobile Genetic Elements Shared across Hosts and Pathogens. Pathogens. 2022; 11(9):1025. https://doi.org/10.3390/pathogens11091025
Chicago/Turabian StyleCrestani, Chiara, Dinah Seligsohn, Taya L. Forde, and Ruth N. Zadoks. 2022. "How GBS Got Its Hump: Genomic Analysis of Group B Streptococcus from Camels Identifies Host Restriction as well as Mobile Genetic Elements Shared across Hosts and Pathogens" Pathogens 11, no. 9: 1025. https://doi.org/10.3390/pathogens11091025
APA StyleCrestani, C., Seligsohn, D., Forde, T. L., & Zadoks, R. N. (2022). How GBS Got Its Hump: Genomic Analysis of Group B Streptococcus from Camels Identifies Host Restriction as well as Mobile Genetic Elements Shared across Hosts and Pathogens. Pathogens, 11(9), 1025. https://doi.org/10.3390/pathogens11091025