
Metagenomic Analysis of RNA Fraction Reveals the Diversity of Swine Oral Virome on South African Backyard Swine Farms in the uMgungundlovu District of KwaZulu-Natal Province



Abstract
:1. Introduction
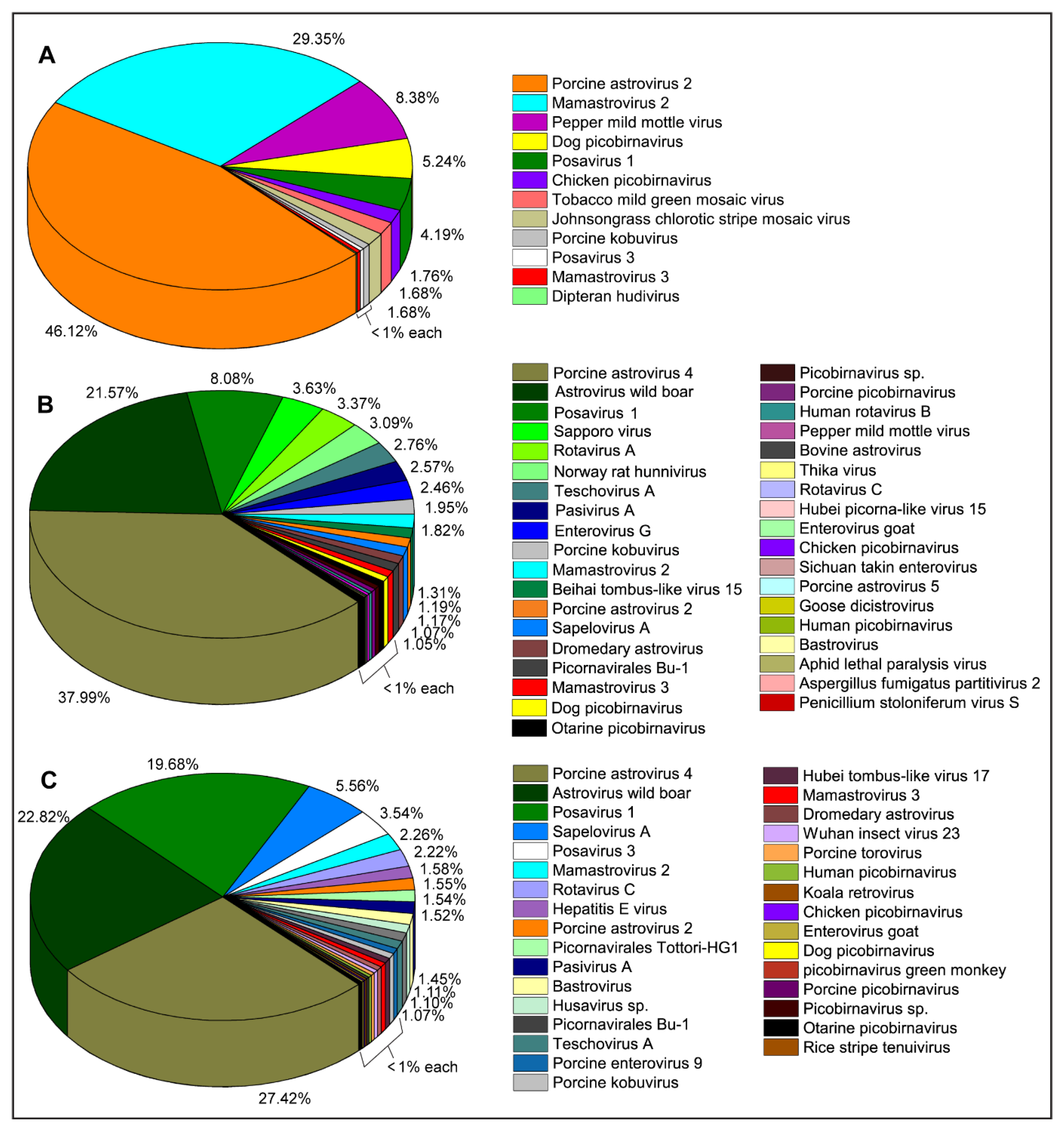
2. Results
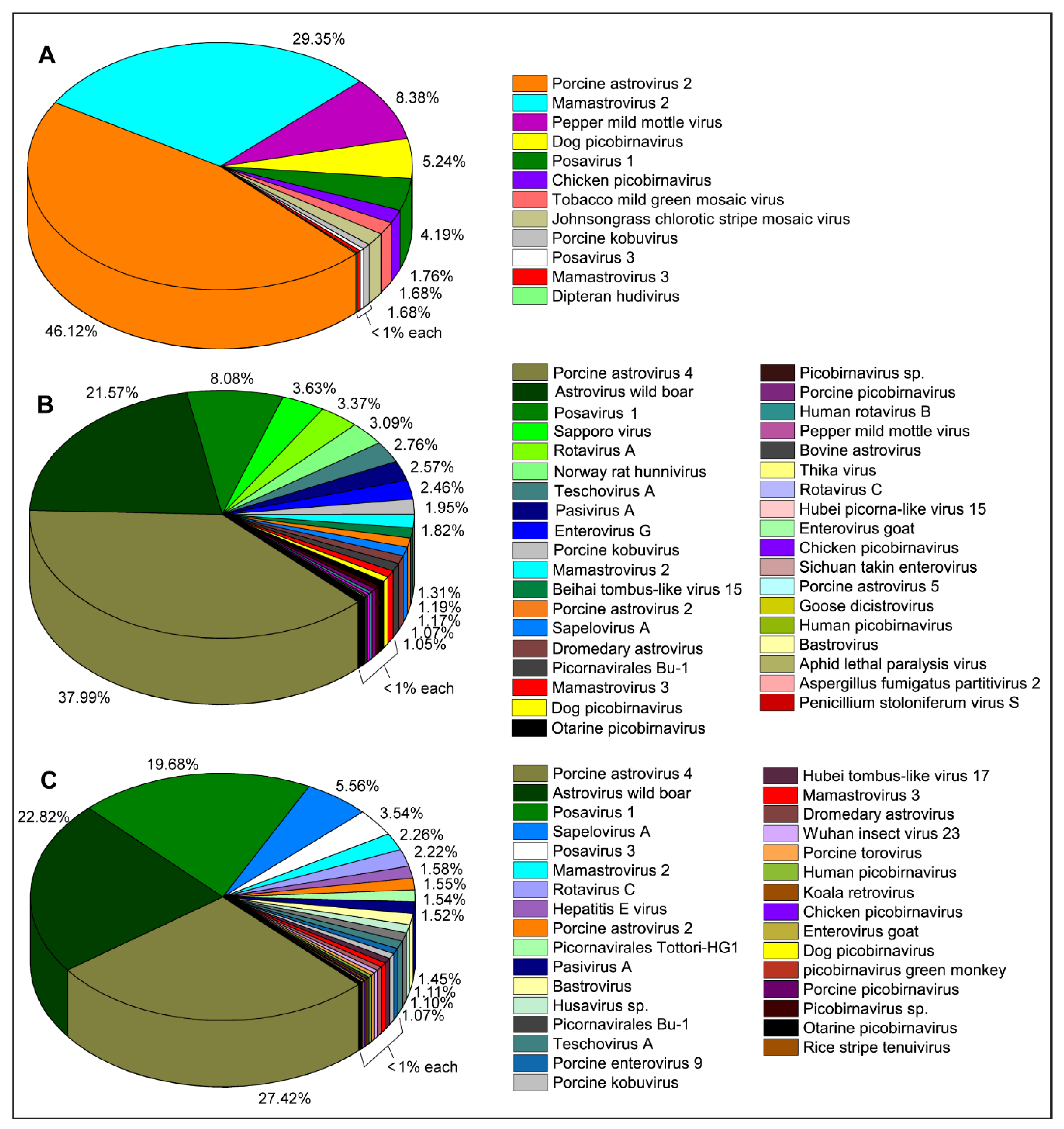
2.1. Identification and Characterization of Swine Viruses
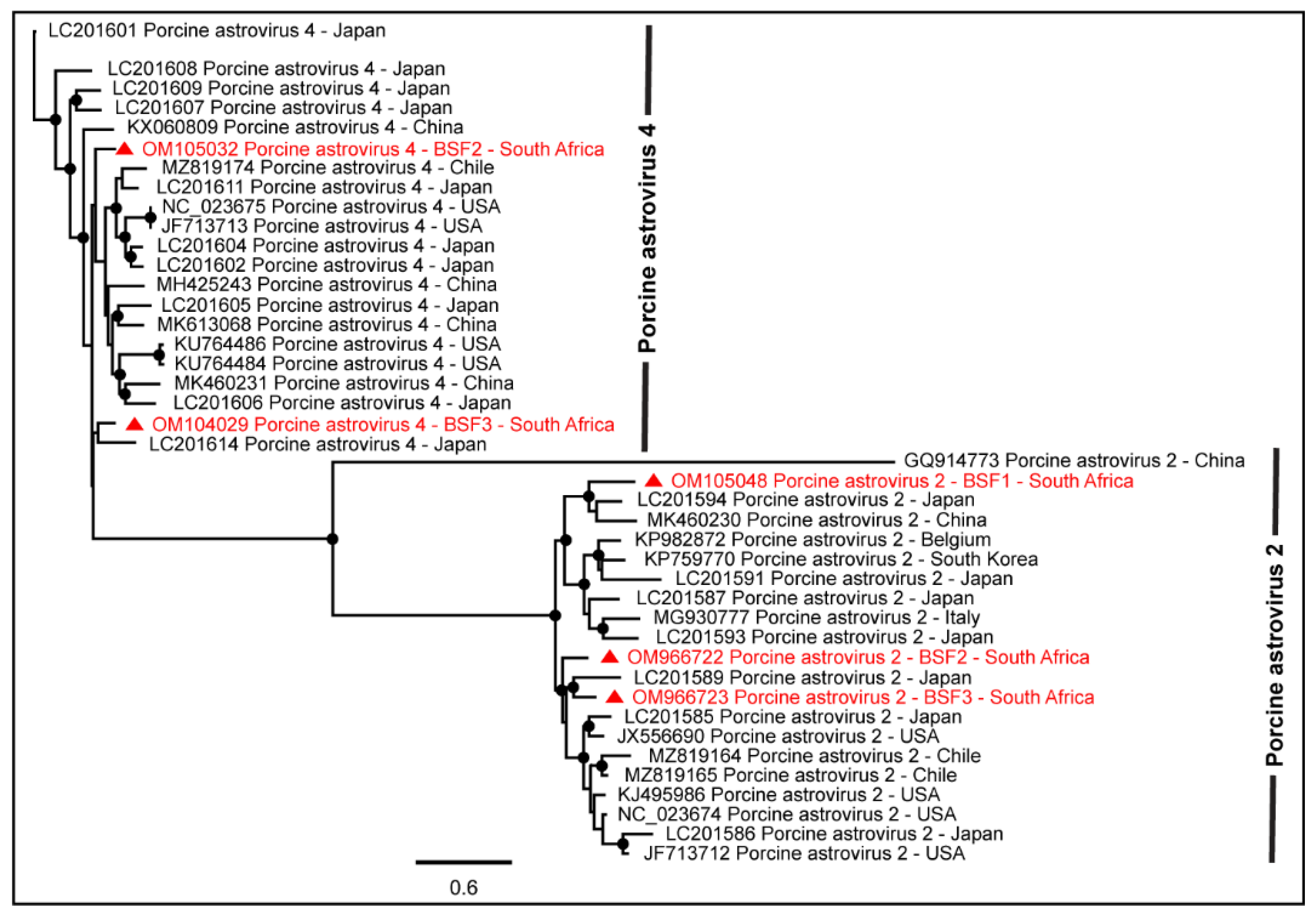
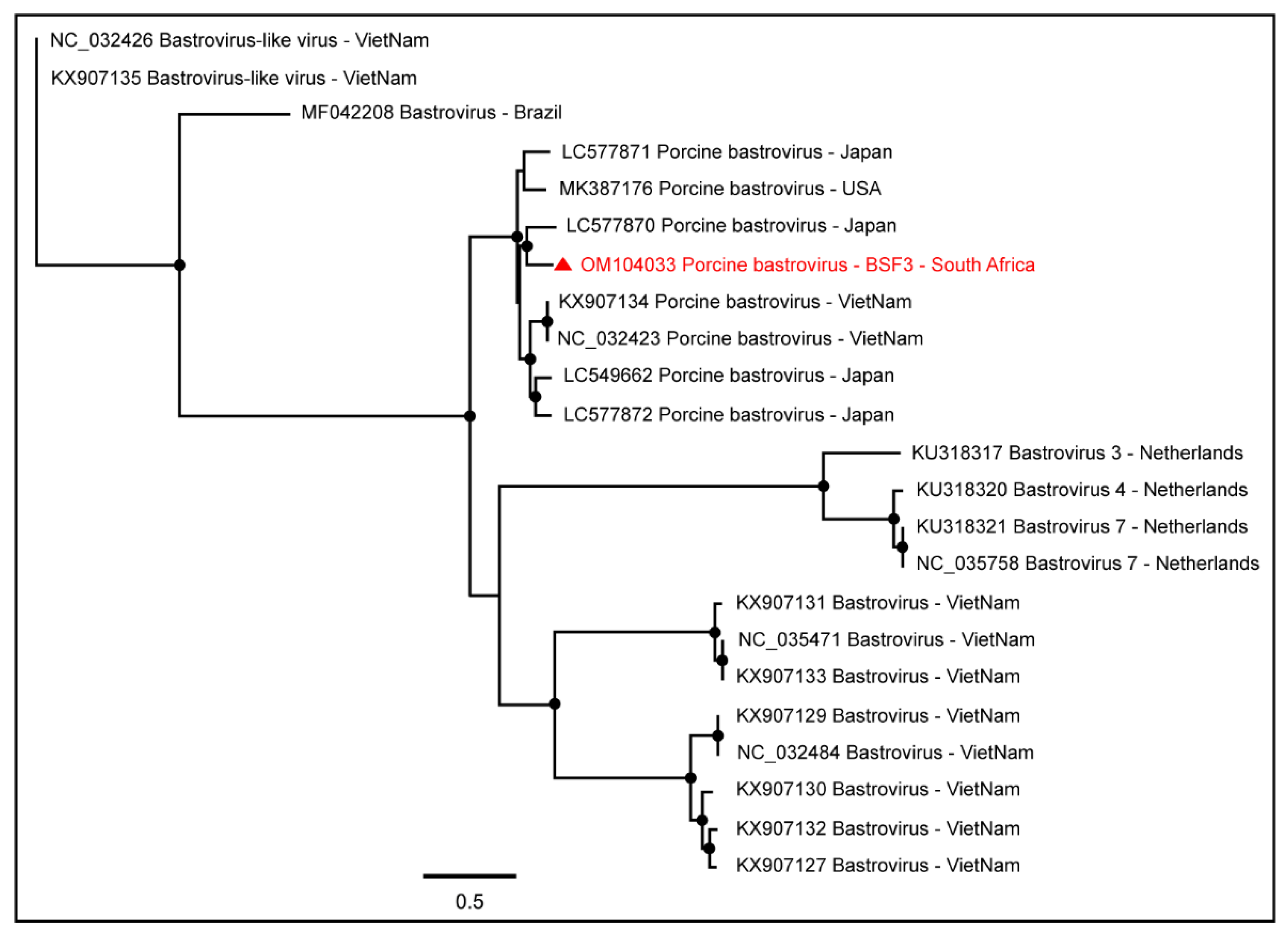
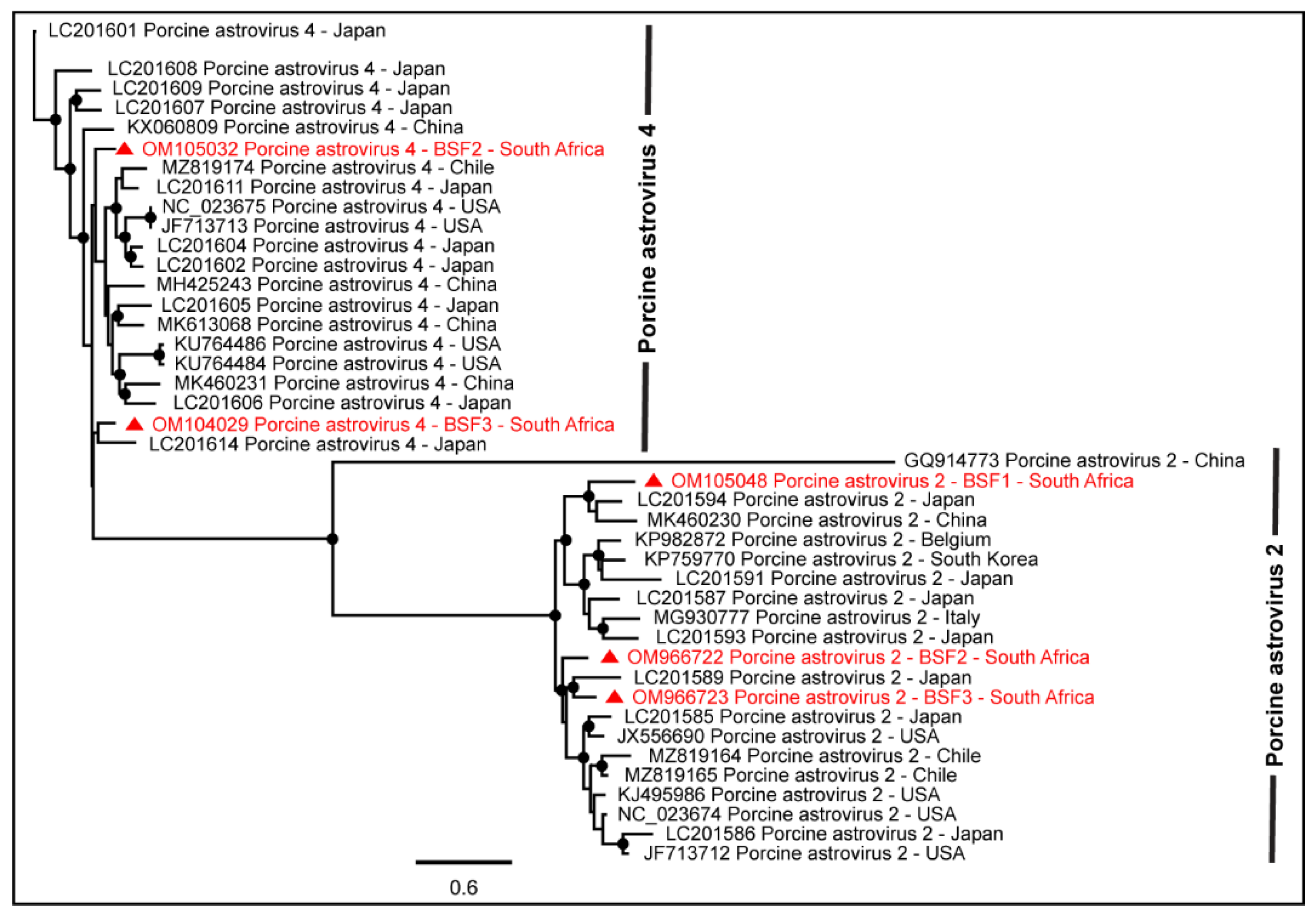
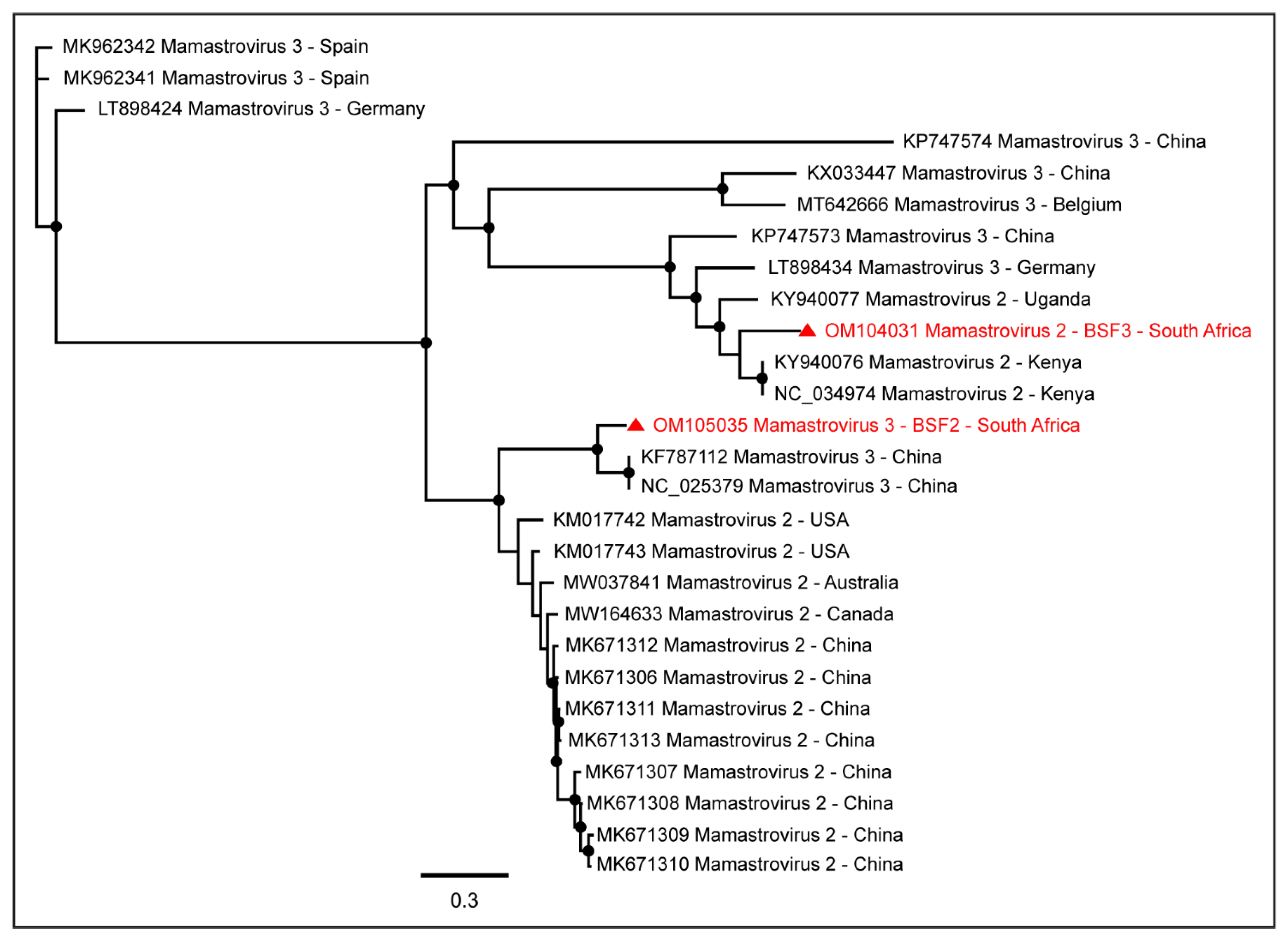
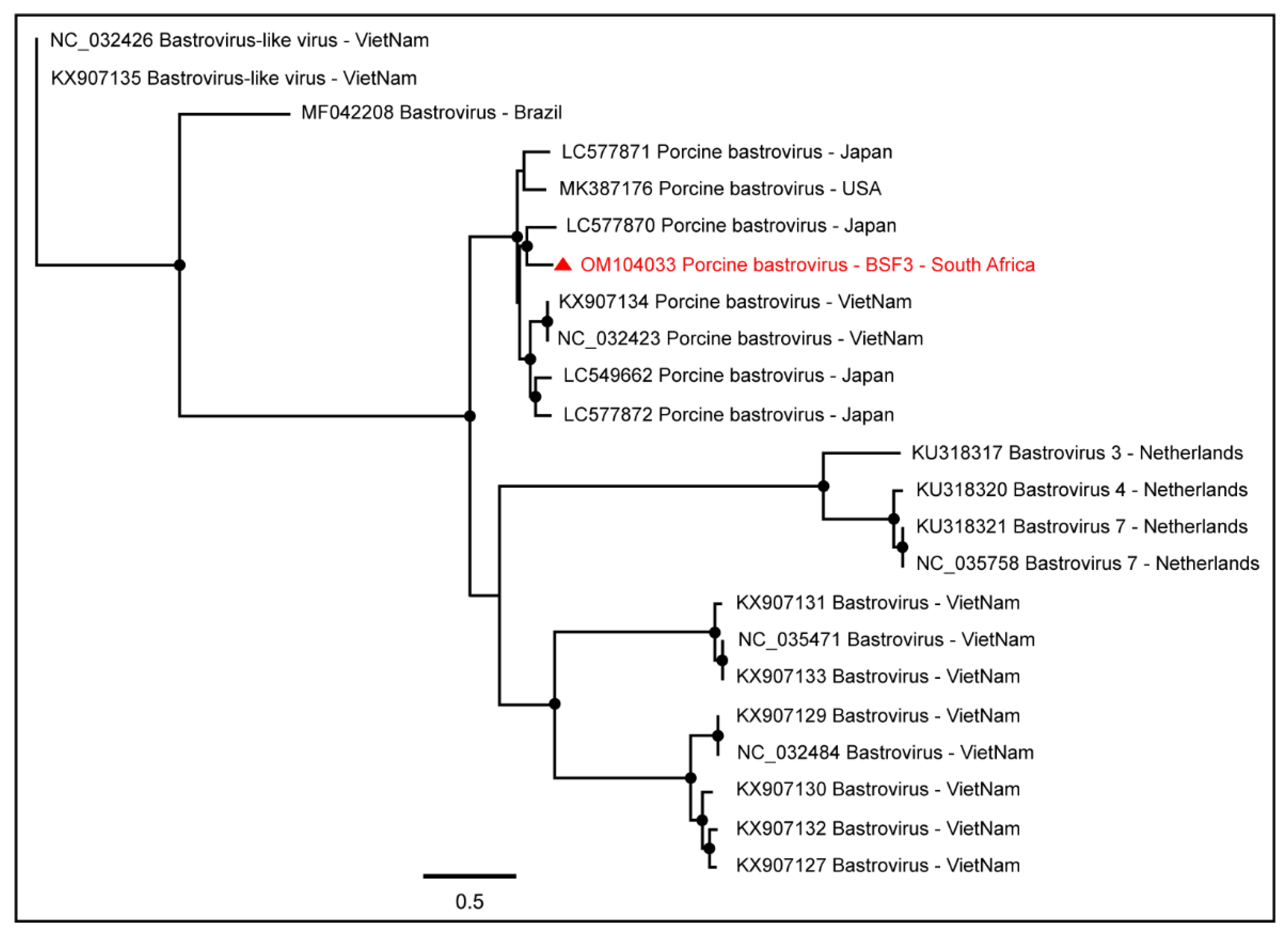
2.1.1. Family Astroviridae
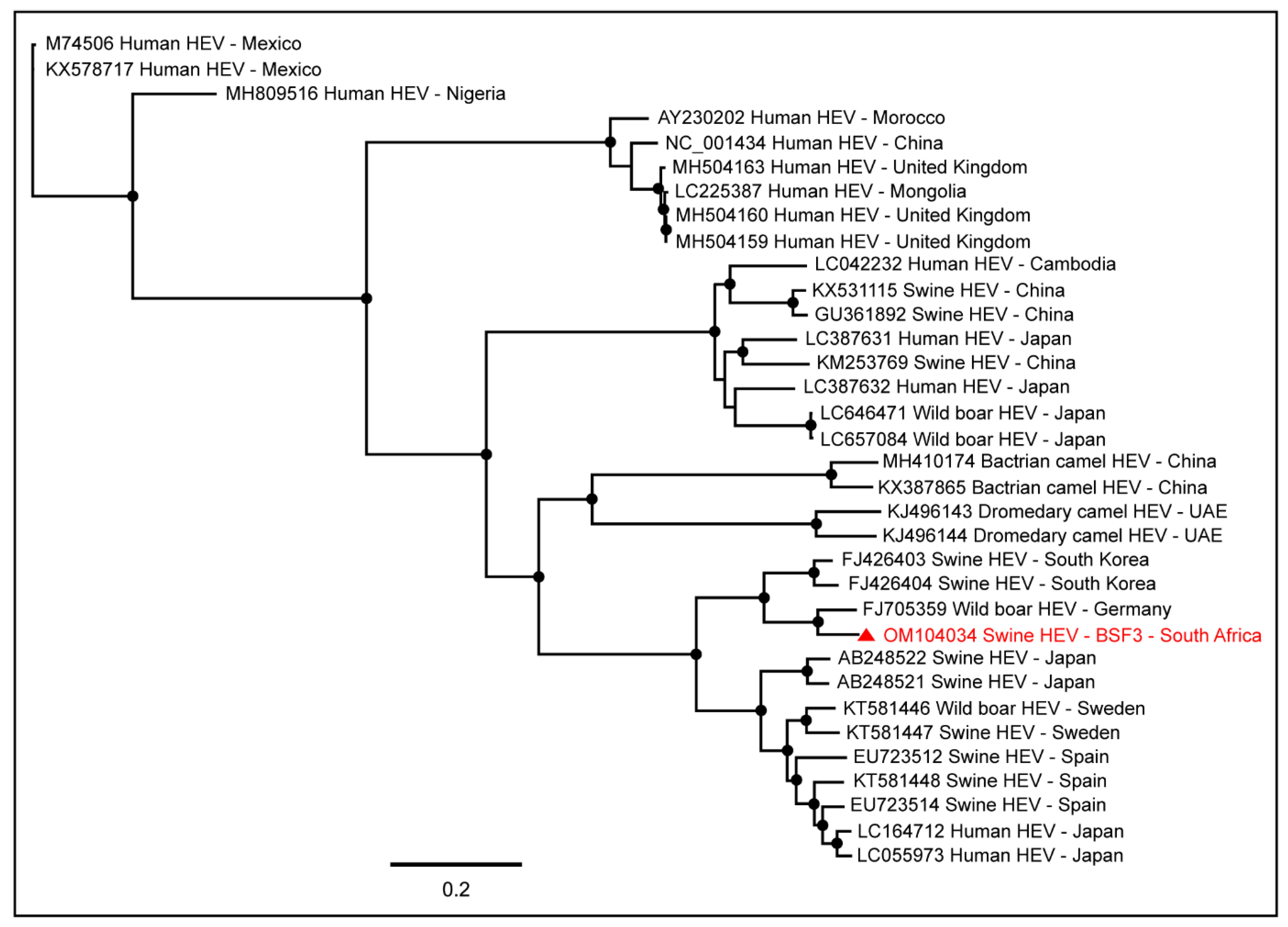
2.1.2. Family Hepeviridae
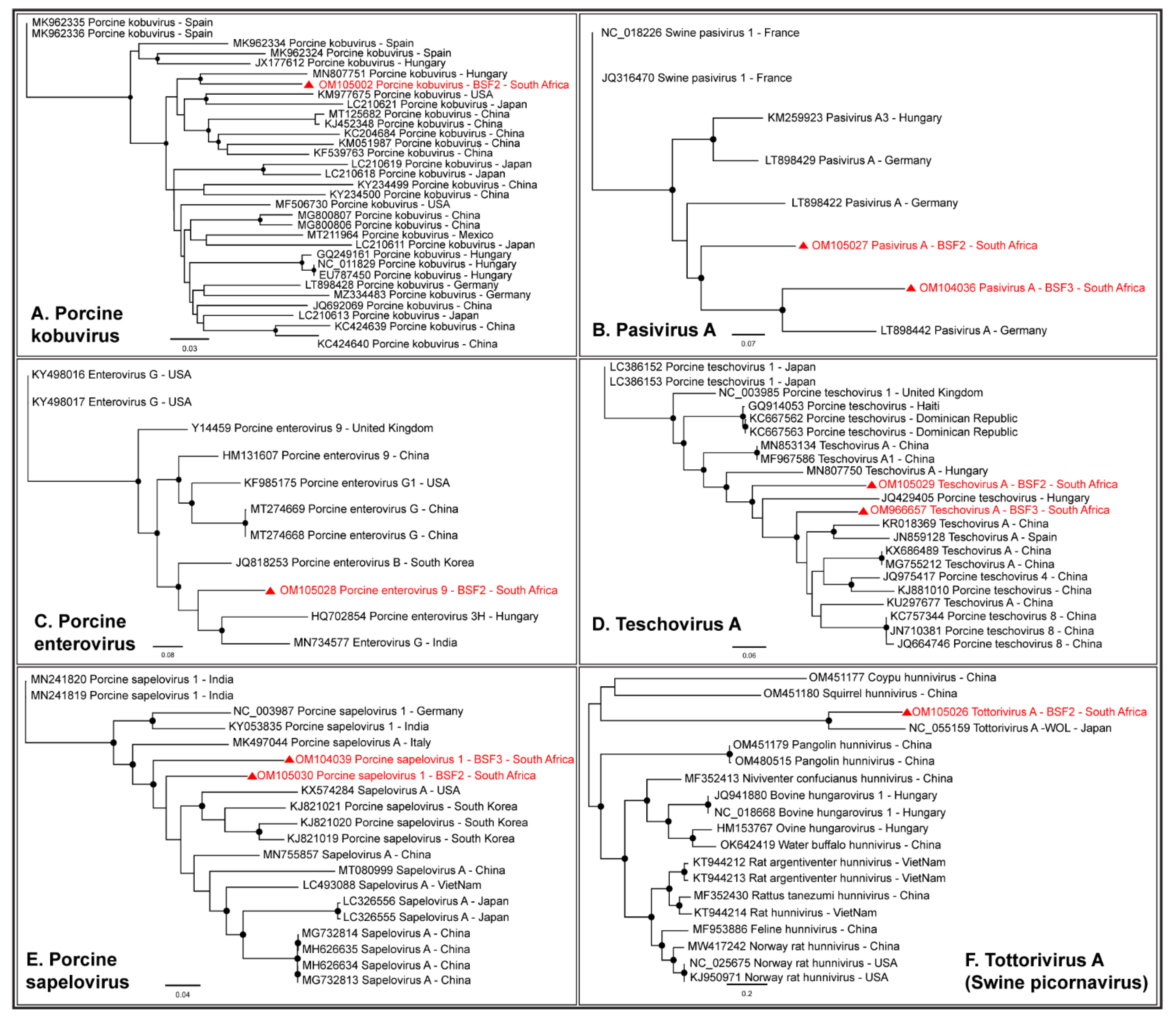
2.1.3. Family Picornaviridae
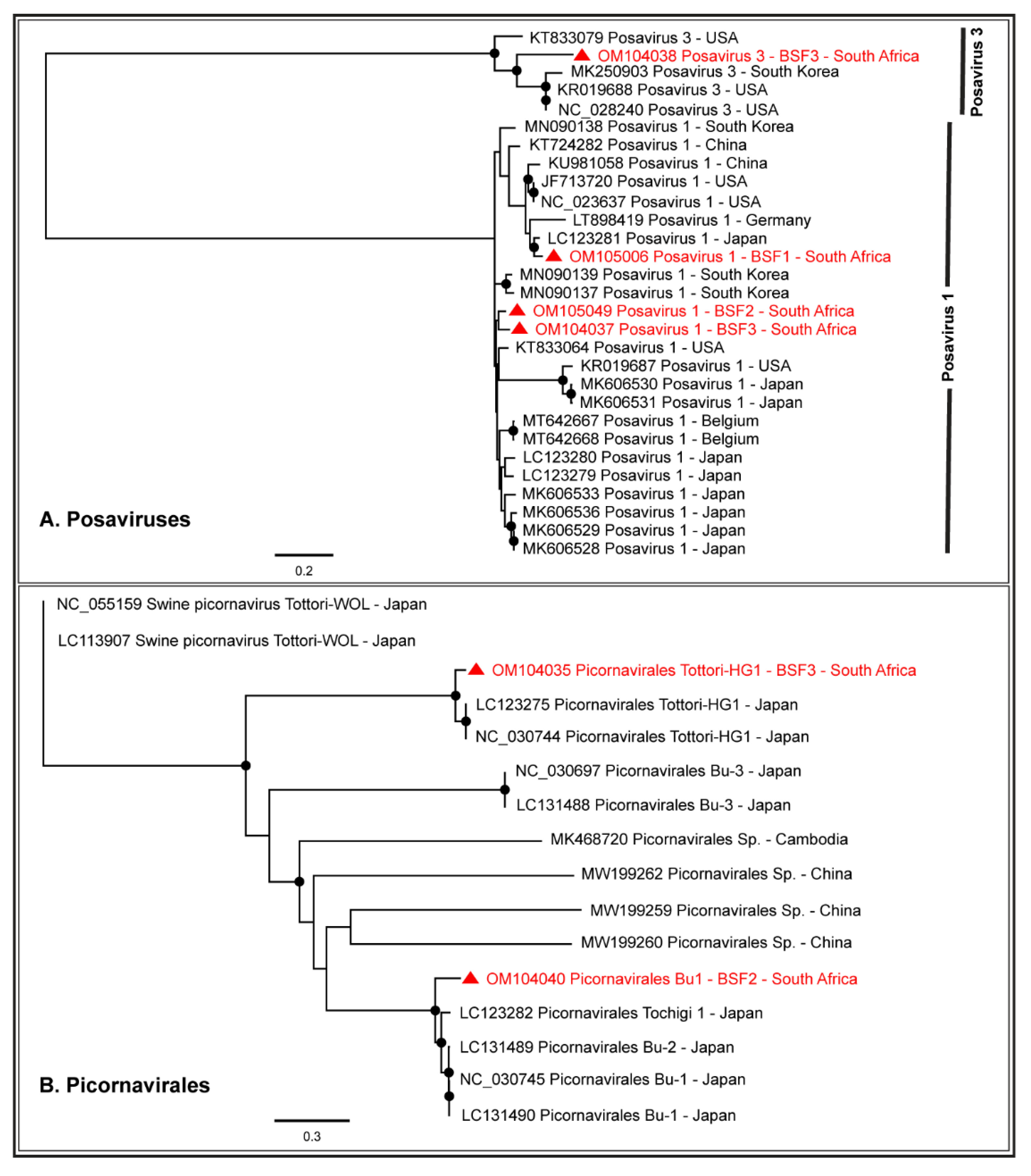
2.1.4. Order Picornavirales
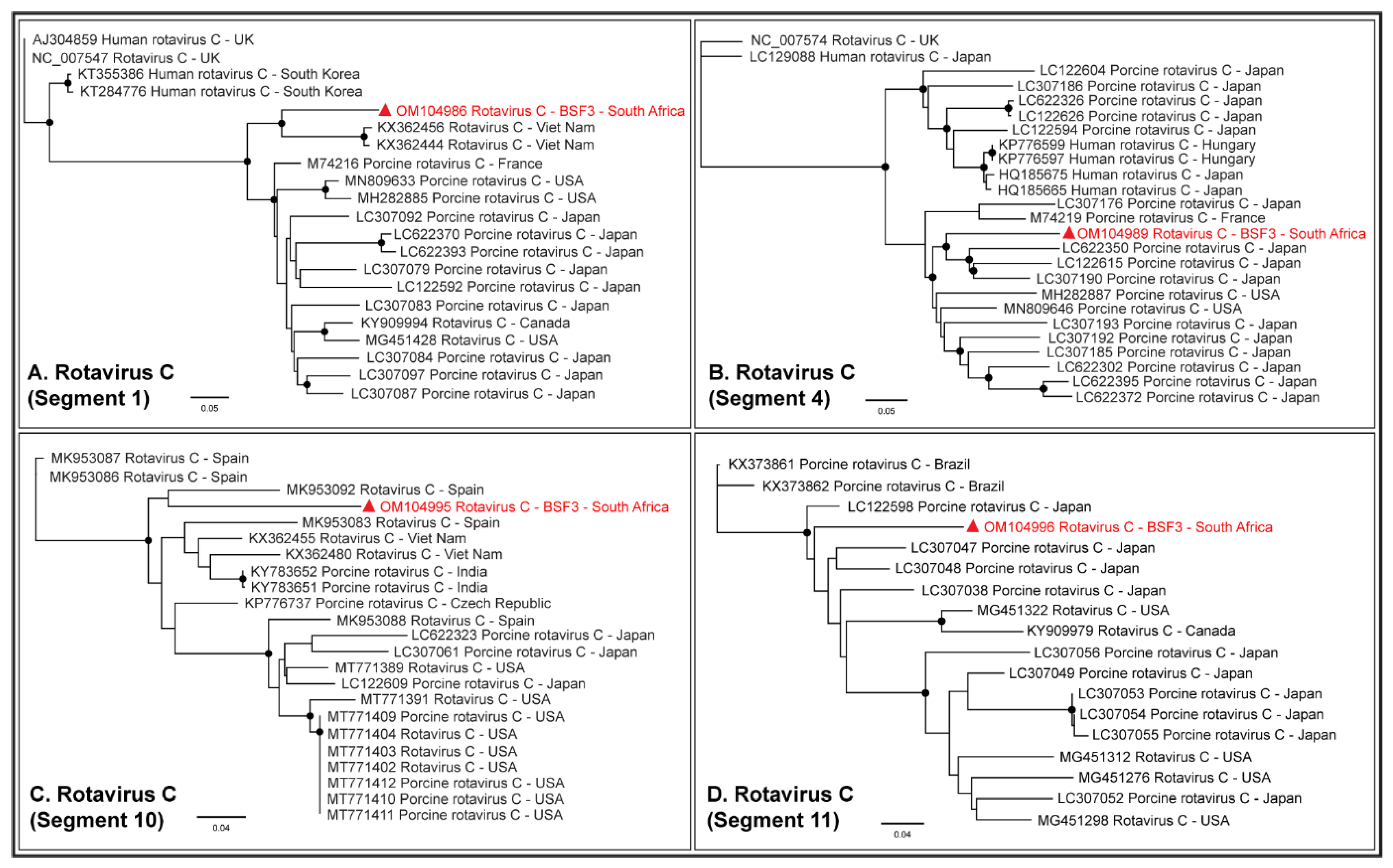
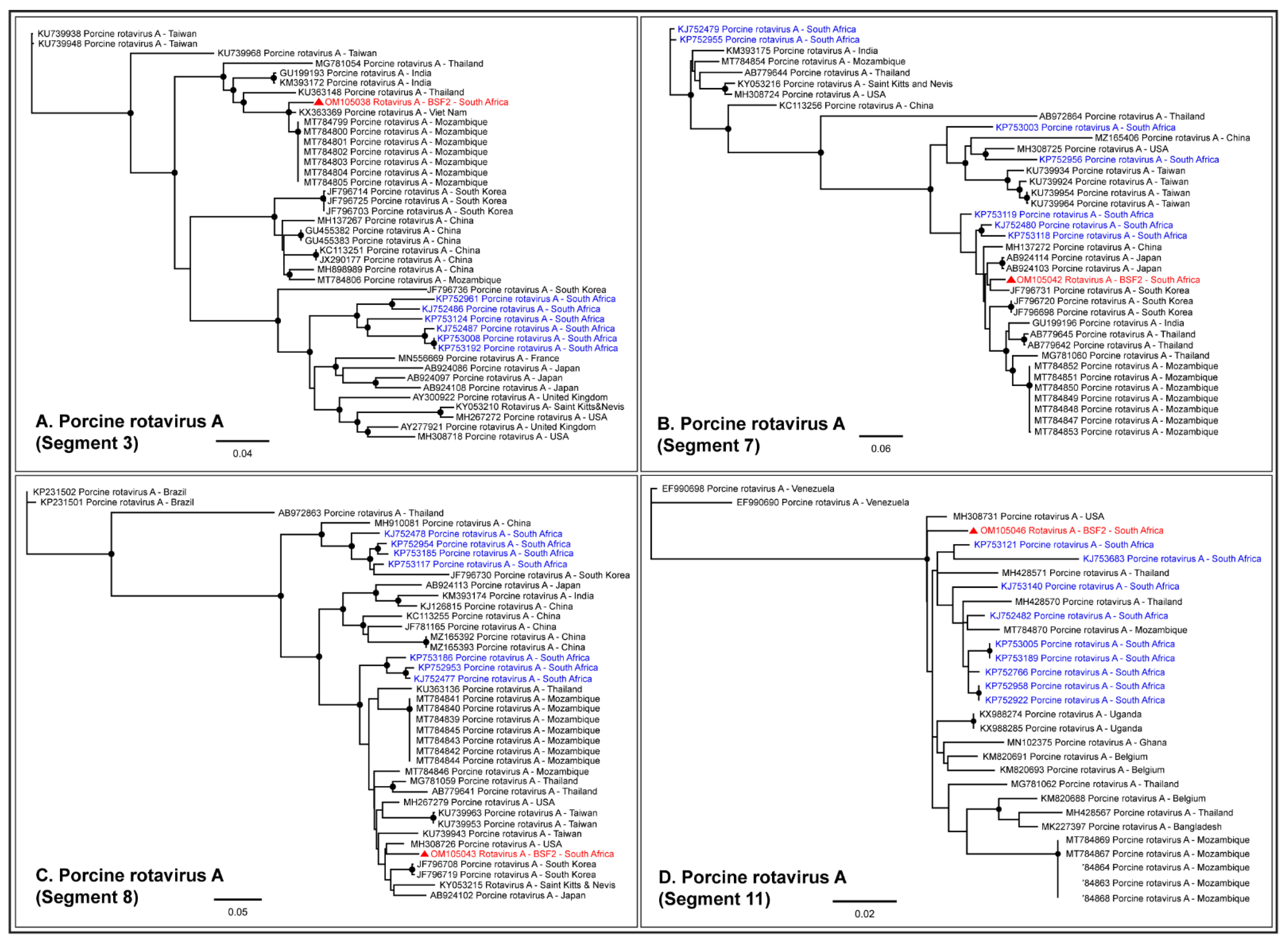
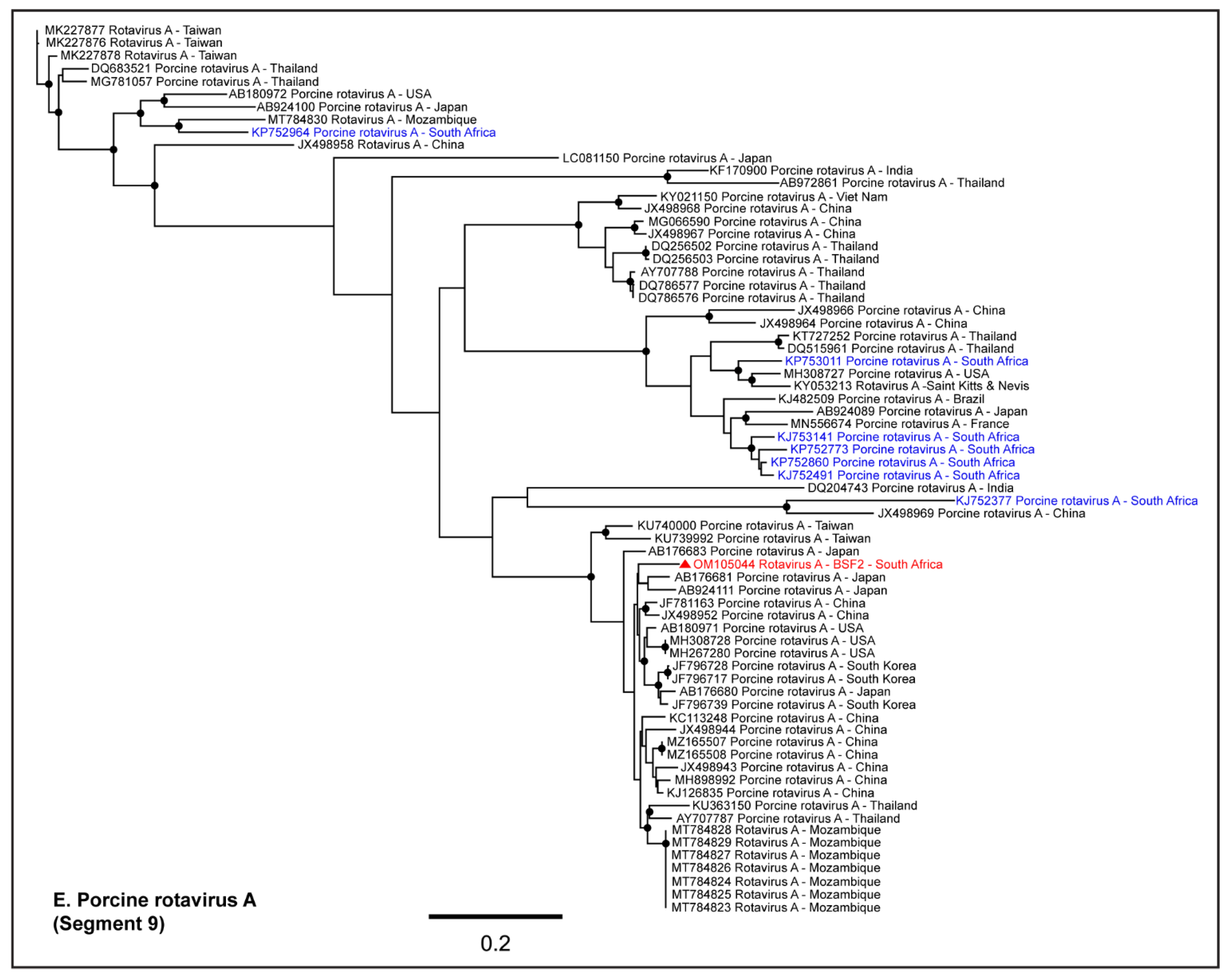
2.1.5. Family Reoviridae
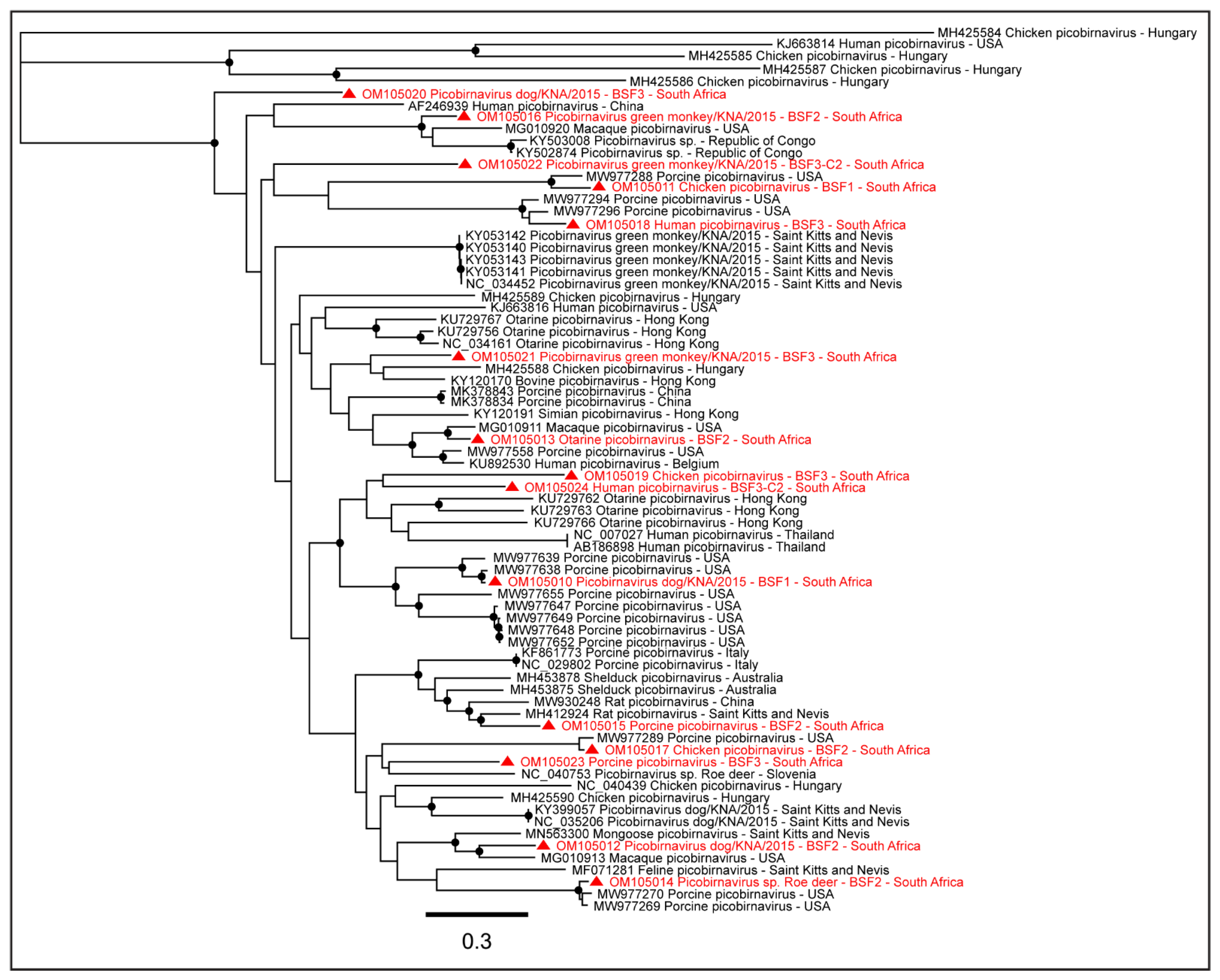
2.1.6. Family Picobirnaviridae
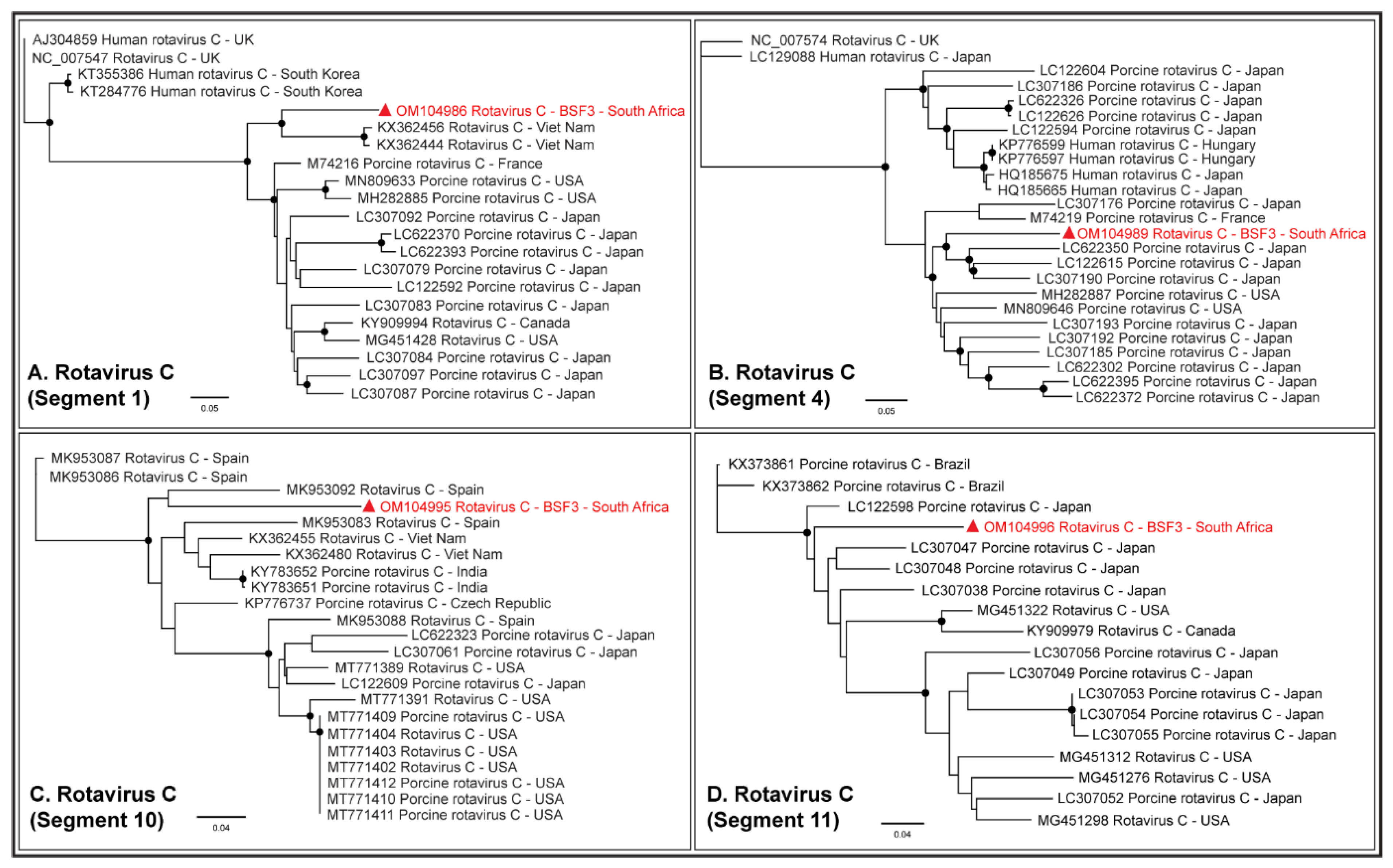
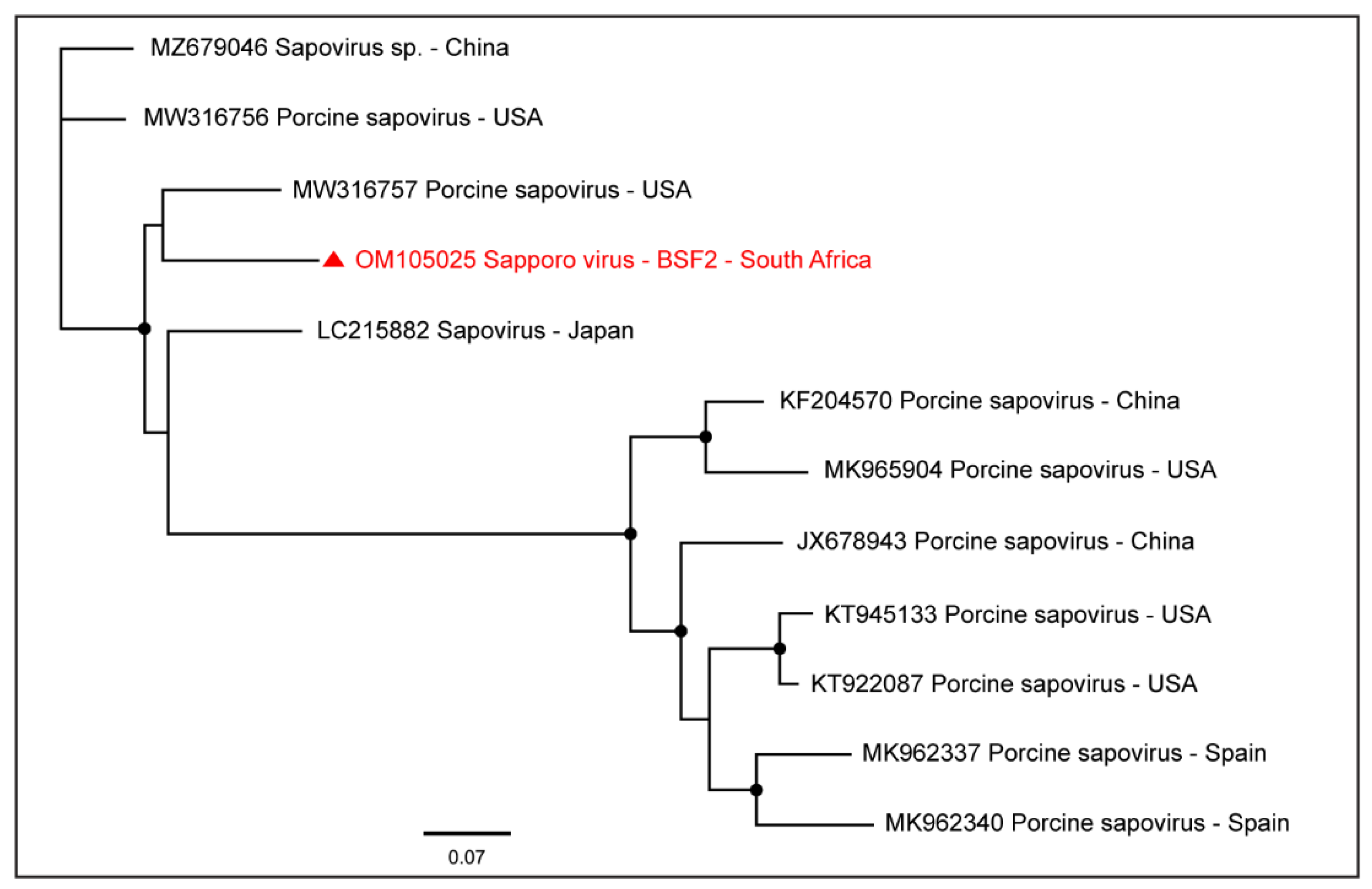
2.1.7. Family Caliciviridae
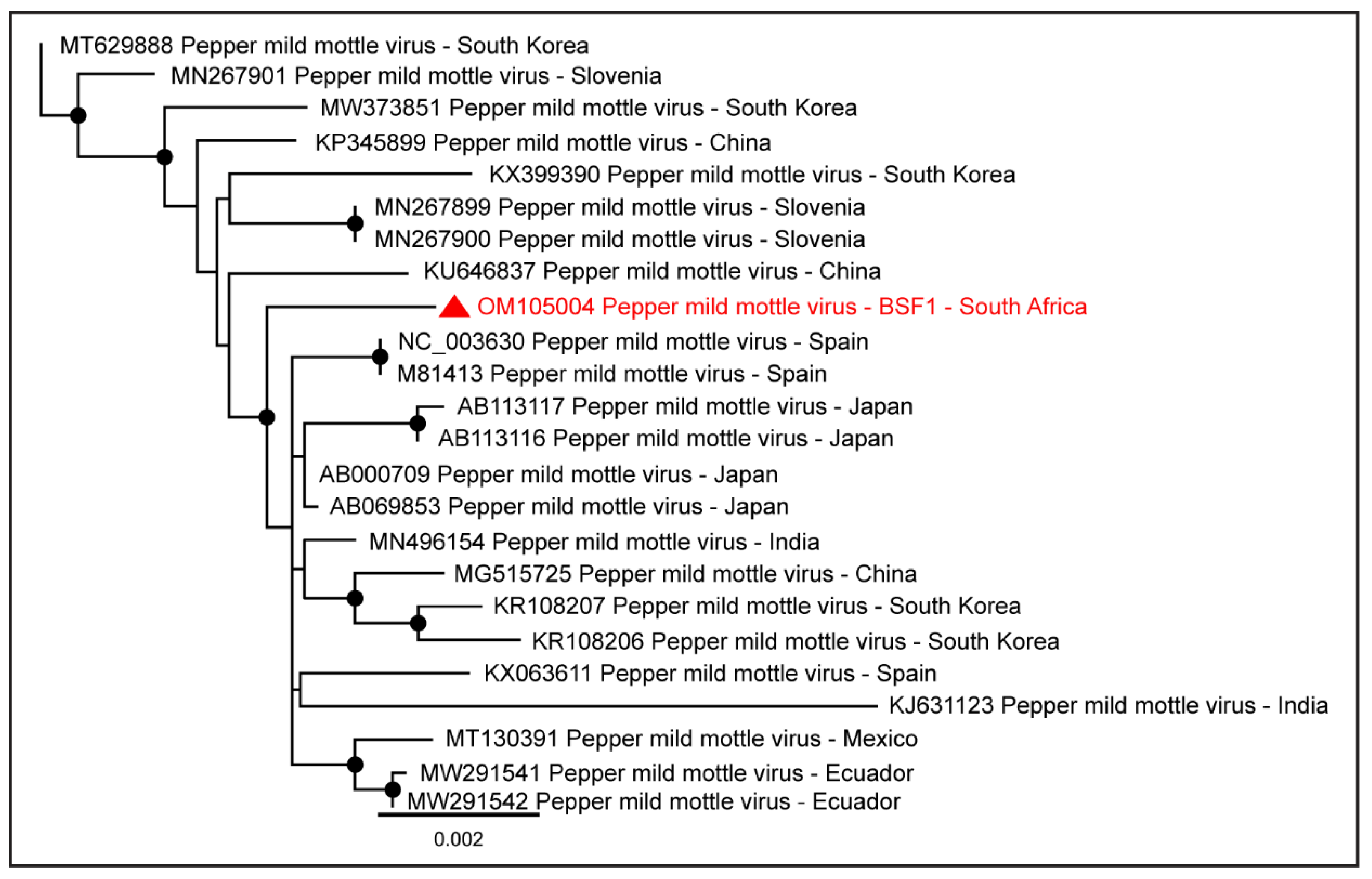
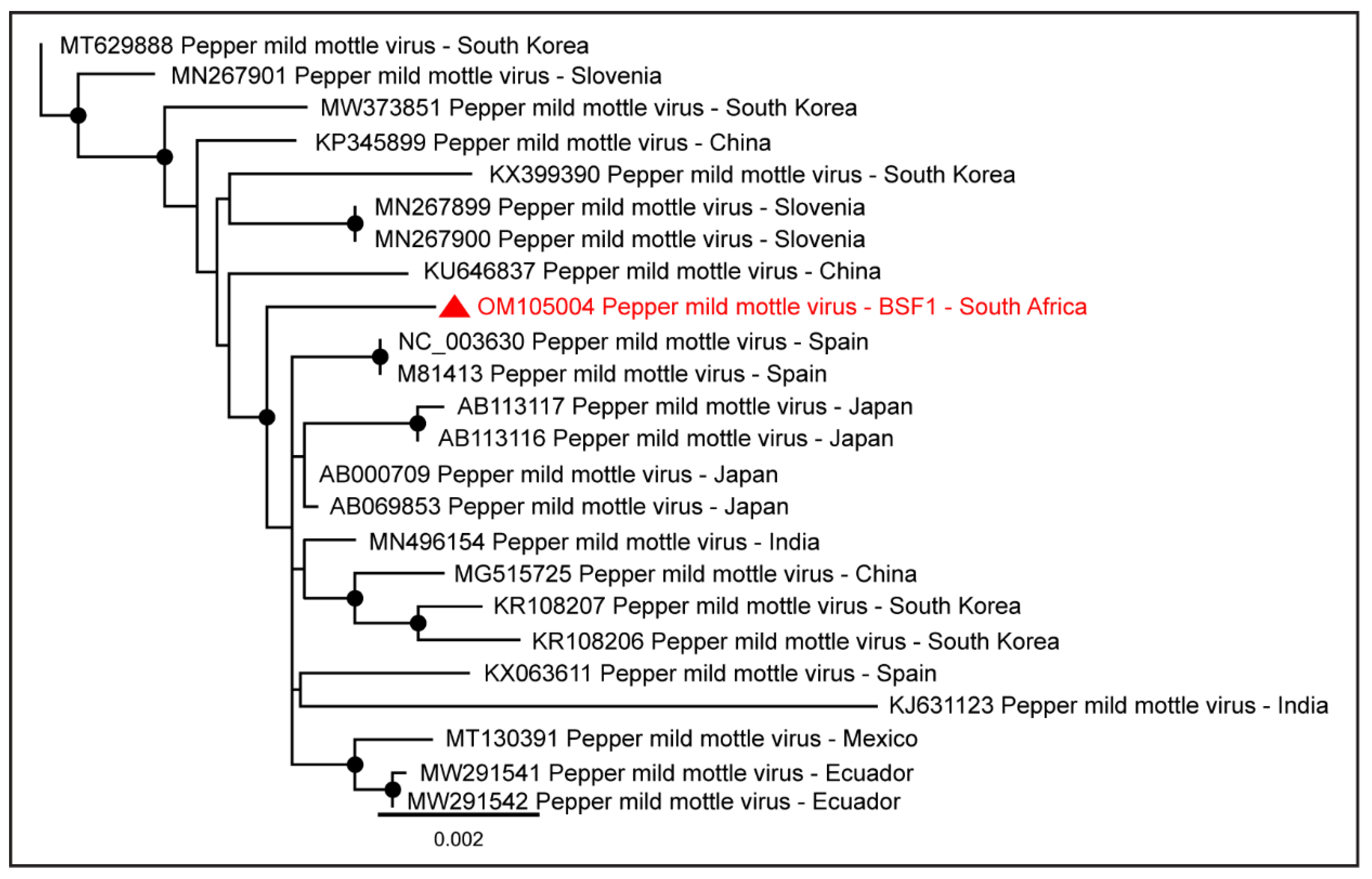
2.1.8. Family Virgaviridae
2.1.9. Family Phenuiviridae
2.1.10. Family Dicistroviridae
2.1.11. Family Partitiviridae
2.1.12. Family Tobaniviridae
2.1.13. Family Retroviridae
2.1.14. Family Tombusviridae
2.1.15. Unclassified RNA Viruses
2.2. NCBI-GenBank Accession Numbers of the Virus Genomes Generated in the Present Study
3. Discussion
4. Materials and Methods
4.1. Ethics Approval
4.2. Sample Collection and Processing
4.3. Metagenomic Sequencing
4.4. Genome Assembly
4.5. Phylogenetic Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gcumisa, S.T.; Oguttu, J.W.; Masafu, M.M. Pig farming in rural South Africa: A case study of uThukela District in KwaZulu-Natal. Indian J. Anim. Res. 2016, 50, 614–620. [Google Scholar] [CrossRef]
- Bravo-Vasquez, N.; Baumberger, C.; Jimenez-Bluhm, P.; Di Pillo, F.; Lazo, A.; Sanhueza, J.; Schultz-Cherry, S.; Hamilton-West, C. Risk factors and spatial relative risk assessment for influenza A virus in poultry and swine in backyard production systems of central Chile. Vet. Med. Sci. 2020, 6, 518–526. [Google Scholar] [CrossRef] [PubMed]
- Mena, I.; Nelson, M.I.; Quezada-Monroy, F.; Dutta, J.; Cortes-Fernández, R.; Lara-Puente, J.H.; Castro-Peralta, F.; Cunha, L.F.; Trovão, N.S.; Lozano-Dubernard, B.; et al. Origins of the 2009 H1N1 influenza pandemic in swine in Mexico. eLife 2016, 5, e16777. [Google Scholar] [CrossRef] [PubMed]
- Chauhan, R.P.; Gordon, M.L. A systematic review of influenza A virus prevalence and transmission dynamics in backyard swine populations globally. Porc. Health Manag. 2022, 8, 10. [Google Scholar] [CrossRef] [PubMed]
- Chauhan, R.P.; Gordon, M.L. A Systematic Review Analyzing the Prevalence and Circulation of Influenza Viruses in Swine Population Worldwide. Pathogens 2020, 9, 355. [Google Scholar] [CrossRef]
- Smith, G.J.; Vijaykrishna, D.; Bahl, J.; Lycett, S.J.; Worobey, M.; Pybus, O.G.; Ma, S.K.; Cheung, C.L.; Raghwani, J.; Bhatt, S.; et al. Origins and evolutionary genomics of the 2009 swine-origin H1N1 influenza A epidemic. Nature 2009, 459, 1122–1125. [Google Scholar] [CrossRef]
- Chauhan, R.P.; Gordon, M.L. An overview of influenza A virus genes, protein functions, and replication cycle highlighting important updates. Virus Genes 2022, 58, 255–269. [Google Scholar] [CrossRef]
- Chauhan, R.P.; Gordon, M.L. Review of genome sequencing technologies in molecular characterization of influenza A viruses in swine. J. Vet. Diagn. Investig. 2022, 34, 177–189. [Google Scholar] [CrossRef]
- Rockx, B.; Winegar, R.; Freiberg, A.N. Recent progress in henipavirus research: Molecular biology, genetic diversity, animal models. Antivir. Res 2012, 95, 135–149. [Google Scholar] [CrossRef]
- Chua, K.B.; Goh, K.J.; Wong, K.T.; Kamarulzaman, A.; Tan, P.S.; Ksiazek, T.G.; Zaki, S.R.; Paul, G.; Lam, S.K.; Tan, C.T. Fatal encephalitis due to Nipah virus among pig-farmers in Malaysia. Lancet 1999, 354, 1257–1259. [Google Scholar] [CrossRef]
- Chua, K.B.; Bellini, W.J.; Rota, P.A.; Harcourt, B.H.; Tamin, A.; Lam, S.K.; Ksiazek, T.G.; Rollin, P.E.; Zaki, S.R.; Shieh, W.; et al. Nipah virus: A recently emergent deadly paramyxovirus. Science 2000, 288, 1432–1435. [Google Scholar] [CrossRef] [PubMed]
- Black, P.F.; Cronin, J.P.; Morrissy, C.J.; Westbury, H.A. Serological examination for evidence of infection with Hendra and Nipah viruses in Queensland piggeries. Aust. Vet. J. 2001, 79, 424–426. [Google Scholar] [CrossRef] [PubMed]
- Chowdhury, S.; Khan, S.U.; Crameri, G.; Epstein, J.H.; Broder, C.C.; Islam, A.; Peel, A.J.; Barr, J.; Daszak, P.; Wang, L.-F.; et al. Serological Evidence of Henipavirus Exposure in Cattle, Goats and Pigs in Bangladesh. PLOS Negl. Trop. Dis. 2014, 8, e3302. [Google Scholar] [CrossRef] [PubMed]
- Hayman, D.T.S.; Wang, L.-F.; Barr, J.; Baker, K.S.; Suu-Ire, R.; Broder, C.C.; Cunningham, A.A.; Wood, J.L.N. Antibodies to Henipavirus or Henipa-Like Viruses in Domestic Pigs in Ghana, West Africa. PLoS ONE 2011, 6, e25256. [Google Scholar] [CrossRef]
- Atherstone, C.; Diederich, S.; Weingartl, H.M.; Fischer, K.; Balkema-Buschmann, A.; Grace, D.; Alonso, S.; Dhand, N.K.; Ward, M.P.; Mor, S.M. Evidence of exposure to henipaviruses in domestic pigs in Uganda. Transbound. Emerg. Dis. 2019, 66, 921–928. [Google Scholar] [CrossRef]
- Reveles-Félix, S.; Carreón-Nápoles, R.; Mendoza-Elvira, S.; Quintero-Ramírez, V.; García-Sánchez, J.; Martínez-Bautista, R.; Saavedra-Montañez, M.; Mosqueda Gualito, J.J.; Sánchez-Betancourt, J.I. Emerging strains of porcine epidemic diarrhoea virus (PEDv) in Mexico. Transbound. Emerg. Dis. 2020, 67, 1035–1041. [Google Scholar] [CrossRef]
- Lee, C. Porcine epidemic diarrhea virus: An emerging and re-emerging epizootic swine virus. Virol. J. 2015, 12, 193. [Google Scholar] [CrossRef]
- Wang, D.; Fang, L.; Xiao, S. Porcine epidemic diarrhea in China. Virus Res. 2016, 226, 7–13. [Google Scholar] [CrossRef]
- Stevenson, G.W.; Hoang, H.; Schwartz, K.J.; Burrough, E.R.; Sun, D.; Madson, D.; Cooper, V.L.; Pillatzki, A.; Gauger, P.; Schmitt, B.J.; et al. Emergence of Porcine epidemic diarrhea virus in the United States: Clinical signs, lesions, and viral genomic sequences. J. Vet. Diagn. Investig. 2013, 25, 649–654. [Google Scholar] [CrossRef]
- Blome, S.; Staubach, C.; Henke, J.; Carlson, J.; Beer, M. Classical Swine Fever-An Updated Review. Viruses 2017, 9, 86. [Google Scholar] [CrossRef]
- Brown, V.R.; Bevins, S.N. A Review of Classical Swine Fever Virus and Routes of Introduction into the United States and the Potential for Virus Establishment. Front. Vet. Sci. 2018, 5, 31. [Google Scholar] [CrossRef] [PubMed]
- Montaner-Tarbes, S.; Del Portillo, H.A.; Montoya, M.; Fraile, L. Key Gaps in the Knowledge of the Porcine Respiratory Reproductive Syndrome Virus (PRRSV). Front. Vet. Sci. 2019, 6, 38. [Google Scholar] [CrossRef] [PubMed]
- Plagemann, P.G. Porcine reproductive and respiratory syndrome virus: Origin hypothesis. Emerg. Infect. Dis. 2003, 9, 903–908. [Google Scholar] [CrossRef] [PubMed]
- Raymond, P.; Bellehumeur, C.; Nagarajan, M.; Longtin, D.; Ferland, A.; Müller, P.; Bissonnette, R.; Simard, C. Porcine reproductive and respiratory syndrome virus (PRRSV) in pig meat. Can. J. Vet. Res. 2017, 81, 162–170. [Google Scholar]
- Stenfeldt, C.; Diaz-San Segundo, F.; de Los Santos, T.; Rodriguez, L.L.; Arzt, J. The Pathogenesis of Foot-and-Mouth Disease in Pigs. Front. Vet. Sci. 2016, 3, 41. [Google Scholar] [CrossRef]
- Knowles, N.J.; Wilsden, G.; Reid, S.M.; Ferris, N.P.; King, D.P.; Paton, D.J.; Fevereiro, M.; Brocchi, E. Reappearance of swine vesicular disease virus in Portugal. Vet. Rec. 2007, 161, 71. [Google Scholar] [CrossRef]
- Bellini, S.; Santucci, U.; Zanardi, G.; Brocchi, E.; Marabelli, R. Swine vesicular disease surveillance and eradication activities in Italy. Rev. Sci. Tech. 2007, 26, 585–593. [Google Scholar] [CrossRef]
- Almeida, P.R.; Lorenzetti, E.; Cruz, R.S.; Watanabe, T.T.; Zlotowski, P.; Alfieri, A.A.; Driemeier, D. Diarrhea caused by rotavirus A, B, and C in suckling piglets from southern Brazil: Molecular detection and histologic and immunohistochemical characterization. J. Vet. Diagn. Investig. 2018, 30, 370–376. [Google Scholar] [CrossRef]
- Yan, N.; Yue, H.; Wang, Y.; Zhang, B.; Tang, C. Genomic analysis reveals G3P[13] porcine rotavirus A interspecific transmission to human from pigs in a swine farm with diarrhoea outbreak. J. Gen. Virol. 2021, 102, 001532. [Google Scholar] [CrossRef]
- Wandera, E.A.; Hatazawa, R.; Tsutsui, N.; Kurokawa, N.; Kathiiko, C.; Mumo, M.; Waithira, E.; Wachira, M.; Mwaura, B.; Nyangao, J.; et al. Genomic characterization of an African G4P[6] human rotavirus strain identified in a diarrheic child in Kenya: Evidence for porcine-to-human interspecies transmission and reassortment. Infect. Genet. Evol. 2021, 96, 105133. [Google Scholar] [CrossRef]
- Wu, F.-T.; Bányai, K.; Jiang, B.; Liu, L.T.-C.; Marton, S.; Huang, Y.-C.; Huang, L.-M.; Liao, M.-H.; Hsiung, C.A. Novel G9 rotavirus strains co-circulate in children and pigs, Taiwan. Sci. Rep. 2017, 7, 40731. [Google Scholar] [CrossRef] [PubMed]
- Ianiro, G.; Micolano, R.; Conte, M.; Labianca, M.; Vaccari, G.; Monini, M. Detection of an animal-derived G4P[6] group A rotavirus strain in a symptomatic child, in Italy. Virus Res. 2019, 260, 7–11. [Google Scholar] [CrossRef] [PubMed]
- Roczo-Farkas, S.; Dunlop, R.H.; Donato, C.M.; Kirkwood, C.D.; McOrist, S. Rotavirus group C infections in neonatal and grower pigs in Australia. Vet. Rec. 2021, 188, e296. [Google Scholar] [CrossRef] [PubMed]
- Flores, P.S.; Costa, F.B.; Amorim, A.R.; Mendes, G.S.; Rojas, M.; Santos, N. Rotavirus A, C, and H in Brazilian pigs: Potential for zoonotic transmission of RVA. J. Vet. Diagn. Investig. 2021, 33, 129–135. [Google Scholar] [CrossRef]
- Lahon, A.; Ingle, V.C.; Birade, H.S.; Raut, C.G.; Chitambar, S.D. Molecular characterization of group B rotavirus circulating in pigs from India: Identification of a strain bearing a novel VP7 genotype, G21. Vet. Microbiol. 2014, 174, 342–352. [Google Scholar] [CrossRef]
- Prozesky, L.; Theodoridis, A. Diarrhoea in pigs induced by rotavirus. Onderstepoort J. Vet. Res. 1977, 44, 275–277. [Google Scholar]
- Geyer, A.; Sebata, T.; Peenze, I.; Steele, A. A molecular epidemiological study of porcine rotaviruses. J. S. Afr. Vet. Assoc. 1995, 66, 202–205. [Google Scholar]
- Geyer, A.; Sebata, T.; Peenze, I.; Steele, A.D. Group B and C porcine rotaviruses identified for the first time in South Africa. J. S. Afr. Vet. Assoc. 1996, 67, 115–116. [Google Scholar]
- Sangare, O.; Bastos, A.D.S.; Marquardt, O.; Venter, E.H.; Vosloo, W.; Thomson, G.R. Molecular Epidemiology of Serotype O Foot-and-Mouth Disease Virus with Emphasis on West and South Africa. Virus Genes 2001, 22, 345–351. [Google Scholar] [CrossRef]
- Sandvik, T.; Crooke, H.; Drew, T.W.; Blome, S.; Greiser-Wilke, I.; Moennig, V.; Gous, T.A.; Gers, S.; Kitching, J.A.; Bührmann, G.; et al. Classical swine fever in South Africa after 87 years’ absence. Vet. Rec. 2005, 157, 267. [Google Scholar] [CrossRef]
- Madzimure, J.; Chimonyo, M.; Zander, K.K.; Dzama, K. Potential for using indigenous pigs in subsistence-oriented and market-oriented small-scale farming systems of Southern Africa. Trop. Anim. Health Prod. 2013, 45, 135–142. [Google Scholar] [CrossRef] [PubMed]
- Adelabu, O.A.; Chuks Iweriebor, B.; Nwodo, U.U.; Obi, L.C.; Okoh, A.I. Incidence and Molecular Characterization of Hepatitis E Virus from Swine in Eastern Cape, South Africa. Adv. Virol. 2017, 2017, 1073253. [Google Scholar] [CrossRef] [PubMed]
- Geyer, A.; Steele, A.D.; Peenze, I.; Lecatsas, G. Astrovirus-like particles, adenoviruses and rotaviruses associated with diarrhoea in piglets. J. S. Afr. Vet. Assoc. 1994, 65, 164–166. [Google Scholar] [PubMed]
- Mor, S.K.; Chander, Y.; Marthaler, D.; Patnayak, D.P.; Goyal, S.M. Detection and molecular characterization of Porcine astrovirus strains associated with swine diarrhea. J. Vet. Diagn. Investig. 2012, 24, 1064–1067. [Google Scholar] [CrossRef]
- Reuter, G.; Pankovics, P.; Boros, A. Identification of a novel astrovirus in a domestic pig in Hungary. Arch. Virol. 2011, 156, 125–128. [Google Scholar] [CrossRef]
- Luo, Z.; Roi, S.; Dastor, M.; Gallice, E.; Laurin, M.A.; L’Homme, Y. Multiple novel and prevalent astroviruses in pigs. Vet. Microbiol. 2011, 149, 316–323. [Google Scholar] [CrossRef]
- Kattoor, J.J.; Malik, Y.S.; Saurabh, S.; Sircar, S.; Vinodhkumar, O.R.; Bora, D.P.; Dhama, K.; Ghosh, S.; Banyai, K.; Touil, N.; et al. First report and genetic characterization of porcine astroviruses of lineage 4 and 2 in diarrhoeic pigs in India. Transbound. Emerg. Dis. 2019, 66, 47–53. [Google Scholar] [CrossRef]
- Lee, M.H.; Jeoung, H.Y.; Park, H.R.; Lim, J.A.; Song, J.Y.; An, D.J. Phylogenetic analysis of porcine astrovirus in domestic pigs and wild boars in South Korea. Virus Genes 2013, 46, 175–181. [Google Scholar] [CrossRef]
- Qin, Y.; Fang, Q.; Li, X.; Li, F.; Liu, H.; Wei, Z.; Ouyang, K.; Chen, Y.; Huang, W. Molecular epidemiology and viremia of porcine astrovirus in pigs from Guangxi province of China. BMC Vet. Res. 2019, 15, 471. [Google Scholar] [CrossRef]
- Padmanabhan, A.; Hause, B.M. Detection and characterization of a novel genotype of porcine astrovirus 4 from nasal swabs from pigs with acute respiratory disease. Arch. Virol. 2016, 161, 2575–2579. [Google Scholar] [CrossRef]
- Stäubli, T.; Rickli, C.I.; Torgerson, P.R.; Fraefel, C.; Lechmann, J. Porcine teschovirus, sapelovirus, and enterovirus in Swiss pigs: Multiplex RT-PCR investigation of viral frequencies and disease association. J. Vet. Diagn. Investig. 2021, 33, 864–874. [Google Scholar] [CrossRef]
- Salamunova, S.; Jackova, A.; Mandelik, R.; Novotny, J.; Vlasakova, M.; Vilcek, S. Molecular detection of enteric viruses and the genetic characterization of porcine astroviruses and sapoviruses in domestic pigs from Slovakian farms. BMC Vet. Res. 2018, 14, 313. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Shen, Q.; Zhang, W.; Zhao, T.; Li, Y.; Jiang, J.; Yu, X.; Guo, Z.; Cui, L.; Hua, X. Genomic organization and recombination analysis of a porcine sapovirus identified from a piglet with diarrhea in China. Virol. J. 2017, 14, 57. [Google Scholar] [CrossRef] [PubMed]
- Oude Munnink, B.B.; Phan, M.V.T.; Simmonds, P.; Koopmans, M.P.G.; Kellam, P.; van der Hoek, L.; Cotten, M. Characterization of Posa and Posa-like virus genomes in fecal samples from humans, pigs, rats, and bats collected from a single location in Vietnam. Virus Evol. 2017, 3, vex022. [Google Scholar] [CrossRef]
- Chung, H.-C.; Nguyen, V.-G.; Huynh, T.-M.-L.; Do, H.-Q.; Vo, D.-C.; Park, Y.-H.; Park, B.-K. Molecular-based investigation and genetic characterization of porcine stool-associated RNA virus (posavirus) lineages 1 to 3 in pigs in South Korea from 2017 to 2019. Res. Vet. Sci. 2020, 128, 286–292. [Google Scholar] [CrossRef] [PubMed]
- Hause, B.M.; Palinski, R.; Hesse, R.; Anderson, G. Highly diverse posaviruses in swine faeces are aquatic in origin. J. Gen. Virol. 2016, 97, 1362–1367. [Google Scholar] [CrossRef]
- Aoki, H.; Sunaga, F.; Ochiai, H.; Masuda, T.; Ito, M.; Akagami, M.; Naoi, Y.; Sano, K.; Katayama, Y.; Omatsu, T.; et al. Phylogenetic analysis of novel posaviruses detected in feces of Japanese pigs with posaviruses and posa-like viruses of vertebrates and invertebrates. Arch. Virol. 2019, 164, 2147–2151. [Google Scholar] [CrossRef]
- Ghosh, S.; Malik, Y.S. The True Host/s of Picobirnaviruses. Front. Vet. Sci. 2020, 7, 615293. [Google Scholar] [CrossRef]
- Ganesh, B.; Masachessi, G.; Mladenova, Z. Animal picobirnavirus. Virusdisease 2014, 25, 223–238. [Google Scholar] [CrossRef]
- Malik, Y.S.; Kumar, N.; Sharma, K.; Dhama, K.; Shabbir, M.Z.; Ganesh, B.; Kobayashi, N.; Banyai, K. Epidemiology, phylogeny, and evolution of emerging enteric Picobirnaviruses of animal origin and their relationship to human strains. BioMed Res. Int. 2014, 2014, 780752. [Google Scholar] [CrossRef]
- Joycelyn, S.J.; Ng, A.; Kleymann, A.; Malik, Y.S.; Kobayashi, N.; Ghosh, S. High detection rates and genetic diversity of picobirnaviruses (PBVs) in pigs on St. Kitts Island: Identification of a porcine PBV strain closely related to simian and human PBVs. Infect. Genet. Evol. 2020, 84, 104383. [Google Scholar] [CrossRef] [PubMed]
- Chandra, R. Picobirnavirus, a novel group of undescribed viruses of mammals and birds: A minireview. Acta Virol. 1997, 41, 59–62. [Google Scholar] [PubMed]
- Masachessi, G.; Martinez, L.C.; Ganesh, B.; Giordano, M.O.; Barril, P.A.; Isa, M.B.; Ibars, A.; Pavan, J.V.; Nates, S.V. Establishment and maintenance of persistent infection by picobirnavirus in greater rhea (Rhea Americana). Arch Virol. 2012, 157, 2075–2082. [Google Scholar] [CrossRef] [PubMed]
- Woo, P.C.; Teng, J.L.; Bai, R.; Wong, A.Y.; Martelli, P.; Hui, S.W.; Tsang, A.K.; Lau, C.C.; Ahmed, S.S.; Yip, C.C.; et al. High Diversity of Genogroup I Picobirnaviruses in Mammals. Front. Microbiol. 2016, 7, 1886. [Google Scholar] [CrossRef] [PubMed]
- Woo, P.C.Y.; Lau, S.K.P.; Bai, R.; Teng, J.L.L.; Lee, P.; Martelli, P.; Hui, S.-W.; Yuen, K.-Y. Complete genome sequence of a novel picobirnavirus, otarine picobirnavirus, discovered in California sea lions. J. Virol. 2012, 86, 6377–6378. [Google Scholar] [CrossRef] [PubMed]
- Chauhan, R.P.; Gordon, M.L. Deciphering transmission dynamics and spillover of avian influenza viruses from avian species to swine populations globally. Virus Genes 2021, 57, 541–555. [Google Scholar] [CrossRef]
- Jankowski, M.D.; Williams, C.J.; Fair, J.M.; Owen, J.C. Birds Shed RNA-Viruses According to the Pareto Principle. PLoS ONE 2013, 8, e72611. [Google Scholar] [CrossRef]
- Rahman, M.M.; Talukder, A.; Chowdhury, M.M.H.; Talukder, R.; Akter, R. Coronaviruses in wild birds—A potential and suitable vector for global distribution. Vet. Med. Sci. 2021, 7, 264–272. [Google Scholar] [CrossRef]
- Papp, H.; Borzák, R.; Farkas, S.; Kisfali, P.; Lengyel, G.; Molnár, P.; Melegh, B.; Matthijnssens, J.; Jakab, F.; Martella, V.; et al. Zoonotic transmission of reassortant porcine G4P[6] rotaviruses in Hungarian pediatric patients identified sporadically over a 15 year period. Infect. Genet. Evol. 2013, 19, 71–80. [Google Scholar] [CrossRef]
- Tacharoenmuang, R.; Guntapong, R.; Upachai, S.; Singchai, P.; Fukuda, S.; Ide, T.; Hatazawa, R.; Sutthiwarakom, K.; Kongjorn, S.; Onvimala, N.; et al. Full genome-based characterization of G4P[6] rotavirus strains from diarrheic patients in Thailand: Evidence for independent porcine-to-human interspecies transmission events. Virus Genes 2021, 57, 338–357. [Google Scholar] [CrossRef]
- Gabbay, Y.B.; Borges, A.A.; Oliveira, D.S.; Linhares, A.C.; Mascarenhas, J.D.; Barardi, C.R.; Simões, C.M.; Wang, Y.; Glass, R.I.; Jiang, B. Evidence for zoonotic transmission of group C rotaviruses among children in Belém, Brazil. J. Med. Virol. 2008, 80, 1666–1674. [Google Scholar] [CrossRef] [PubMed]
- Bouwknegt, M.; Rutjes, S.A.; Reusken, C.B.E.M.; Stockhofe-Zurwieden, N.; Frankena, K.; de Jong, M.C.M.; de Roda Husman, A.M.; van der Poel, W.H.M. The course of hepatitis E virus infection in pigs after contact-infection and intravenous inoculation. BMC Vet. Res. 2009, 5, 7. [Google Scholar] [CrossRef] [PubMed]
- Krog, J.S.; Larsen, L.E.; Breum, S. Tracing Hepatitis E Virus in Pigs From Birth to Slaughter. Front. Vet. Sci. 2019, 6, 50. [Google Scholar] [CrossRef] [PubMed]
- Andraud, M.; Dumarest, M.; Cariolet, R.; Aylaj, B.; Barnaud, E.; Eono, F.; Pavio, N.; Rose, N. Direct contact and environmental contaminations are responsible for HEV transmission in pigs. Vet. Res. 2013, 44, 102. [Google Scholar] [CrossRef] [PubMed]
- Meester, M.; Tobias, T.J.; Bouwknegt, M.; Kusters, N.E.; Stegeman, J.A.; van der Poel, W.H.M. Infection dynamics and persistence of hepatitis E virus on pig farms—A review. Porc. Health Manag. 2021, 7, 16. [Google Scholar] [CrossRef] [PubMed]
- Barrón-Rodríguez, R.J.; Rojas-Anaya, E.; Ayala-Sumuano, J.T.; Romero-Espinosa JÁ, I.; Vázquez-Pérez, J.A.; Cortés-Cruz, M.; García-Espinosa, G.; Loza-Rubio, E. Swine virome on rural backyard farms in Mexico: Communities with different abundances of animal viruses and phages. Arch. Virol. 2021, 166, 475–489. [Google Scholar] [CrossRef]
- Prickett, J.R.; Kim, W.; Simer, R. Oral-fluid samples for surveillance of commercial growing pigs for porcine reproductive and respiratory syndrome virus and porcine circovirus type 2 infections. J. Swine Health Prod. 2008, 16, 86–91. [Google Scholar]
- Henao-Diaz, A.; Giménez-Lirola, L.; Baum, D.H.; Zimmerman, J. Guidelines for oral fluid-based surveillance of viral pathogens in swine. Porc. Health Manag. 2020, 6, 28. [Google Scholar] [CrossRef]
- Vilsker, M.; Moosa, Y.; Nooij, S.; Fonseca, V.; Ghysens, Y.; Dumon, K.; Pauwels, R.; Alcantara, L.C.; Vanden Eynden, E.; Vandamme, A.-M.; et al. Genome Detective: An automated system for virus identification from high-throughput sequencing data. Bioinformatics 2018, 35, 871–873. [Google Scholar] [CrossRef]
- Deforche, K. An alignment method for nucleic acid sequences against annotated genomes. bioRxiv 2017, 200394. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef] [PubMed]
- Guindon, S.; Dufayard, J.-F.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New Algorithms and Methods to Estimate Maximum-Likelihood Phylogenies: Assessing the Performance of PhyML 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Virus ID | Percent Coverage of Virus Genomes | ||
|---|---|---|---|
| BSF1 | BSF2 | BSF3 | |
| Hepatitis E virus | - | - | 97.7% |
| Mamastrovirus 3 | - | 97.9% | - |
| Pasivirus A | - | 99.3% | 96.3% |
| Pepper mild mottle virus | 98.0% | - | - |
| Picornavirales Tottori-HG1 | - | - | 99.2% |
| Porcine astrovirus 2 | 99.3% | 99.3% | 96.3% |
| Porcine bastrovirus | - | - | 97.3% |
| Porcine enterovirus 9 | - | 99.3% | - |
| Porcine enteric sapovirus | - | 99.5% | - |
| Porcine kobuvirus | - | 98.9% | - |
| Posavirus 1 | - | 99.2% | 99.2% |
| Posavirus 3 | - | - | 99.5% |
| Sapelovirus A | - | 98.9% | 98.9% |
| Teschovirus A | - | 98.3% | 99.4% |
| Name of Virus | Backyard Farm ID | Genome Length (# Nucleotides) * | GenBank Accession Numbers (This Study) |
|---|---|---|---|
| Porcine astrovirus 4 | BSF3 | 4492 | OM104029 |
| Astrovirus wild boar/WBAstV-1/2011/HUN | BSF3 | 4854 | OM104030 |
| Mamastrovirus 2 | BSF3 | 5338 | OM104031 |
| Dromedary astrovirus | BSF3 | 2542 | OM104032 |
| Porcine bastrovirus | BSF3 | 5848 | OM104033 |
| Hepatitis E virus | BSF3 | 7040 | OM104034 |
| Picornavirales Tottori-HG1 | BSF3 | 9787 | OM104035 |
| Pasivirus A | BSF3 | 6672 | OM104036 |
| Posavirus 1 | BSF3 | 9782 | OM104037 |
| Posavirus 3 | BSF3 | 8845 | OM104038 |
| Porcine sapelovirus 1 | BSF3 | 7438 | OM104039 |
| Picornavirales Bu-1 | BSF3 | 4499 | OM104040 |
| Rotavirus C—segment 1 | BSF3 | 3279 | OM104986 |
| Rotavirus C—segment 2 | BSF3 | 1305 | OM104987 |
| Rotavirus C—segment 3 | BSF3 | 1001 | OM104988 |
| Rotavirus C—segment 4 | BSF3 | 2105 | OM104989 |
| Rotavirus C—segment 5 | BSF3 | 653 | OM104990 |
| Rotavirus C—segment 6 | BSF3 | 937 | OM104991 |
| Rotavirus C—segment 7 | BSF3 | 1023 | OM104992 |
| Rotavirus C—segment 8 | BSF3 | 365 | OM104993 |
| Rotavirus C—segment 9 | BSF3 | 439 | OM104994 |
| Rotavirus C—segment 10 | BSF3 | 615 | OM104995 |
| Rotavirus C—segment 11 | BSF3 | 432 | OM104996 |
| Rotavirus C—segment 1 | BSF2 | 835 | OM104997 |
| Rotavirus C—segment 4 | BSF2 | 845 | OM104998 |
| Rotavirus C—segment 6 | BSF2 | 376 | OM104999 |
| Rotavirus C—segment 7 | BSF2 | 302 | OM105000 |
| Rotavirus C—segment 8 | BSF2 | 294 | OM105001 |
| Porcine kobuvirus | BSF2 | 8123 | OM105002 |
| Porcine astrovirus 2 (contig 2) | BSF1 | 3874 | OM105003 |
| Pepper mild mottle virus | BSF1 | 6233 | OM105004 |
| Johnsongrass chlorotic stripe mosaic virus | BSF1 | 1643 | OM105005 |
| Posavirus 1 | BSF1 | 4873 | OM105006 |
| Tobacco mild green mosaic virus | BSF1 | 1220 | OM105007 |
| Aspergillus fumigatus partitivirus 2 | BSF2 | 1391 | OM105008 |
| Penicillium stoloniferum virus S | BSF2 | 356 | OM105009 |
| Picobirnavirus dog/KNA/2015—segment 2 | BSF1 | 1594 | OM105010 |
| Chicken picobirnavirus—segment 2 | BSF1 | 1496 | OM105011 |
| Picobirnavirus dog/KNA/2015—segment 2 | BSF2 | 1651 | OM105012 |
| Otarine picobirnavirus—segment 2 | BSF2 | 1655 | OM105013 |
| Picobirnavirus sp. Roe deer—segment 2 | BSF2 | 1644 | OM105014 |
| Porcine picobirnavirus—segment S | BSF2 | 1669 | OM105015 |
| Picobirnavirus green monkey/KNA/2015—segment 2 | BSF2 | 679 | OM105016 |
| Chicken picobirnavirus—segment 2 | BSF2 | 707 | OM105017 |
| Human picobirnavirus—segment 2 | BSF3 | 1428 | OM105018 |
| Chicken picobirnavirus—segment 2 | BSF3 | 1276 | OM105019 |
| Picobirnavirus dog/KNA/2015—segment 2 | BSF3 | 597 | OM105020 |
| Picobirnavirus green monkey/KNA/2015—segment 2 | BSF3 | 467 | OM105021 |
| Picobirnavirus green monkey/KNA/2015—segment 2 (contig 2) | BSF3 | 611 | OM105022 |
| Porcine picobirnavirus—segment S | BSF3 | 748 | OM105023 |
| Human picobirnavirus—segment 2 (contig 2) | BSF3 | 574 | OM105024 |
| Sapporo virus | BSF2 | 7290 | OM105025 |
| Tottorivirus A | BSF2 | 6746 | OM105026 |
| Pasivirus A | BSF2 | 6876 | OM105027 |
| Porcine enterovirus 9 | BSF2 | 7351 | OM105028 |
| Teschovirus A | BSF2 | 7001 | OM105029 |
| Porcine sapelovirus 1 | BSF2 | 7473 | OM105030 |
| Picornavirales Bu-1 | BSF2 | 8877 | OM105031 |
| Porcine astrovirus 4 | BSF2 | 4508 | OM105032 |
| Astrovirus wild boar/WBAstV-1/2011/HUN | BSF2 | 4451 | OM105033 |
| Dromedary astrovirus | BSF2 | 2580 | OM105034 |
| Mamastrovirus 3 | BSF2 | 6562 | OM105035 |
| Rotavirus A—segment 1 | BSF2 | 1388 | OM105036 |
| Rotavirus A—segment 2 | BSF2 | 1054 | OM105037 |
| Rotavirus A—segment 3 | BSF2 | 2552 | OM105038 |
| Rotavirus A—segment 4 | BSF2 | 734 | OM105039 |
| Rotavirus A—segment 5 | BSF2 | 599 | OM105040 |
| Rotavirus A—segment 6 | BSF2 | 689 | OM105041 |
| Rotavirus A—segment 7 | BSF2 | 999 | OM105042 |
| Rotavirus A—segment 8 | BSF2 | 990 | OM105043 |
| Rotavirus A—segment 9 | BSF2 | 1024 | OM105044 |
| Rotavirus A—segment 10 | BSF2 | 485 | OM105045 |
| Rotavirus A—segment 11 | BSF2 | 585 | OM105046 |
| Mamastrovirus 2 | BSF1 | 3401 | OM105047 |
| Porcine astrovirus 2 | BSF1 | 6364 | OM105048 |
| Posavirus 1 | BSF2 | 9789 | OM105049 |
| Teschovirus A | BSF3 | 7078 | OM966657 |
| Porcine astrovirus 2 | BSF2 | 6313 | OM966722 |
| Porcine astrovirus 2 | BSF3 | 6127 | OM966723 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chauhan, R.P.; San, J.E.; Gordon, M.L. Metagenomic Analysis of RNA Fraction Reveals the Diversity of Swine Oral Virome on South African Backyard Swine Farms in the uMgungundlovu District of KwaZulu-Natal Province. Pathogens 2022, 11, 927. https://doi.org/10.3390/pathogens11080927
Chauhan RP, San JE, Gordon ML. Metagenomic Analysis of RNA Fraction Reveals the Diversity of Swine Oral Virome on South African Backyard Swine Farms in the uMgungundlovu District of KwaZulu-Natal Province. Pathogens. 2022; 11(8):927. https://doi.org/10.3390/pathogens11080927
Chicago/Turabian StyleChauhan, Ravendra P., James E. San, and Michelle L. Gordon. 2022. "Metagenomic Analysis of RNA Fraction Reveals the Diversity of Swine Oral Virome on South African Backyard Swine Farms in the uMgungundlovu District of KwaZulu-Natal Province" Pathogens 11, no. 8: 927. https://doi.org/10.3390/pathogens11080927
APA StyleChauhan, R. P., San, J. E., & Gordon, M. L. (2022). Metagenomic Analysis of RNA Fraction Reveals the Diversity of Swine Oral Virome on South African Backyard Swine Farms in the uMgungundlovu District of KwaZulu-Natal Province. Pathogens, 11(8), 927. https://doi.org/10.3390/pathogens11080927