
CD4+ Cytotoxic T Cells Involved in the Development of EBV-Associated Diseases


Abstract
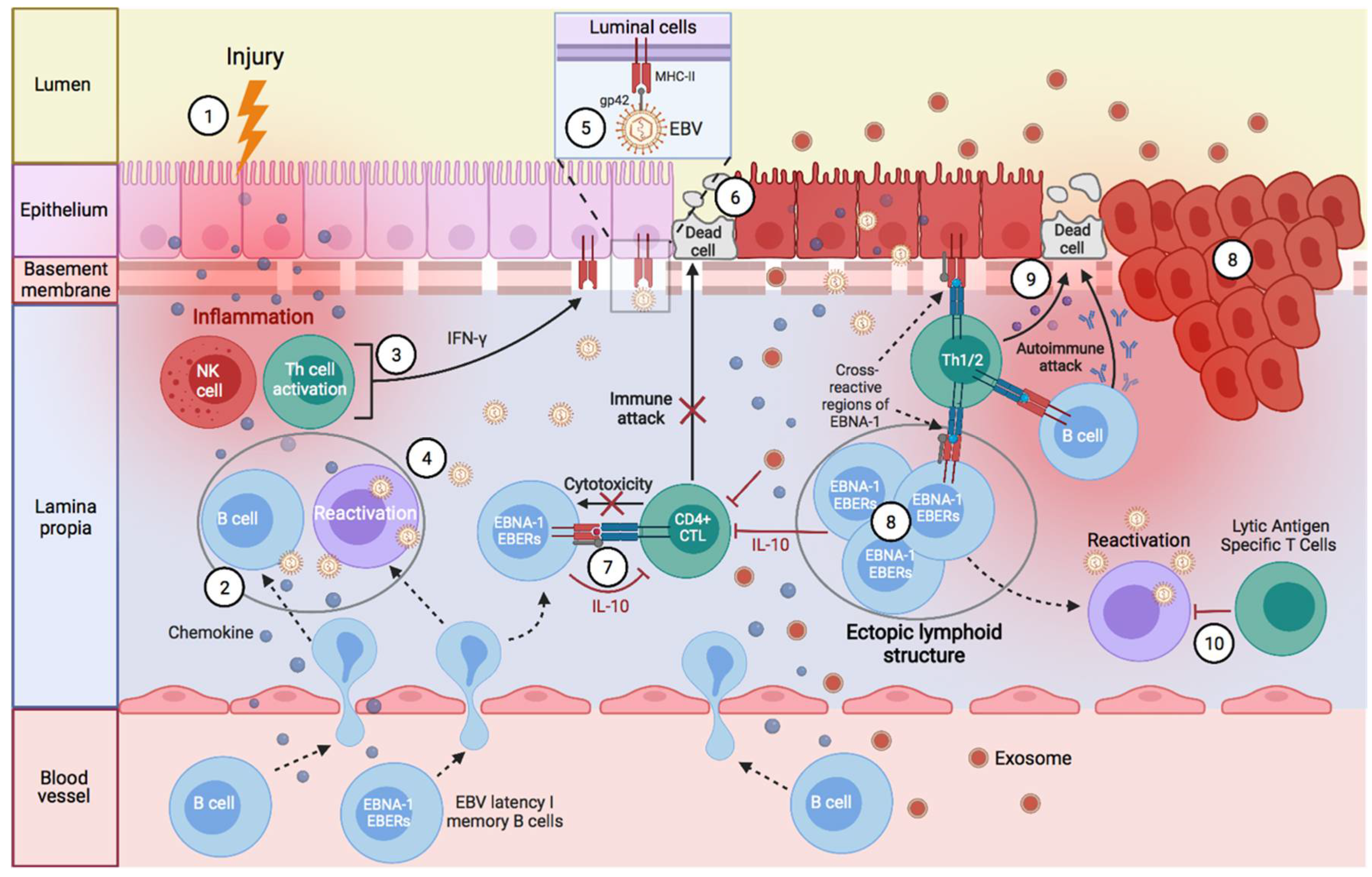
1. Introduction
Factors Predisposing towards the Development of EBV-Associated Diseases
2. EBV-Associated Diseases
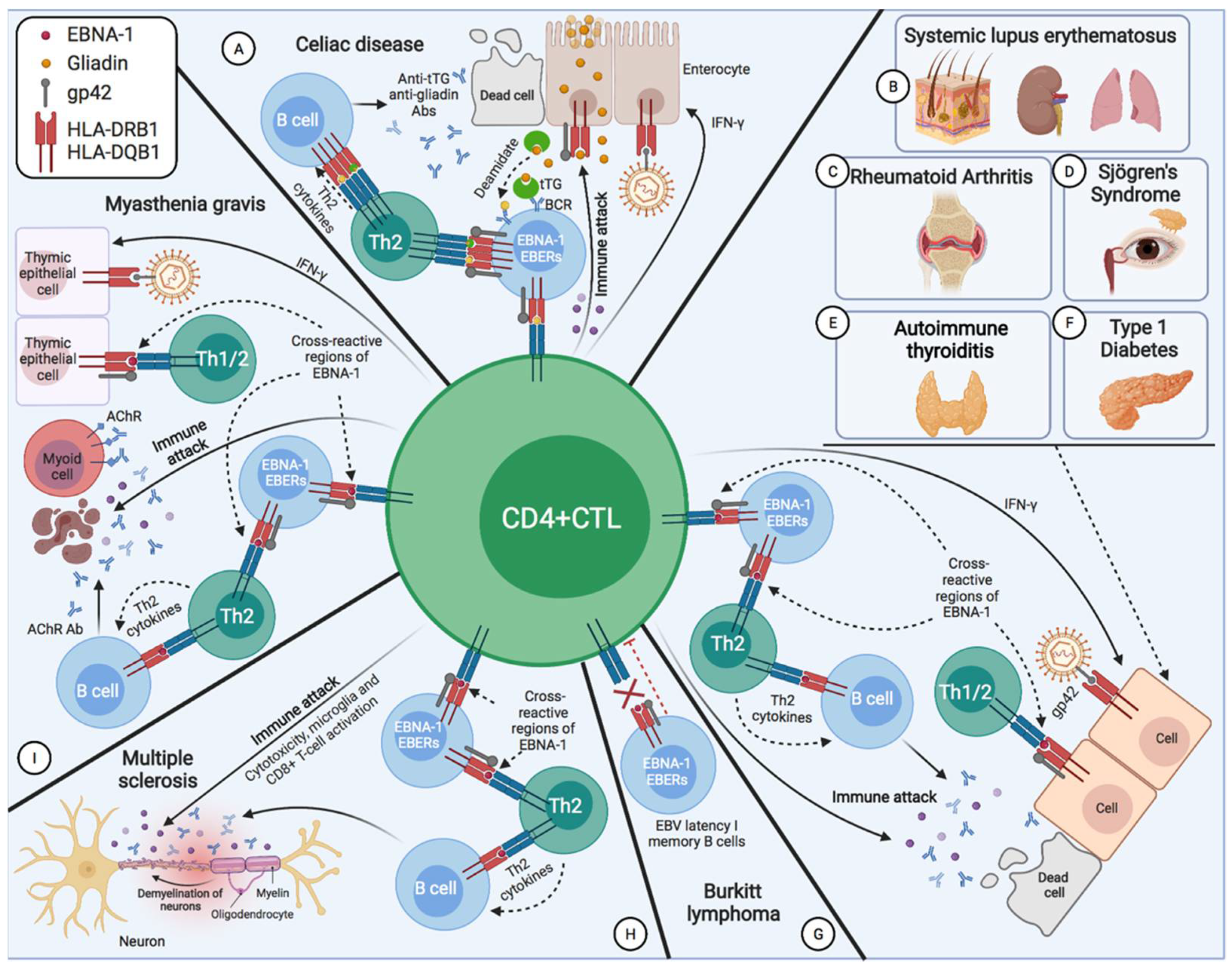
2.1. Burkitt’s Disease
2.2. X-Linked Lymphoproliferative Disease
2.3. Systemic Lupus Erythematosus
2.4. Sjögren’s Syndrome
2.5. Multiple Sclerosis
2.6. Myasthenia Gravis
2.7. Rheumatoid Arthritis
2.8. Type 1 Diabetes Mellitus
2.9. Fulminant Type Diabetes
2.10. Celiac Disease
2.11. Autoimmune Thyroiditis
3. Discussion
4. Conclusions and Future Directions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tian, Y.; Sette, A.; Weiskopf, D. Cytotoxic CD4 T Cells: Differentiation, Function, and Application to Dengue Virus Infection. Front. Immunol. 2016, 7, 531. [Google Scholar] [CrossRef] [PubMed]
- Marshall, N.B.; Swain, S.L. Cytotoxic CD4 T Cells in Antiviral Immunity. J. Biomed. Biotechnol. 2011, 2011, 954602. [Google Scholar] [CrossRef]
- Hintzen, R.Q.; de Jong, R.; Lens, S.M.; Brouwer, M.; Baars, P.; van Lier, R.A. Regulation of CD27 Expression on Subsets of Mature T-Lymphocytes. J. Immunol. 1993, 151, 2426–2435. Available online: https://pubmed.ncbi.nlm.nih.gov/7689607/ (accessed on 8 January 2021). [PubMed]
- Globerson, A.; Effros, R.B. Ageing of Lymphocytes and Lymphocytes in the Aged. Immunol. Today 2000, 21, 515–521. [Google Scholar] [CrossRef]
- van de Berg, P.J.; van Leeuwen, E.M.; ten Berge, I.J.; van Lier, R. Cytotoxic Human CD4+ T Cells. Curr. Opin. Immunol. 2008, 20, 339–343. [Google Scholar] [CrossRef] [PubMed]
- Libri, V.; Azevedo, R.I.; Jackson, S.E.; Di Mitri, D.; Lachmann, R.; Fuhrmann, S.; Vukmanovic-Stejic, M.; Yong, K.; Battistini, L.; Kern, F.; et al. Cytomegalovirus Infection Induces the Accumulation of Short-Lived, Multifunctional CD4+ CD45RA+ CD27− T Cells: The Potential Involvement of Interleukin-7 in This Process. Immunology 2011, 132, 326–339. [Google Scholar] [CrossRef]
- Bruns, H.; Meinken, C.; Schauenberg, P.; Härter, G.; Kern, P.; Modlin, R.L.; Antoni, C.; Stenger, S. Anti-TNF Immunotherapy Reduces CD8+ T Cell-Mediated Antimicrobial Activity against Mycobacterium Tuberculosis in Humans. J. Clin. Investig. 2009, 119, 1167–1177. [Google Scholar] [CrossRef]
- Muraro, E.; Merlo, A.; Martorelli, D.; Cangemi, M.; Santa, S.D.; Dolcetti, R.; Rosato, A. Fighting Viral Infections and Virus-Driven Tumors with Cytotoxic CD4+ T Cells. Front. Immunol. 2017, 8, 197. [Google Scholar] [CrossRef]
- Appay, V.; Zaunders, J.J.; Papagno, L.; Sutton, J.; Jaramillo, A.; Waters, A.; Easterbrook, P.; Grey, P.; Smith, D.; McMichael, A.J.; et al. Characterization of CD4+ CTLs Ex Vivo. J. Immunol. 2002, 168, 5954–5958. [Google Scholar] [CrossRef]
- Mucida, D.; Husain, M.M.; Muroi, S.; Van Wijk, F.; Shinnakasu, R.; Naoe, Y.; Reis, B.S.; Huang, Y.; Lambolez, F.; Docherty, M.; et al. Transcriptional Reprogramming of Mature CD4+ Helper T Cells Generates Distinct MHC Class II-Restricted Cytotoxic T Lymphocytes. Nat. Immunol. 2013, 14, 281–289. [Google Scholar] [CrossRef]
- Mahnke, Y.D.; Brodie, T.M.; Sallusto, F.; Roederer, M.; Lugli, E. The Who’s Who of T-Cell Differentiation: Human Memory T-Cell Subsets. Eur. J. Immunol. 2013, 43, 2797–2809. [Google Scholar] [CrossRef] [PubMed]
- Cheroutre, H.; Husain, M.M. CD4 CTL: Living up to the Challenge. Semin. Immunol. 2013, 25, 273–281. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Perdomo, M.F.; Kantele, A.; Hedman, L.; Hedman, K.; Franssila, R. Granzyme B Mediated Function of Parvovirus B19-Specific CD4+ T Cells. Clin. Transl. Immunol. 2015, 4, e39. [Google Scholar] [CrossRef] [PubMed]
- Ressing, M.E.; Horst, D.; Griffin, B.D.; Tellam, J.; Zuo, J.; Khanna, R.; Rowe, M.; Wiertz, E.J. Epstein-Barr Virus Evasion of CD8+ and CD4+ T Cell Immunity via Concerted Actions of Multiple Gene Products. Semin. Cancer Biol. 2008, 18, 397–408. [Google Scholar] [CrossRef] [PubMed]
- Hatton, O.L.; Harris-Arnold, A.; Schaffert, S.; Krams, S.M.; Martinez, O.M. The Interplay between Epstein-Barr Virus and B Lymphocytes: Implications for Infection, Immunity, and Disease. Immunol. Res. 2014, 58, 268–276. [Google Scholar] [CrossRef]
- Mbiribindi, B.; Pena, J.K.; Arvedson, M.P.; Moreno Romero, C.; McCarthy, S.R.; Hatton, O.L.; Esquivel, C.O.; Martinez, O.M.; Krams, S.M. Epstein–Barr Virus Peptides Derived from Latent Cycle Proteins Alter NKG2A + NK Cell Effector Function. Sci. Rep. 2020, 10, 19973. [Google Scholar] [CrossRef]
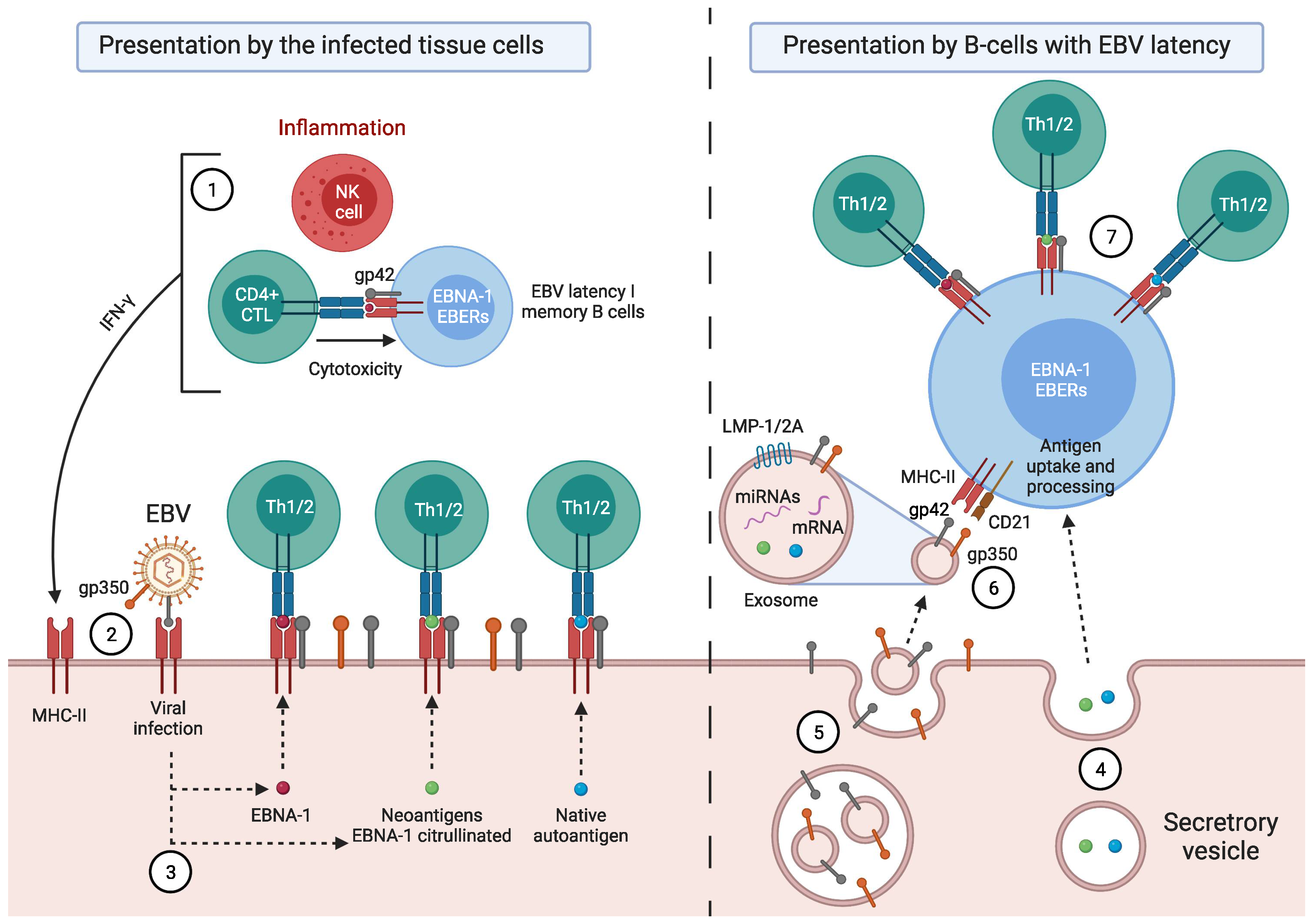
- Münz, C.; Bickham, K.L.; Subklewe, M.; Tsang, M.L.; Chahroudi, A.; Kurilla, M.G.; Zhang, D.; O’Donnell, M.; Steinman, R.M. Human CD4+ T Lymphocytes Consistently Respond to the Latent Epstein- Barr Virus Nuclear Antigen EBNA1. J. Exp. Med. 2000, 191, 1649–1660. [Google Scholar] [CrossRef]
- Tsao, S.W.; Tsang, C.M.; Lo, K.W. Epstein-Barr Virus Infection and Nasopharyngeal Carcinoma. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2017, 372, 20160270. [Google Scholar] [CrossRef]
- Bhattacharjee, S.; Bose, P.; Patel, K.; Roy, S.G.; Gain, C.; Gowda, H.; Robertson, E.S.; Saha, A. Transcriptional and Epigenetic Modulation of Autophagy Promotes EBV Oncoprotein EBNA3C Induced B-Cell Survival Article. Cell Death Dis. 2018, 9, 605. [Google Scholar] [CrossRef]
- You, L.; Jin, S.; Zhu, L.; Qian, W. Autophagy, Autophagy-Associated Adaptive Immune Responses and Its Role in Hematologic Malignancies. Oncotarget 2017, 8, 12374–12388. [Google Scholar] [CrossRef]
- Leung, C.S.; Haigh, T.A.; Mackay, L.K.; Rickinson, A.B.; Taylor, G.S. Nuclear Location of an Endogenously Expressed Antigen, EBNA1, Restricts Access to Macroautophagy and the Range of CD4 Epitope Display. Proc. Natl. Acad. Sci. USA 2010, 107, 2165–2170. [Google Scholar] [CrossRef] [PubMed]
- Münz, C. Antigen Processing for MHC Class II Presentation via Autophagy. Front. Immunol. 2012, 3, 9. [Google Scholar] [CrossRef]
- Trier, N.; Izarzugaza, J.; Chailyan, A.; Marcatili, P.; Houen, G. Human MHC-II with Shared Epitope Motifs Are Optimal Epstein-Barr Virus Glycoprotein 42 Ligands—Relation to Rheumatoid Arthritis. Int. J. Mol. Sci. 2018, 19, 317. [Google Scholar] [CrossRef] [PubMed]
- Fingeroth, J.D.; Clabby, M.L.; Strominger, J.D. Characterization of a T-Lymphocyte Epstein-Barr Virus/C3d Receptor (CD21). J. Virol. 1988, 62, 1442–1447. [Google Scholar] [CrossRef] [PubMed]
- Fingeroth, J.D.; Weis, J.J.; Tedder, T.F.; Strominger, J.L.; Biro, P.A.; Fearon, D.T. Epstein-Barr Virus Receptor of Human B Lymphocytes Is the C3d Receptor CR2. Proc. Natl. Acad. Sci. USA 1984, 81, 4510–4514. [Google Scholar] [CrossRef]
- Tanner, J.; Weis, J.; Fearon, D.; Whang, Y.; Kieff, E. Epstein-Barr Virus Gp350/220 Binding to the B Lymphocyte C3d Receptor Mediates Adsorption, Capping, and Endocytosis. Cell 1987, 50, 203–213. [Google Scholar] [CrossRef]
- Ressing, M.E.; Van Leeuwen, D.; Verreck, F.A.W.; Gomez, R.; Heemskerk, B.; Toebes, M.; Mullen, M.M.; Jardetzky, T.S.; Longnecker, R.; Schilham, M.W.; et al. Interference with T Cell Receptor-HLA-DR Interactions by Epstein-Barr Virus Gp42 Results in Reduced T Helper Cell Recognition. Proc. Natl. Acad. Sci. USA 2003, 100, 11583–11588. [Google Scholar] [CrossRef]
- Spriggs, M.K.; Armitage, R.J.; Comeau, M.R.; Strockbine, L.; Farrah, T.; Macduff, B.; Ulrich, D.; Alderson, M.R.; Müllberg, J.; Cohen, J.I. The Extracellular Domain of the Epstein-Barr Virus BZLF2 Protein Binds the HLA-DR Beta Chain and Inhibits Antigen Presentation. J. Virol. 1996, 70, 5557–5563. [Google Scholar] [CrossRef]
- Ressing, M.E.; van Leeuwen, D.; Verreck, F.A.W.; Keating, S.; Gomez, R.; Franken, K.L.M.C.; Ottenhoff, T.H.M.; Spriggs, M.; Schumacher, T.N.; Hutt-Fletcher, L.M.; et al. Epstein-Barr Virus Gp42 Is Posttranslationally Modified to Produce Soluble Gp42 That Mediates HLA Class II Immune Evasion. J. Virol. 2005, 79, 841–852. [Google Scholar] [CrossRef]
- Mullen, M.M.; Haan, K.M.; Longnecker, R.; Jardetzky, T.S. Structure of the Epstein-Barr Virus Gp42 Protein Bound to the MHC Class II Receptor HLA-DR1. Mol. Cell 2002, 9, 375–385. [Google Scholar] [CrossRef]
- Zuo, J.; Rowe, M. Herpesviruses Placating the Unwilling Host: Manipulation of the MHC Class II Antigen Presentation Pathway. Viruses 2012, 4, 1335–1353. [Google Scholar] [CrossRef] [PubMed]
- Bontrop, R.E.; Otting, N.; De Groot, N.G.; Doxiadis, G.G.M. Major Histocompatibility Complex Class II Polymorphisms in Primates. Immunol. Rev. 1999, 167, 339–350. [Google Scholar] [CrossRef]
- McShane, M.P.; Mullen, M.M.; Haan, K.M.; Jardetzky, T.S.; Longnecker, R. Mutational Analysis of the HLA Class II Interaction with Epstein-Barr Virus Glycoprotein 42. J. Virol. 2003, 77, 7655–7662. [Google Scholar] [CrossRef] [PubMed]
- Haan, K.M.; Longnecker, R. Coreceptor Restriction within the HLA-DQ Locus for Epstein-Barr Virus Infection. Proc. Natl. Acad. Sci. USA 2000, 97, 9252–9257. [Google Scholar] [CrossRef]
- Al-Motwee, S.; Jawdat, D.; Jehani, G.S.; Anazi, H.; Shubaili, A.; Sutton, P.; Uyar, A.F.; Hajeer, A.H. Association of HLA-DRB1*15 and HLA-DQB1* 06 with SLE in Saudis. Ann. Saudi Med. 2013, 33, 229–234. [Google Scholar] [CrossRef] [PubMed]
- Liphaus, B.L.; Kiss, M.H.B.; Goldberg, A.C. HLA-DRB1 Alleles in Juvenile-Onset Systemic Lupus Erythematosus: Renal Histologic Class Correlations. Braz. J. Med. Biol. Res. 2007, 40, 591–597. [Google Scholar] [CrossRef]
- Shimane, K.; Kochi, Y.; Suzuki, A.; Okada, Y.; Ishii, T.; Horita, T.; Saito, K.; Okamoto, A.; Nishimoto, N.; Myouzen, K.; et al. An Association Analysis of HLA-DRB1 with Systemic Lupus Erythematosus and Rheumatoid Arthritis in a Japanese Population: Effects of *09:01 Allele on Disease Phenotypes. Rheumatology 2013, 52, 1172–1182. [Google Scholar] [CrossRef]
- Morris, D.L.; Taylor, K.E.; Fernando, M.M.A.; Nititham, J.; Alarcón-Riquelme, M.E.; Barcellos, L.F.; Behrens, T.W.; Cotsapas, C.; Gaffney, P.M.; Graham, R.R.; et al. Unraveling Multiple MHC Gene Associations with Systemic Lupus Erythematosus: Model Choice Indicates a Role for HLA Alleles and Non-HLA Genes in Europeans. Am. J. Hum. Genet. 2012, 91, 778–793. [Google Scholar] [CrossRef]
- Cobb, B.L.; Lessard, C.J.; Harley, J.B.; Moser, K.L. Genes and Sjögren’s Syndrome. Rheum. Dis. Clin. N. Am. 2008, 34, 847–868. [Google Scholar] [CrossRef]
- Guggenbuhl, P.; Jean, S.; Jego, P.; Grosbois, B.; Chalès, G.; Semana, G.; Lancien, G.; Veillard, E.; Pawlotsky, Y.; Perdriger, A. Primary Sjogren’s Syndrome: Role of the HLA-DRB1 0301-1501 Heterozygotes. J. Rheumatol. 1998, 25, 900–905. Available online: https://europepmc.org/article/med/9598888 (accessed on 8 January 2021).
- Lünemann, J.D.; Jelčić, I.; Roberts, S.; Lutterotti, A.; Tackenberg, B.; Martin, R.; Münz, C. EBNA1-Specific T Cells from Patients with Multiple Sclerosis Cross React with Myelin Antigens and Co-Produce IFN-γ and IL-2. J. Exp. Med. 2008, 205, 1763–1773. [Google Scholar] [CrossRef] [PubMed]
- Hollenbach, J.A.; Oksenberg, J.R. The Immunogenetics of Multiple Sclerosis: A Comprehensive Review. J. Autoimmun. 2015, 64, 13–25. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.; Fritz, E.L.; Kerr, J.R.; Cleare, A.J.; Wessely, S.; Mattey, D.L. Association of Chronic Fatigue Syndrome with Human Leucocyte Antigen Class II Alleles. J. Clin. Pathol. 2005, 58, 860–863. [Google Scholar] [CrossRef] [PubMed]
- Underhill, J.A.; Mahalingam, M.; Peakman, M.; Wessely, S. Lack of Association between HLA Genotype and Chronic Fatigue Syndrome. Eur. J. Immunogenet. 2001, 28, 425–428. [Google Scholar] [CrossRef]
- Maniaol, A.H.; Elsais, A.; Lorentzen, Å.R.; Owe, J.F.; Viken, M.K.; Sæther, H.; Flåm, S.T.; Bråthen, G.; Kampman, M.T.; Midgard, R.; et al. Late Onset Myasthenia Gravis Is Associated with HLA DRB1*15:01 in the Norwegian Population. PLoS ONE 2012, 7, e36603. [Google Scholar] [CrossRef]
- Misra, M.K.; Damotte, V.; Hollenbach, J.A. The Immunogenetics of Neurological Disease. Immunology 2018, 153, 399–414. [Google Scholar] [CrossRef]
- Fujiya, A.; Ochiai, H.; Mizukoshi, T.; Kiyota, A.; Shibata, T.; Suzuki, A.; Ohashi, N.; Sobajima, H. Fulminant Type 1 Diabetes Mellitus Associated with a Reactivation of Epstein-Barr Virus That Developed in the Course of Chemotherapy of Multiple Myeloma. J. Diabetes Investig. 2010, 1, 286–289. [Google Scholar] [CrossRef]
- McAulay, K.A.; Jarrett, R.F. Human Leukocyte Antigens and Genetic Susceptibility to Lymphoma. Tissue Antigens 2015, 86, 98–113. [Google Scholar] [CrossRef] [PubMed]
- Klitz, W.; Aldrich, C.L.; Fildes, N.; Horning, S.J.; Begovich, A.B. Localization of Predisposition to Hodgkin Disease in the HLA Class II Region. Am. J. Hum. Genet. 1994, 54, 497. Available online: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC1918115/ (accessed on 11 April 2022). [PubMed]
- Harty, L.C.; Lin, A.Y.; Goldstein, A.M.; Jaffe, E.S.; Carrington, M.; Tucker, M.A.; Modi, W.S. HLA-DR, HLA-DQ, and TAP Genes in Familial Hodgkin Disease. Blood 2002, 99, 690–693. [Google Scholar] [CrossRef]
- Huang, X.; Kushekhar, K.; Nolte, I.; Kooistra, W.; Visser, L.; Bouwman, I.; Kouprie, N.; Veenstra, R.; van Imhoff, G.; Olver, B.; et al. HLA Associations in Classical Hodgkin Lymphoma: EBV Status Matters. PLoS ONE 2012, 7, e39986. [Google Scholar] [CrossRef] [PubMed]
- Noble, J.A.; Valdes, A.M. Genetics of the HLA Region in the Prediction of Type 1 Diabetes. Curr. Diab. Rep. 2011, 11, 533–542. [Google Scholar] [CrossRef] [PubMed]
- Badenhoop, K.; Walfish, P.G.; Rau, H.; Fischer, S.; Nicolay, A.; Bogner, U.; Schleusener, H.; Usadel, K.H. Susceptibility and Resistance Alleles of Human Leukocyte Antigen (HLA) DQA1 and HLA DQB1 Are Shared in Endocrine Autoimmune Disease. J. Clin. Endocrinol. Metab. 1995, 80, 2112–2117. [Google Scholar] [CrossRef] [PubMed]
- Luckey, D.; Bastakoty, D.; Mangalam, A.K. Role of HLA Class II Genes in Susceptibility and Resistance to Multiple Sclerosis: Studies Using HLA Transgenic Mice. J. Autoimmun. 2011, 37, 122–128. [Google Scholar] [CrossRef]
- Pociot, F.; Lernmark, Å. Genetic Risk Factors for Type 1 Diabetes. Lancet 2016, 387, 2331–2339. [Google Scholar] [CrossRef]
- Burrack, A.L.; Martinov, T.; Fife, B.T. T Cell-Mediated Beta Cell Destruction: Autoimmunity and Alloimmunity in the Context of Type 1 Diabetes. Front. Endocrinol. 2017, 8, 343. [Google Scholar] [CrossRef]
- Camarca, M.E.; Mozzillo, E.; Nugnes, R.; Zito, E.; Falco, M.; Fattorusso, V.; Mobilia, S.; Buono, P.; Valerio, G.; Troncone, R.; et al. Celiac Disease in Type 1 Diabetes Mellitus. Ital. J. Pediatr. 2012, 38, 10. [Google Scholar] [CrossRef]
- Smigoc Schweiger, D.; Mendez, A.; Kunilo Jamnik, S.; Bratanic, N.; Bratina, N.; Battelino, T.; Brecelj, J.; Vidan-Jeras, B. High-Risk Genotypes HLA-DR3-DQ2/DR3-DQ2 and DR3-DQ2/DR4-DQ8 in Co-Occurrence of Type 1 Diabetes and Celiac Disease. Autoimmunity 2016, 49, 240–247. [Google Scholar] [CrossRef]
- Liu, E.; Lee, H.-S.; Aronsson, C.A.; Hagopian, W.A.; Koletzko, S.; Rewers, M.J.; Eisenbarth, G.S.; Bingley, P.J.; Bonifacio, E.; Simell, V.; et al. Risk of Pediatric Celiac Disease According to HLA Haplotype and Country. N. Engl. J. Med. 2014, 371, 42–49. [Google Scholar] [CrossRef]
- El-Ahwal, L.; AbdEL-Bar, E. The Frequency of HLA DRB1-DQB1 Alleles in Autoimmune Type 1 Diabetes with or without Autoimmune Thyroid Disease. Tanta Med. J. 2015, 43, 66. [Google Scholar] [CrossRef]
- Zeitlin, A.A.; Heward, J.M.; Newby, P.R.; Carr-Smith, J.D.; Franklyn, J.A.; Gough, S.C.L.; Simmonds, M.J. Analysis of HLA Class II Genes in Hashimoto’s Thyroiditis Reveals Differences Compared to Graves’ Disease. Genes Immun. 2008, 9, 358–363. [Google Scholar] [CrossRef] [PubMed]
- Zagoriti, Z.; Kambouris, M.E.; Patrinos, G.P.; Tzartos, S.J.; Poulas, K. Recent Advances in Genetic Predisposition of Myasthenia Gravis. BioMed Res. Int. 2013, 2013, 404053. [Google Scholar] [CrossRef] [PubMed]
- Hjelmström, P.; Giscombe, R.; Lefvert, A.K.; Pirskanen, R.; Kockum, I.; Landin-Olsson, M.; Sanjeevi, C.B. Different HLA-DQ Are Positively and Negatively Associated in Swedish Patients with Myasthenia Gravis. Autoimmunity 1995, 22, 59–65. [Google Scholar] [CrossRef] [PubMed]
- Chemin, K.; Gerstner, C.; Malmström, V. Effector Functions of CD4+ T Cells at the Site of Local Autoimmune Inflammation-Lessons from Rheumatoid Arthritis. Front. Immunol. 2019, 10, 353. [Google Scholar] [CrossRef]
- Voskuhl, R. Sex Differences in Autoimmune Diseases. Biol. Sex Differ. 2011, 2, 1. [Google Scholar] [CrossRef]
- Ngo, S.T.; Steyn, F.J.; McCombe, P.A. Gender Differences in Autoimmune Disease. Front. Neuroendocrinol. 2014, 35, 347–369. [Google Scholar] [CrossRef]
- Azar, S.T.; Tamim, H.; Beyhum, H.N.; Zouhair Habbal, M.; Almawi, W.Y. Type I (Insulin-Dependent) Diabetes Is a Th1- and Th2-Mediated Autoimmune Disease. Clin. Diagn. Lab. Immunol. 1999, 6, 306–310. [Google Scholar] [CrossRef]
- Moldovan, I.R.; Cotleur, A.C.; Zamor, N.; Butler, R.S.; Pelfrey, C.M. Multiple Sclerosis Patients Show Sexual Dimorphism in Cytokine Responses to Myelin Antigens. J. Neuroimmunol. 2008, 193, 161–169. [Google Scholar] [CrossRef]
- Bao, M.; Yang, Y.; Jun, H.-S.; Yoon, J.-W. Molecular Mechanisms for Gender Differences in Susceptibility to T Cell-Mediated Autoimmune Diabetes in Nonobese Diabetic Mice. J. Immunol. 2002, 168, 5369–5375. [Google Scholar] [CrossRef]
- Vaseghi, H.; Jadali, Z. Th1/Th2 Cytokines in Type 1 Diabetes: Relation to Duration of Disease and Gender. Indian J. Endocrinol. Metab. 2016, 20, 312–316. [Google Scholar] [CrossRef]
- Silverman, M.N.; Pearce, B.D.; Biron, C.A.; Miller, A.H. Immune Modulation of the Hypothalamic-Pituitary-Adrenal (HPA) Axis during Viral Infection. Viral Immunol. 2005, 18, 41–78. [Google Scholar] [CrossRef] [PubMed]
- López-Montañés, M.; Alari-Pahissa, E.; Sintes, J.; Martínez-Rodríguez, J.E.; Muntasell, A.; López-Botet, M. Antibody-Dependent NK Cell Activation Differentially Targets EBV-Infected Cells in Lytic Cycle and Bystander B Lymphocytes Bound to Viral Antigen–Containing Particles. J. Immunol. 2017, 199, 656–665. [Google Scholar] [CrossRef] [PubMed]
- Paludan, C.; Schmid, D.; Landthaler, M.; Vockerodt, M.; Kube, D.; Tuschl, T.; Münz, C. Endogenous MHC Class II Processing of a Viral Nuclear Antigen After Autophagy. Science 2005, 307, 593–596. [Google Scholar] [CrossRef]
- Fu, T.; Kui, S.V.; Wang, R.F. Critical Role of EBNA1-Specific CD4+ T Colls in the Control of Mouse Burkitt Lymphoma in Vivo. J. Clin. Investig. 2004, 114, 542–550. [Google Scholar] [CrossRef]
- Futagbi, G.; Gyan, B.; Nunoo, H.; Tetteh, J.; Welbeck, J.; Renner, L.; Ofori, M.; Dodoo, D.; Edoh, D.; Akanmori, B. High Levels of IL-10 and CD4+ CD25hi+ Treg Cells in Endemic Burkitt’s Lymphoma Patients. Biomedicines 2015, 3, 224–236. [Google Scholar] [CrossRef] [PubMed]
- Roncarolo, M.G.; Bacchetta, R.; Bordignon, C.; Narula, S.; Levings, M.K. Type 1 T Regulatory Cells. Immunol. Rev. 2001, 182, 68–79. [Google Scholar] [CrossRef]
- Burdin, N.; Péronne, C.; Banchereau, J.; Rousset, F. Epstein-Barr Virus Transformation Induces B Lymphocytes to Produce Human Interleukin 10. J. Exp. Med. 1993, 177, 295–304. [Google Scholar] [CrossRef]
- Veronese, M.L.; Veronesi, A.; D’andrea, E.; Del Mistro, A.; Indraccolo, S.; Mazza, M.R.; Mion, M.; Zamarchi, R.; Menin, C.; Panozzo, M.; et al. Lymphoproliferative Disease in Human Peripheral Blood Mononuclear Cell-Injected SCID Mice. I. T Lymphocyte Requirement for B Cell Tumor Generation. J. Exp. Med. 1992, 176, 1763–1767. [Google Scholar] [CrossRef]
- MacArthur, G.J.; Wilson, A.D.; Birchall, M.A.; Morgan, A.J. Primary CD4+ T-Cell Responses Provide Both Helper and Cytotoxic Functions during Epstein-Barr Virus Infection and Transformation of Fetal Cord Blood B Cells. J. Virol. 2007, 81, 4766–4775. [Google Scholar] [CrossRef]
- God, J.M.; Zhao, D.; Cameron, C.A.; Amria, S.; Bethard, J.R.; Haque, A. Disruption of HLA Class II Antigen Presentation in Burkitt Lymphoma: Implication of a 47 000 MW Acid Labile Protein in CD4+ T-Cell Recognition. Immunology 2014, 142, 492–505. [Google Scholar] [CrossRef]
- Hossain, A.; God, J.M.; Radwan, F.F.Y.; Amria, S.; Zhao, D.; Bethard, J.R.; Haque, A. HLA Class II Defects in Burkitt Lymphoma: Bryostatin-1-Induced 17kDa Protein Restores CD4+ T-Cell Recognition. Clin. Dev. Immunol. 2011, 2011, 780839. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Burton, E.M.; Bhaduri-McIntosh, S. Chloroquine Triggers Epstein-Barr Virus Replication through Phosphorylation of KAP1/TRIM28 in Burkitt Lymphoma Cells. PLoS Pathog. 2017, 13, e1006249. [Google Scholar] [CrossRef]
- Karmali, R.A.; Horrobin, D.F.; Menezes, J.; Patel, P.; Musto, J. Chloroquine Enhances Epstein-Barr Virus Expression. Nature 1978, 275, 444–445. [Google Scholar] [CrossRef] [PubMed]
- Sadoff, L. Letter: Antimalarial Drugs and Burkitt’s Lymphoma. Lancet 1973, 302, 1262–1263. [Google Scholar] [CrossRef]
- Zhou, D.; Dai, S.-M.; Tong, Q. COVID-19: A Recommendation to Examine the Effect of Hydroxychloroquine in Preventing Infection and Progression. J. Antimicrob. Chemother. 2020, 75, 1667–1670. [Google Scholar] [CrossRef] [PubMed]
- Levine, A.M. Acquired Immunodeficiency Syndrome-Related Lymphoma. Blood 1992, 80, 8–20. [Google Scholar] [CrossRef] [PubMed]
- Saifuddin, M.; Spear, G.T.; Chang, C.H.; Roebuck, K.A. Expression of MHC Class II in T Cells Is Associated with Increased HIV-1 Expression. Clin. Exp. Immunol. 2000, 121, 324–331. [Google Scholar] [CrossRef]
- Bibas, M.; Antinori, A. EBV and HIV-Related Lymphoma. Mediterr. J. Hematol. Infect. Dis. 2009, 1, 2009032. [Google Scholar] [CrossRef]
- Lee, E.; Bacchetti, P.; Milush, J.; Shao, W.; Boritz, E.; Douek, D.; Fromentin, R.; Liegler, T.; Hoh, R.; Deeks, S.G.; et al. Memory CD4+ T-Cells Expressing HLA-DR Contribute to HIV Persistence during Prolonged Antiretroviral Therapy. Front. Microbiol. 2019, 10, 2214. [Google Scholar] [CrossRef]
- Geldmacher, C.; Koup, R.A. Pathogen-Specific T Cell Depletion and Reactivation of Opportunistic Pathogens in HIV Infection. Trends Immunol. 2012, 33, 207–214. [Google Scholar] [CrossRef]
- Shindiapina, P.; Ahmed, E.H.; Mozhenkova, A.; Abebe, T.; Baiocchi, R.A. Immunology of EBV-Related Lymphoproliferative Disease in HIV-Positive Individuals. Front. Oncol. 2020, 10, 1723. [Google Scholar] [CrossRef]
- Cocks, B.G.; Chang, C.C.J.; Carballido, J.M.; Yssel, H.; de Vries, J.E.; Aversa, G. A Novel Receptor Involved in T–Cell Activation. Nature 1995, 376, 260–263. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Satoskar, A.; Faubion, W.; Howie, D.; Okamoto, S.; Feske, S.; Gullo, C.; Clarke, K.; Sosa, M.R.; Sharpe, A.H.; et al. The Cell Surface Receptor SLAM Controls T Cell and Macrophage Functions. J. Exp. Med. 2004, 199, 1255–1264. [Google Scholar] [CrossRef] [PubMed]
- Gaspar, H.B.; Sharifi, R.; Gilmour, K.C.; Thrasher, A.J. X-Linked Lymphoproliferative Disease: Clinical, Diagnostic and Molecular Perspective. Br. J. Haematol. 2002, 119, 585–595. [Google Scholar] [CrossRef] [PubMed]
- Vilar, M.L.L.V.; Frutuoso, M.S.; Arruda, S.M.; Lima, D.M.; Bezerra, C.S.; Pompeu, M.M.L. The Role of the SLAM-SAP Signaling Pathway in the Modulation of CD4+ T Cell Responses. Braz. J. Med. Biol. Res. 2011, 44, 276–282. [Google Scholar] [CrossRef]
- Wu, C.; Nguyen, K.B.; Pien, G.C.; Wang, N.; Gullo, C.; Howie, D.; Sosa, M.R.; Edwards, M.J.; Borrow, P.; Satoskar, A.R.; et al. SAP Controls T Cell Responses to Virus and Terminal Differentiation of TH2 Cells. Nat. Immunol. 2001, 2, 410–414. [Google Scholar] [CrossRef]
- Cannons, J.L.; Yu, L.J.; Hill, B.; Mijares, L.A.; Dombroski, D.; Nichols, K.E.; Antonellis, A.; Koretzky, G.A.; Gardner, K.; Schwartzberg, P.L. SAP Regulates TH2 Differentiation and PKC-θ-Mediated Activation of NF-ΚB1. Immunity 2004, 21, 693–706. [Google Scholar] [CrossRef]
- Chuang, H.C.; Lay, J.D.; Hsieh, W.C.; Wang, H.C.; Chang, Y.; Chuang, S.E.; Su, I.J. Epstein-Barr Virus LMP1 Inhibits the Expression of SAP Gene and Upregulates Th1 Cytokines in the Pathogenesis of Hemophagocytic Syndrome. Blood 2005, 106, 3090–3096. [Google Scholar] [CrossRef]
- Palendira, U.; Low, C.; Chan, A.; Hislop, A.D.; Ho, E.; Phan, T.G.; Deenick, E.; Cook, M.C.; Riminton, D.S.; Choo, S.; et al. Molecular Pathogenesis of EBV Susceptibility in XLP as Revealed by Analysis of Female Carriers with Heterozygous Expression of SAP. PLoS Biol. 2011, 9, e1001187. [Google Scholar] [CrossRef]
- Veillette, A.; Zhang, S.; Shi, X.; Dong, Z.; Davidson, D.; Zhong, M.C. SAP Expression in T Cells, Not in B Cells, Is Required for Humoral Immunity. Proc. Natl. Acad. Sci. USA 2008, 105, 1273–1278. [Google Scholar] [CrossRef]
- Dong, Z.; Veillette, A. How Do SAP Family Deficiencies Compromise Immunity? Trends Immunol. 2010, 31, 295–302. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.; Kim, S.T.; Craft, J. The Pathogenesis of Systemic Lupus Erythematosus-an Update. Curr. Opin. Immunol. 2012, 24, 651–657. [Google Scholar] [CrossRef] [PubMed]
- James, J.A.; Robertson, J.M. Lupus and Epstein-Barr. Curr. Opin. Rheumatol. 2012, 24, 383–388. [Google Scholar] [CrossRef] [PubMed]
- Kang, I.; Quan, T.; Nolasco, H.; Park, S.-H.; Hong, M.S.; Crouch, J.; Pamer, E.G.; Howe, J.G.; Craft, J. Defective Control of Latent Epstein-Barr Virus Infection in Systemic Lupus Erythematosus. J. Immunol. 2004, 172, 1287–1294. [Google Scholar] [CrossRef] [PubMed]
- Carli, L.; Tani, C.; Vagnani, S.; Signorini, V.; Mosca, M. Leukopenia, Lymphopenia, and Neutropenia in Systemic Lupus Erythematosus: Prevalence and Clinical Impact-A Systematic Literature Review. Semin. Arthritis Rheum. 2015, 45, 190–194. [Google Scholar] [CrossRef]
- Gross, A.J.; Hochberg, D.; Rand, W.M.; Thorley-Lawson, D.A. EBV and Systemic Lupus Erythematosus: A New Perspective. J. Immunol. 2005, 174, 6599–6607. [Google Scholar] [CrossRef]
- Draborg, A.H.; Duus, K.; Houen, G. Epstein-Barr Virus and Systemic Lupus Erythematosus. Clin. Dev. Immunol. 2012, 2012, 370516. [Google Scholar] [CrossRef]
- Esen, B.A.; YIlmaz, G.; Uzun, S.; Özdamar, M.; Aksözek, A.; KamalI, S.; Türkoǧlu, S.; Gül, A.; Öcal, L.; Aral, O.; et al. Serologic Response to Epstein-Barr Virus Antigens in Patients with Systemic Lupus Erythematosus: A Controlled Study. Rheumatol. Int. 2012, 32, 79–83. [Google Scholar] [CrossRef]
- Berner, B.R.; Tary-Lehmann, M.; Yonkers, N.L.; Askari, A.D.; Lehmann, P.V.; Anthony, D.D. Phenotypic and Functional Analysis of EBV-Specific Memory CD8 Cells in SLE. Cell. Immunol. 2005, 235, 29–38. [Google Scholar] [CrossRef]
- Cassaniti, I.; Cavagna, L.; Calarota, S.A.; Adzasehoun, K.M.G.; Comolli, G.; Montecucco, C.; Baldanti, F. Evaluation of EBV- and HCMV-Specific T Cell Responses in Systemic Lupus Erythematosus (SLE) Patients Using a Normalized Enzyme-Linked Immunospot (ELISPOT) Assay. J. Immunol. Res. 2019, 2019, 4236503. [Google Scholar] [CrossRef]
- Draborg, A.H.; Jacobsen, S.; Westergaard, M.; Mortensen, S.; Larsen, J.L.; Houen, G.; Duus, K. Reduced Response to Epstein-Barr Virus Antigens by T-Cells in Systemic Lupus Erythematosus Patients. Lupus Sci. Med. 2014, 1, 15. [Google Scholar] [CrossRef] [PubMed]
- Lourenco, E.; Cava, A. Cytokines in Systemic Lupus Erythematosus. Curr. Mol. Med. 2009, 9, 242–254. [Google Scholar] [CrossRef] [PubMed]
- Beebe, A.M.; Cua, D.J.; De Waal Malefyt, R. The Role of Interleukin-10 in Autoimmune Disease: Systemic Lupus Erythematosus (SLE) and Multiple Sclerosis (MS). Cytokine Growth Factor Rev. 2002, 13, 403–412. [Google Scholar] [CrossRef]
- Llorente, L.; Richaud-Patin, Y.; Fior, R.; Alcocer-Varela, J.; Wijdenes, J.; Fourrier, B.M.; Galanaud, P.; Emilie, D. In Vivo Production of Interleukin-10 by Non–t Cells in Rheumatoid Arthritis, Sjöugren’s Syndrome, and Systemic Lupus Erythematosus. Arthritis Rheum. 1994, 37, 1647–1655. [Google Scholar] [CrossRef] [PubMed]
- Chun, H.Y.; Chung, J.W.; Kim, H.A.; Yun, J.M.; Jeon, J.Y.; Ye, Y.M.; Kim, S.H.; Park, H.S.; Suh, C.H. Cytokine IL-6 and IL-10 as Biomarkers in Systemic Lupus Erythematosus. J. Clin. Immunol. 2007, 27, 461–466. [Google Scholar] [CrossRef]
- Kammer, G.M. Altered Regulation of IL-2 Production in Systemic Lupus Erythematosus: An Evolving Paradigm. J. Clin. Investig. 2005, 115, 836–840. [Google Scholar] [CrossRef]
- Llorente, L.; Richaud-Patin, Y.; Wijdenes, J.; Alcocer-Varela, J.; Maillot, M.C.; Durand-Gasselin, I.; Fourrier, B.M.; Galanaud, P.; Emilie, D. Spontaneous Production of Interleukin-10 by B Lymphocytes and Monocytes in Systemic Lupus Erythematosus. Eur. Cytokine Netw. 1993, 4, 421–427. Available online: https://pubmed.ncbi.nlm.nih.gov/8186374/ (accessed on 8 January 2021).
- Ooi, J.D.; Kitching, A.R. CD4+ Th1 Cells Are Effectors in Lupus Nephritis—But What Are Their Targets? Kidney Int. 2012, 82, 947–949. [Google Scholar] [CrossRef][Green Version]
- Ribeiro, F.M.; Gomez, V.E.; Albuquerque, E.M.N.; Klumb, E.M.; Shoenfeld, Y. Lupus and Leprosy: Beyond the Coincidence. Immunol. Res. 2014, 61, 160–163. [Google Scholar] [CrossRef]
- Boddu, P.; Mohammed, A.S.; Annem, C.; Sequeira, W. SLE and Non-Hodgkin’s Lymphoma: A Case Series and Review of the Literature. Case Rep. Rheumatol. 2017, 2017, 1658473. [Google Scholar] [CrossRef]
- Bernatsky, S.; Ramsey-Goldman, R.; Labrecque, J.; Joseph, L.; Boivin, J.F.; Petri, M.; Zoma, A.; Manzi, S.; Urowitz, M.B.; Gladman, D.; et al. Cancer Risk in Systemic Lupus: An Updated International Multi-Centre Cohort Study. J. Autoimmun. 2013, 42, 130–135. [Google Scholar] [CrossRef] [PubMed]
- Bernatsky, S.; Ramsey-Goldman, R.; Rajan, R.; Boivin, J.F.; Joseph, L.; Lachance, S.; Cournoyer, D.; Zoma, A.; Manzi, S.; Ginzler, E.; et al. Non-Hodgkin’s Lymphoma in Systemic Lupus Erythematosus. Ann. Rheum. Dis. 2005, 64, 1507–1509. [Google Scholar] [CrossRef] [PubMed]
- Karadeniz, A.; Lally, L.; Magro, C.; Levy, R.; Erkan, D.; Lockshin, M.D. Lepromatous Leprosy Mimicking Systemic Lupus Erythematosus: A Clinical Pathology Conference Held by the Division of Rheumatology at Hospital for Special Surgery. HSS J. 2014, 10, 286–291. [Google Scholar] [CrossRef]
- da Silva, S.A.; Mazini, P.S.; Reis, P.G.; Sell, A.M.; Tsuneto, L.T.; Peixoto, P.R.; Visentainer, J. EL HLA-DR and HLA-DQ Alleles in Patients from the South of Brazil: Markers for Leprosy Susceptibility and Resistance. BMC Infect. Dis. 2009, 9, 134. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Liu, H.; Chen, S.; Wang, C.; Zhu, C.; Zhang, L.; Chu, T.; Liu, D.; Yan, X.; Liu, J. Evidence for an Association of HLA-DRB115 and DRB109 with Leprosy and the Impact of DRB109 on Disease Onset in a Chinese Han Population. BMC Med. Genet. 2009, 10, 133. [Google Scholar] [CrossRef]
- Papageorgiou, P.S.; Sorokin, C.F.; Kouzoutzakoglou, K.; Bonforte, R.J.; Workman, P.L.; Glade, P.R. Host Responses to Epstein Barr Virus and Cytomegalovirus Infection in Leprosy. Infect. Immun. 1973, 7, 620–624. [Google Scholar] [CrossRef]
- Papageorgiou, P.S.; Sorokin, C.; Kouzoutzakoglou, K.; Glade, P.R. Herpes-like Epstein-Barr Virus in Leprosy. Nature 1971, 231, 47–49. [Google Scholar] [CrossRef]
- Yadav, P.; Tran, H.; Ebegbe, R.; Gottlieb, P.; Wei, H.; Lewis, R.H.; Mumbey-Wafula, A.; Kaplan, A.; Kholdarova, E.; Spatz, L. Antibodies Elicited in Response to EBNA-1 May Cross-React with DsDNA. PLoS ONE 2011, 6, e14488. [Google Scholar] [CrossRef]
- Sundar, K.; Jacques, S.; Gottlieb, P.; Villars, R.; Benito, M.E.; Taylor, D.K.; Spatz, L.A. Expression of the Epstein-Barr Virus Nuclear Antigen-1 (EBNA-1) in the Mouse Can Elicit the Production of Anti-DsDNA and Anti-Sm Antibodies. J. Autoimmun. 2004, 23, 127–140. [Google Scholar] [CrossRef]
- Poole, B.D.; Scofield, R.H.; Harley, J.B.; James, J.A. Epstein-Barr Virus and Molecular Mimicry in Systemic Lupus Erythematosus. Autoimmunity 2006, 39, 63–70. [Google Scholar] [CrossRef]
- Jarduli, L.R.; Sell, A.M.; Reis, P.G.; Sippert, E.Â.; Ayo, C.M.; Mazini, P.S.; Alves, H.V.; Teixeira, J.J.V.; Visentainer, J.E.L. Role of HLA, KIR, MICA, and Cytokines Genes in Leprosy. BioMed Res. Int. 2013, 2013, 17. [Google Scholar] [CrossRef] [PubMed]
- Krause-Kyora, B.; Nutsua, M.; Boehme, L.; Pierini, F.; Pedersen, D.D.; Kornell, S.C.; Drichel, D.; Bonazzi, M.; Möbus, L.; Tarp, P.; et al. Ancient DNA Study Reveals HLA Susceptibility Locus for Leprosy in Medieval Europeans. Nat. Commun. 2018, 9, 1569. [Google Scholar] [CrossRef]
- Fox, R.I.; Luppi, M.; Kang, H.I.; Pisa, P. Reactivation of Epstein-Barr Virus in Sjögren’s Syndrome. Springer Semin. Immunopathol. 1991, 13, 217–231. [Google Scholar] [CrossRef] [PubMed]
- Baer, A.N.; Walitt, B. Sjögren Syndrome and Other Causes of Sicca in Older Adults. Clin. Geriatr. Med. 2017, 33, 87–103. [Google Scholar] [CrossRef] [PubMed]
- Tucci, M.; Quatraro, C.; Silvestris, F. Sjögren’s Syndrome: An Autoimmune Disorder with Otolaryngological Involvement. Acta Otorhinolaryngol. Ital. 2005, 25, 139–144. Available online: https://pubmed.ncbi.nlm.nih.gov/16450767/ (accessed on 24 January 2021).
- Draborg, A.H.; Duus, K.; Houen, G. Epstein-Barr Virus in Systemic Autoimmune Diseases. Clin. Dev. Immunol. 2013, 2013, 535738. [Google Scholar] [CrossRef]
- Croia, C.; Astorri, E.; Murray-Brown, W.; Willis, A.; Brokstad, K.A.; Sutcliffe, N.; Piper, K.; Jonsson, R.; Tappuni, A.R.; Pitzalis, C.; et al. Implication of Epstein-Barr Virus Infection in Disease-Specific Autoreactive B Cell Activation in Ectopic Lymphoid Structures of Sjögren’s Syndrome. Arthritis Rheumatol. 2014, 66, 2545–2557. [Google Scholar] [CrossRef]
- Harley, J.B.; Zoller, E.E. What Caused All These Troubles, Anyway? Epstein-Barr Virus in Sjögren’s Syndrome Reevaluated. Arthritis Rheumatol. 2014, 66, 2328–2330. [Google Scholar] [CrossRef]
- Poole, B.D.; Gross, T.; Maier, S.; Harley, J.B.; James, J.A. Lupus-like Autoantibody Development in Rabbits and Mice after Immunization with EBNA-1 Fragments. J. Autoimmun. 2008, 31, 362–371. [Google Scholar] [CrossRef]
- Yamaoka, K.; Miyasaka, N.; Yamamoto, K. Possible Involvement of Epstein-Barr Virus in Polyclonal B Cell Activation in Sjögren’s Syndrome. Arthritis Rheum. 1988, 31, 1014–1021. [Google Scholar] [CrossRef]
- Inoue, N.; Harada, S.; Miyasaka, N.; Oya, A.; Yanagi, K. Analysis of Antibody Titers to Epstein-Barr Virus Nuclear Antigens in Sera of Patients with Sjögren’s Syndrome and with Rheumatoid Arthritis. J. Infect. Dis. 1991, 164, 22–28. [Google Scholar] [CrossRef] [PubMed]
- Lerner, M.R.; Andrews, N.C.; Miller, G.; Steitz, J.A. Two Small RNAs Encoded by Epstein-Barr Virus and Complexed with Protein Are Precipitated by Antibodies from Patients with Systemic Lupus Erythematosus. Proc. Natl. Acad. Sci. USA 1981, 78, 805–809. [Google Scholar] [CrossRef] [PubMed]
- Navone, R.; Lunardi, C.; Gerli, R.; Tinazzi, E.; Peterlana, D.; Bason, C.; Corrocher, R.; Puccetti, A. Identification of Tear Lipocalin as a Novel Autoantigen Target in Sjögren’s Syndrome. J. Autoimmun. 2005, 25, 229–234. [Google Scholar] [CrossRef]
- Wang, Q.; Che, N.; Lu, C.; Sun, X.; Wang, Y.; Wang, Q.; Tan, W.; Zhou, L.; Zhang, X.; Xu, D.; et al. Correlation of Peripheral CD4+ GranzB+ CTLs with Disease Severity in Patients with Primary Sjögren’s Syndrome. Arthritis Res. Ther. 2021, 23, 257. [Google Scholar] [CrossRef]
- Maślińska, M. The Role of Epstein–Barr Virus Infection in Primary Sjögren’s Syndrome. Curr. Opin. Rheumatol. 2019, 31, 475–483. [Google Scholar] [CrossRef] [PubMed]
- Manoussakis, M.N.; Georgopoulou, C.; Zintzaras, E.; Spyropoulou, M.; Stavropoulou, A.; Skopouli, F.N.; Moutsopoulos, H.M. Sjögren’s Syndrome Associated with Systemic Lupus Erythematosus: Clinical and Laboratory Profiles and Comparison with Primary Sjögren’s Syndrome. Arthritis Rheum. 2004, 50, 882–891. [Google Scholar] [CrossRef]
- Scofield, R.H.; Bruner, G.R.; Harley, J.B.; Namjou, B. Autoimmune Thyroid Disease Is Associated with a Diagnosis of Secondary Sjögren’s Syndrome in Familial Systemic Lupus. Ann. Rheum. Dis. 2007, 66, 410–413. [Google Scholar] [CrossRef]
- Sun, X.; Lu, L.; Li, Y.; Yang, R.; Shan, L.; Wang, Y. Increased Risk of Thyroid Disease in Patients with Sjogren’s Syndrome: A Systematic Review and Meta-Analysis. PeerJ 2019, 2019, e6737. [Google Scholar] [CrossRef]
- Baldini, C.; Ferro, F.; Mosca, M.; Fallahi, P.; Antonelli, A. The Association of Sjögren Syndrome and Autoimmune Thyroid Disorders. Front. Endocrinol. 2018, 9, 121. [Google Scholar] [CrossRef]
- Bjornevik, K.; Cortese, M.; Healy, B.C.; Kuhle, J.; Mina, M.J.; Leng, Y.; Elledge, S.J.; Niebuhr, D.W.; Scher, A.I.; Munger, K.L.; et al. Longitudinal Analysis Reveals High Prevalence of Epstein-Barr Virus Associated with Multiple Sclerosis. Science 2022, 375, 296–301. [Google Scholar] [CrossRef]
- Fraser, K.B.; Millar, J.H.D.; Haire, M.; Mccrea, S. Increased Tendency to Spontaneous In-Vitro Lymphocyte Transformation in Clinically Active Multiple Sclerosis. Lancet 1979, 314, 715–717. [Google Scholar] [CrossRef]
- Tørring, C.; Andreasen, C.; Gehr, N.; Bjerg, L.; Petersen, T.; Höllsberg, P. Higher Incidence of Epstein-Barr Virus-Induced Lymphocyte Transformation in Multiple Sclerosis. Acta Neurol. Scand. 2014, 130, 90–96. [Google Scholar] [CrossRef] [PubMed]
- Kreft, K.L.; Van Nierop, G.P.; Scherbeijn, S.M.J.; Janssen, M.; Verjans, G.M.G.M.; Hintzen, R.Q. Elevated EBNA-1 IgG in MS Is Associated with Genetic MS Risk Variants. Neurol. Neuroimmunol. NeuroInflamm. 2017, 4, 406. [Google Scholar] [CrossRef] [PubMed]
- Sumaya, C.V.; Myers, L.W.; Ellison, G.W.; Ench, Y. Increased Prevalence and Titer of Epstein-Barr Virus Antibodies in Patients with Multiple Sclerosis. Ann. Neurol. 1985, 17, 371–377. [Google Scholar] [CrossRef] [PubMed]
- Riverol, M.; Sepulcre, J.; Fernandez-Alonso, M.; Uccelli, A.; Brieva, L.; Rubio, M.; Rodriguez, A.; Fernandez-Diez, B.; Villoslada, P. Antibodies against Epstein-Barr Virus and Herpesvirus Type 6 Are Associated with the Early Phases of Multiple Sclerosis. J. Neuroimmunol. 2007, 192, 184–185. [Google Scholar] [CrossRef]
- Buljevac, D.; Van Doornum, G.J.J.; Flach, H.Z.; Groen, J.; Osterhaus, A.D.M.E.; Hop, W.; Van Doorn, P.A.; Van Der Meché, F.G.A.; Hintzen, R.Q. Epstein-Barr Virus and Disease Activity in Multiple Sclerosis. J. Neurol. Neurosurg. Psychiatry 2005, 76, 1377–1381. [Google Scholar] [CrossRef]
- Myhr, K.M.; Riise, T.; Barrett-Connor, E.; Myrmel, H.; Vedeler, C.; Grønning, M.; Kalvenes, M.B.; Nyland, H. Altered Antibody Pattern to Epstein-Barr Virus but Not to Other Herpesviruses in Multiple Sclerosis: A Population Based Case-Control Study from Western Norway. J. Neurol. Neurosurg. Psychiatry 1998, 64, 539–542. [Google Scholar] [CrossRef]
- Wandinger, K.P.; Jabs, W.; Siekhaus, A.; Bubel, S.; Trillenberg, P.; Wagner, H.J.; Wessel, K.; Kirchner, H.; Hennig, H. Association between Clinical Disease Activity and Epstein-Barr Virus Reactivation in MS. Neurology 2000, 55, 178–184. [Google Scholar] [CrossRef]
- Tzartos, J.S.; Khan, G.; Vossenkamper, A.; Cruz-Sadaba, M.; Lonardi, S.; Sefia, E.; Meager, A.; Elia, A.; Middeldorp, J.M.; Clemens, M.; et al. Association of Innate Immune Activation with Latent Epstein-Barr Virus in Active MS Lesions. Neurology 2012, 78, 15–23. [Google Scholar] [CrossRef]
- Cepok, S.; Zhou, D.; Srivastava, R.; Nessler, S.; Stei, S.; Büssow, K.; Sommer, N.; Hemmer, B. Identification of Epstein-Barr Virus Proteins as Putative Targets of the Immune Response in Multiple Sclerosis. J. Clin. Investig. 2005, 115, 1352–1360. [Google Scholar] [CrossRef]
- Jilek, S.; Schluep, M.; Meylan, P.; Vingerhoets, F.; Guignard, L.; Monney, A.; Kleeberg, J.; Le Goff, G.; Pantaleo, G.; Du Pasquier, R.A. Strong EBV-Specific CD8+ T-Cell Response in Patients with Early Multiple Sclerosis. Brain 2008, 131, 1712–1721. [Google Scholar] [CrossRef] [PubMed]
- Serafini, B.; Rosicarelli, B.; Veroni, C.; Mazzola, G.A.; Aloisi, F. Epstein-Barr Virus-Specific CD8 T Cells Selectively Infiltrate the Brain in Multiple Sclerosis and Interact Locally with Virus-Infected Cells: Clue for a Virus-Driven Immunopathological Mechanism. J. Virol. 2019, 93, 980–999. [Google Scholar] [CrossRef] [PubMed]
- Lünemann, J.D.; Edwards, N.; Muraro, P.A.; Hayashi, S.; Cohen, J.I.; Münz, C.; Martin, R. Increased Frequency and Broadened Specificity of Latent EBV Nuclear Antigen-1-Specific T Cells in Multiple Sclerosis. Brain 2006, 129, 1493–1506. [Google Scholar] [CrossRef] [PubMed]
- Jelcic, I.; Al Nimer, F.; Wang, J.; Lentsch, V.; Planas, R.; Jelcic, I.; Madjovski, A.; Ruhrmann, S.; Faigle, W.; Frauenknecht, K.; et al. Memory B Cells Activate Brain-Homing, Autoreactive CD4+ T Cells in Multiple Sclerosis. Cell 2018, 175, 85–100.e23. [Google Scholar] [CrossRef]
- Pozzilli, C.; Tomassini, V.; Marinelli, F.; Paolillo, A.; Gasperini, C.; Bastianello, S. “Gender Gap” in Multiple Sclerosis: Magnetic Resonance Imaging Evidence. Eur. J. Neurol. 2003, 10, 95–97. [Google Scholar] [CrossRef]
- Planas, R.; Metz, I.; Ortiz, Y.; Vilarrasa, N.; Jelčić, I.; Salinas-Riester, G.; Heesen, C.; Brück, W.; Martin, R.; Sospedra, M. Central Role of Th2/Tc2 Lymphocytes in Pattern II Multiple Sclerosis Lesions. Ann. Clin. Transl. Neurol. 2015, 2, 875–893. [Google Scholar] [CrossRef]
- Costanza, M. Type 2 Inflammatory Responses in Autoimmune Demyelination of the Central Nervous System: Recent Advances. J. Immunol. Res. 2019, 2019, 4204512. [Google Scholar] [CrossRef]
- Lafaille, J.J.; Van De Keere, F.; Hsu, A.L.; Baron, J.L.; Haas, W.; Raine, C.S.; Tonegawa, S. Myelin Basic Protein-Specific T Helper 2 (Th2) Cells Cause Experimental Autoimmune Encephalomyelitis in Immunodeficient Hosts Rather than Protect Them from the Disease. J. Exp. Med. 1997, 186, 307–312. [Google Scholar] [CrossRef]
- Genain, C.P.; Abel, K.; Belmar, N.; Villinger, F.; Rosenberg, D.P.; Linington, C.; Raine, C.S.; Hauser, S.L. Late Complications of Immune Deviation Therapy in a Nonhuman Primate. Science 1996, 274, 2054–2057. [Google Scholar] [CrossRef]
- Bray, P.F.; Luka, J.; Bray, P.F.; Culp, K.W.; Schlight, J.P. Antibodies against Epstein-Barr Nuclear Antigen (EBNA) in Multiple Sclerosis CSF and Two Pentapepfide Seauence Identities between EBNA and Myelin Basic Protein. Neurology 1992, 42, 1798–1804. [Google Scholar] [CrossRef]
- Mescheriakova, J.Y.; van Nierop, G.P.; van der Eijk, A.A.; Kreft, K.L.; Hintzen, R.Q. EBNA-1 Titer Gradient in Families with Multiple Sclerosis Indicates a Genetic Contribution. Neurol. Neuroimmunol. Neuroinflamm. 2020, 7, 872. [Google Scholar] [CrossRef] [PubMed]
- Ghabaee, M.; Bayati, A.; Amri Saroukolaei, S.; Sahraian, M.A.; Sanaati, M.H.; Karimi, P.; Houshmand, M.; Sadeghian, H.; Hashemi Chelavi, L. Analysis of HLA DR2&DQ6 (DRB1*1501, DQA1*0102, DQB1*0602) Haplotypes in Iranian Patients with Multiple Sclerosis. Cell. Mol. Neurobiol. 2009, 29, 109–114. [Google Scholar] [CrossRef] [PubMed]
- Fernández, O.; Fernández, V.; Alonso, A.; Caballero, A.; Luque, G.; Bravo, M.; Leon, A.; Mayorga, C.; Leyva, L.; De Ramón, E. DQB1*0602 Allele Shows a Strong Association with Multiple Sclerosis in Patients in Malaga, Spain. J. Neurol. 2004, 251, 440–444. [Google Scholar] [CrossRef] [PubMed]
- Grytten, N.; Myhr, K.M.; Celius, E.G.; Benjaminsen, E.; Kampman, M.; Midgard, R.; Vatne, A.; Aarseth, J.H.; Riise, T.; Torkildsen, Ø. Risk of Cancer among Multiple Sclerosis Patients, Siblings, and Population Controls: A Prospective Cohort Study. Mult. Scler. J. 2020, 26, 1569–1580. [Google Scholar] [CrossRef] [PubMed]
- Hjalgrim, H.; Rasmussen, S.; Rostgaard, K.; Nielsen, N.M.; Koch-Henriksen, N.; Munksgaard, L.; Storm, H.H.; Melbye, M. Familial Clustering of Hodgkin Lymphoma and Multiple Sclerosis. JNCI J. Natl. Cancer Inst. 2004, 96, 780–784. [Google Scholar] [CrossRef]
- Bahmanyar, S.; Montgomery, S.M.; Hillert, J.; Ekbom, A.; Olsson, T. Cancer Risk among Patients with Multiple Sclerosis and Their Parents. Neurology 2009, 72, 1170–1177. [Google Scholar] [CrossRef]
- Frampton, M.; Da Silva Filho, M.I.; Broderick, P.; Thomsen, H.; Försti, A.; Vijayakrishnan, J.; Cooke, R.; Enciso-Mora, V.; Hoffmann, P.; Nöthen, M.M.; et al. Variation at 3p24.1 and 6q23.3 Influences the Risk of Hodgkin’s Lymphoma. Nat. Commun. 2013, 4, 2549. [Google Scholar] [CrossRef]
- Nischwitz, S.; Cepok, S.; Kroner, A.; Wolf, C.; Knop, M.; Müller-Sarnowski, F.; Pfister, H.; Roeske, D.; Rieckmann, P.; Hemmer, B.; et al. Evidence for VAV2 and ZNF433 as Susceptibility Genes for Multiple Sclerosis. J. Neuroimmunol. 2010, 227, 162–166. [Google Scholar] [CrossRef]
- Gilhus, N.E.; Skeie, G.O.; Romi, F.; Lazaridis, K.; Zisimopoulou, P.; Tzartos, S. Myasthenia Gravis—Autoantibody Characteristics and Their Implications for Therapy. Nat. Rev. Neurol. 2016, 12, 259–268. [Google Scholar] [CrossRef]
- Cufi, P.; Dragin, N.; Weiss, J.M.; Martinez-Martinez, P.; De Baets, M.H.; Roussin, R.; Fadel, E.; Berrih-Aknin, S.; Le Panse, R. Implication of Double-Stranded RNA Signaling in the Etiology of Autoimmune Myasthenia Gravis. Ann. Neurol. 2013, 73, 281–293. [Google Scholar] [CrossRef]
- Cavalcante, P.; Marcuzzo, S.; Franzi, S.; Galbardi, B.; Maggi, L.; Motta, T.; Ghislandi, R.; Buzzi, A.; Spinelli, L.; Novellino, L.; et al. Epstein-Barr Virus in Tumor-Infiltrating B Cells of Myasthenia Gravis Thymoma: An Innocent Bystander or an Autoimmunity Mediator? Oncotarget 2017, 8, 95432–95449. [Google Scholar] [CrossRef] [PubMed]
- Yin, W.; Allman, W.; Ouyang, S.; Li, Y.; Li, J.; Christadoss, P.; Yang, H. The Increased Expression of CD21 on AchR Specified B Cells in Patients with Myasthenia Gravis. J. Neuroimmunol. 2013, 256, 49–54. [Google Scholar] [CrossRef] [PubMed]
- Thiruppathi, M.; Rowin, J.; Li Jiang, Q.; Sheng, J.R.; Prabhakar, B.S.; Meriggioli, M.N. Functional Defect in Regulatory T Cells in Myasthenia Gravis. Ann. N. Y. Acad. Sci. 2012, 1274, 68–76. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Amezquita, R.A.; Kleinstein, S.H.; Stathopoulos, P.; Nowak, R.J.; O’Connor, K.C. Autoreactive T Cells from Patients with Myasthenia Gravis Are Characterized by Elevated IL-17, IFN-γ, and GM-CSF and Diminished IL-10 Production. J. Immunol. 2016, 196, 2075–2084. [Google Scholar] [CrossRef] [PubMed]
- Yeh, J.H.; Lin, C.C.; Chen, Y.K.; Sung, F.C.; Chiu, H.C.; Kao, C.H. Excessive Risk of Cancer and in Particular Lymphoid Malignancy in Myasthenia Gravis Patients: A Population-Based Cohort Study. Neuromuscul. Disord. 2014, 24, 245–249. [Google Scholar] [CrossRef]
- Levin, N.; Abramsky, O.; Lossos, A.; Karussis, D.; Siegal, T.; Argov, Z.; Hur, T. Ben Extrathymic Malignancies in Patients with Myasthenia Gravis. J. Neurol. Sci. 2005, 237, 39–43. [Google Scholar] [CrossRef]
- Basiri, K.; Etemadifar, M.; Maghzi, A.H.; Zarghami, N. Frequency of Myasthenia Gravis in Multiple Sclerosis: Report of Five Cases from Isfahan, Iran. Neurol. India 2009, 57, 638–640. [Google Scholar] [CrossRef]
- Danikowski, K.M.; Jayaraman, S.; Prabhakar, B.S. Regulatory T Cells in Multiple Sclerosis and Myasthenia Gravis. J. Neuroinflamm. 2017, 14, 117. [Google Scholar] [CrossRef]
- Toussirot, E.; Roudier, J. Pathophysiological Links between Rheumatoid Arthritis and the Epstein-Barr Virus: An Update. Jt. Bone Spine 2007, 74, 418–426. [Google Scholar] [CrossRef]
- Callan, M.F. Epstein-Barr Virus, Arthritis, and the Development of Lymphoma in Arthritis Patients. Curr. Opin. Rheumatol. 2004, 16, 399–405. [Google Scholar] [CrossRef]
- Cohen, J. Epstein-Barr Virus Infection. N. Engl. J. Med. 2000, 343, 481–492. [Google Scholar] [CrossRef] [PubMed]
- Percy, J.S.; Davis, P.; Russell, A.S.; Brisson, E. A Longitudinal Study of in Vitro Tests for Lymphocyte Function in Rheumatoid Arthritis. Ann. Rheum. Dis. 1978, 37, 416–420. [Google Scholar] [CrossRef] [PubMed]
- Horwitz, D.A.; Garrett, M.A. Lymphocyte Reactivity to Mitogens in Subjects with Systemic Lupus Erythematosus, Rheumatoid Arthritis and Scleroderma. Clin. Exp. Immunol. 1977, 27, 92. Available online: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC1540894/ (accessed on 24 January 2021). [PubMed]
- Trier, N.H.; Holm, B.E.; Heiden, J.; Slot, O.; Locht, H.; Lindegaard, H.; Svendsen, A.; Nielsen, C.T.; Jacobsen, S.; Theander, E.; et al. Antibodies to a Strain-Specific Citrullinated Epstein-Barr Virus Peptide Diagnoses Rheumatoid Arthritis. Sci. Rep. 2018, 8, 3684. [Google Scholar] [CrossRef]
- Yazbek, M.A.; de Barros-Mazon, S.; Rossi, C.L.; Londe, A.C.; Costallat, L.T.L.; Bértolo, M.B. Association Analysis of Anti-Epstein-Barr Nuclear Antigen-1 Antibodies, Anti-Cyclic Citrullinated Peptide Antibodies, the Shared Epitope and Smoking Status in Brazilian Patients with Rheumatoid Arthritis. Clinics 2011, 66, 1401–1406. [Google Scholar] [CrossRef]
- Van Der Woude, D.; Catrina, A.I. HLA and Anti-Citrullinated Protein Antibodies: Building Blocks in RA. Best Pract. Res. Clin. Rheumatol. 2015, 29, 692–705. [Google Scholar] [CrossRef]
- Hill, J.A.; Southwood, S.; Sette, A.; Jevnikar, A.M.; Bell, D.A.; Cairns, E. Cutting Edge: The Conversion of Arginine to Citrulline Allows for a High-Affinity Peptide Interaction with the Rheumatoid Arthritis-Associated HLA-DRB1*0401 MHC Class II Molecule. J. Immunol. 2003, 171, 538–541. [Google Scholar] [CrossRef]
- Pratesi, F.; Tommasi, C.; Anzilotti, C.; Chimenti, D.; Migliorini, P. Deiminated Epstein-Barr Virus Nuclear Antigen 1 Is a Target of Anti-Citrullinated Protein Antibodies in Rheumatoid Arthritis. Arthritis Rheum. 2006, 54, 733–741. [Google Scholar] [CrossRef]
- Smith, M.J.; Simmons, K.M.; Cambier, J.C. B Cells in Type 1 Diabetes Mellitus and Diabetic Kidney Disease. Nat. Rev. Nephrol. 2017, 13, 712–720. [Google Scholar] [CrossRef]
- Von Herrath, M.G.; Oldstone, M.B.A. Interferon-γ Is Essential for Destruction of β Cells and Development of Insulin-Dependent Diabetes Mellitus. J. Exp. Med. 1997, 185, 531–539. [Google Scholar] [CrossRef]
- Pujol-Borrell, R.; Todd, I.; Doshi, M.; Bottazzo, G.F.; Sutton, R.; Gary, D.; Adolf, G.R.; Feldmann, M. HLA Class II Induction in Human Islet Cells by Interferon-γ plus Tumour Necrosis Factor or Lymphotoxin. Nature 1987, 326, 304–306. [Google Scholar] [CrossRef]
- Radenkovic, M.; Uvebrant, K.; Skog, O.; Sarmiento, L.; Avartsson, J.; Storm, P.; Vickman, P.; Bertilsson, P.-A.; Fex, M.; Korgsgren, O.; et al. Characterization of Resident Lymphocytes in Human Pancreatic Islets. Clin. Exp. Immunol. 2017, 187, 418. [Google Scholar] [CrossRef]
- Arif, S.; Leete, P.; Nguyen, V.; Marks, K.; Nor, N.M.; Estorninho, M.; Kronenberg-Versteeg, D.; Bingley, P.J.; Todd, J.A.; Guy, C.; et al. Blood and Islet Phenotypes Indicate Immunological Heterogeneity in Type 1 Diabetes. Diabetes 2014, 63, 3835–3845. [Google Scholar] [CrossRef] [PubMed]
- De Marquesini, L.G.P.; Fu, J.; Connor, K.J.; Bishop, A.J.; McLintock, N.E.; Pope, C.; Wong, F.S.; Dayan, C.M. IFN-γ and IL-10 Islet-Antigen-Specific T Cell Responses in Autoantibody-Negative First-Degree Relatives of Patients with Type 1 Diabetes. Diabetologia 2010, 53, 1451–1460. [Google Scholar] [CrossRef] [PubMed]
- Filippi, C.M.; Von Herrath, M.G. Viral Trigger for Type 1 Diabetes: Pros and Cons. Diabetes 2008, 57, 2863–2871. [Google Scholar] [CrossRef] [PubMed]
- Kawasaki, E.; Matsuura, N.; Eguchi, K. Type 1 Diabetes in Japan. Diabetologia 2006, 49, 828–836. [Google Scholar] [CrossRef]
- Carstensen, B.; Read, S.H.; Friis, S.; Sund, R.; Keskimäki, I.; Svensson, A.-M.; Ljung, R.; Wild, S.H.; Kerssens, J.J.; Harding, J.L.; et al. Cancer Incidence in Persons with Type 1 Diabetes: A Five-Country Study of 9,000 Cancers in Type 1 Diabetic Individuals. Diabetologia 2016, 59, 980. [Google Scholar] [CrossRef]
- Tsutsumi, C.; Imagawa, A.; Ikegami, H.; Makino, H.; Kobayashi, T.; Hanafusa, T. Class II HLA Genotype in Fulminant Type 1 Diabetes: A Nationwide Survey with Reference to Glutamic Acid Decarboxylase Antibodies. J. Diabetes Investig. 2012, 3, 62–69. [Google Scholar] [CrossRef]
- Alhassan, E.; Yadav, A.; Kelly, C.P.; Mukherjee, R. Novel Nondietary Therapies for Celiac Disease. Cell. Mol. Gastroenterol. Hepatol. 2019, 8, 335–345. [Google Scholar] [CrossRef]
- Parzanese, I.; Qehajaj, D.; Patrinicola, F.; Aralica, M.; Chiriva-Internati, M.; Stifter, S.; Elli, L.; Grizzi, F. Celiac Disease: From Pathophysiology to Treatment. World J. Gastrointest. Pathophysiol. 2017, 8, 27. [Google Scholar] [CrossRef]
- Jabri, B.; Sollid, L.M. T Cells in Celiac Disease. J. Immunol. 2017, 198, 3005–3014. [Google Scholar] [CrossRef] [PubMed]
- Harley, J.B.; Chen, X.; Pujato, M.; Miller, D.; Maddox, A.; Forney, C.; Magnusen, A.F.; Lynch, A.; Chetal, K.; Yukawa, M.; et al. Transcription Factors Operate across Disease Loci, with EBNA2 Implicated in Autoimmunity. Nat. Genet. 2018, 50, 699–707. [Google Scholar] [CrossRef] [PubMed]
- Wosen, J.E.; Mukhopadhyay, D.; MacAubas, C.; Mellins, E.D. Epithelial MHC Class II Expression and Its Role in Antigen Presentation in the Gastrointestinal and Respiratory Tracts. Front. Immunol. 2018, 9, 2144. [Google Scholar] [CrossRef] [PubMed]
- Eriksen, W. The Spread of EBV to Ectopic Lymphoid Aggregates May Be the Final Common Pathway in the Pathogenesis of ME/CFS. Med. Hypotheses 2017, 102, 8–15. [Google Scholar] [CrossRef]
- Pender, M.P. Infection of Autoreactive B Lymphocytes with EBV, Causing Chronic Autoimmune Diseases. Trends Immunol. 2003, 24, 584–588. [Google Scholar] [CrossRef]
- Han, Y.; Chen, W.; Li, P.; Ye, J. Association between Coeliac Disease and Risk of Any Malignancy and Gastrointestinal Malignancy: A Meta-Analysis. Medicine 2015, 94, e1612. [Google Scholar] [CrossRef]
- Marafini, I.; Monteleone, G.; Stolfi, C. Association between Celiac Disease and Cancer. Int. J. Mol. Sci. 2020, 21, 4155. [Google Scholar] [CrossRef]
- Kahles, H.; Fain, P.R.; Baker, P.; Eisenbarth, G.; Badenhoop, K. Genetics of Autoimmune Thyroiditis in Type 1 Diabetes Reveals a Novel Association with DPB1∗0201: Data from the Type 1 Diabetes Genetics Consortium. Diabetes Care 2015, 38, S21–S28. [Google Scholar] [CrossRef]
- Stiefel, P.; Aparicio, R.; Dolores Nieto, M.; Alfaro, V. Infección Por El Virus de Epstein-Barr y Enfermedad de Graves-Basedow: ¿simple Casualidad o Algo Más? Med. Clin. 2006, 126, 278–279. [Google Scholar] [CrossRef]
- Janegova, A.; Janega, P.; Rychly, B.; Kuracinova, K.; Babal, P. The Role of Epstein-Barr Virus Infection in the Development of Autoimmune Thyroid Diseases. Endokrynol. Pol. 2015, 66, 132–136. [Google Scholar] [CrossRef]
- Arranz, E.; Telleria, J.J.; Sanz, A.; Martin, J.F.; Alonso, M.; Calvo, C.; Blanco-Quirós, A. HLA-DQA1*0501 and DQB1*02 Homozygosity and Disease Susceptibility in Spanish Coeliac Patients. Exp. Clin. Immunogenet. 1997, 14, 286–290. Available online: https://pubmed.ncbi.nlm.nih.gov/9523165/ (accessed on 10 January 2021). [PubMed]
- Kocjan, T.; Wraber, B.; Repnik, U.; Hojker, S. Changes in Th1/Th2 Cytokine Balance in Graves’ Desease. Pflug. Arch. 2000, 440, R094–R095. [Google Scholar] [CrossRef]
- Marique, L.; Van Regemorter, V.; Gérard, A.C.; Craps, J.; Senou, M.; Marbaix, E.; Rahier, J.; Daumerie, C.; Mourad, M.; Lengelé, B.; et al. The Expression of Dual Oxidase, Thyroid Peroxidase, and Caveolin-1 Differs According to the Type of Immune Response (TH1/TH2) Involved in Thyroid Autoimmune Disorders. J. Clin. Endocrinol. Metab. 2014, 99, 1722–1732. [Google Scholar] [CrossRef] [PubMed]
- Rapoport, B.; McLachlan, S.M. Graves’ Hyperthyroidism Is Antibody-Mediated but Is Predominantly a Th1-Type Cytokine Disease. J. Clin. Endocrinol. Metab. 2014, 99, 4060–4061. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, S.M.; Elia, G.; Virili, C.; Centanni, M.; Antonelli, A.; Fallahi, P. Systemic Lupus Erythematosus and Thyroid Autoimmunity. Front. Endocrinol. 2017, 8, 138. [Google Scholar] [CrossRef]
- Biró, E.; Szekanecz, Z.; Czirják, L.; Dankó, K.; Kiss, E.; Szabó, N.A.; Szűcs, G.; Zeher, M.; Bodolay, E.; Szegedi, G.; et al. Association of Systemic and Thyroid Autoimmune Diseases. Clin. Rheumatol. 2006, 25, 240–245. [Google Scholar] [CrossRef]
- Izzedine, H.; Roura, R.; Bourry, E.; Georgin-Lavialle, S.; Cacoub, P.; Deray, G. Systemic Lupus Erythematous and Graves Disease. Endocrinologist 2005, 15, 289–291. Available online: https://journals.lww.com/theendocrinologist/Abstract/2005/09000/Systemic_Lupus_Erythematous_and_Graves_Disease.6.aspx (accessed on 10 March 2021). [CrossRef]
- Meiss, F.; Fischer, M.; Hädecke, J.; Knorrn, M.; Marsch, W.C. Morbus Basedow. Eine Auch Dermatologisch Wichtige Differenzial-Diagnose Des Systemischen Lupus Erythematodes. Hautarzt 2004, 55, 475–479. [Google Scholar] [CrossRef]
- Pender, M.P.; Burrows, S.R. Epstein–Barr Virus and Multiple Sclerosis: Potential Opportunities for Immunotherapy. Clin. Transl. Immunol. 2014, 3, e27. [Google Scholar] [CrossRef]
- Pender, M.P. CD8+ T-Cell Deficiency, Epstein-Barr Virus Infection, Vitamin D Deficiency, and Steps to Autoimmunity: A Unifying Hypothesis. Autoimmune Dis. 2012, 2012, 189096. [Google Scholar] [CrossRef]
- Lin, Z.; Huang, L.; Li, S.L.; Gu, J.; Cui, X.; Zhou, Y. Pan-Cancer Analysis of Genomic Properties and Clinical Outcome Associated with Tumor Tertiary Lymphoid Structure. Sci. Rep. 2020, 10, 21530. [Google Scholar] [CrossRef] [PubMed]
- Pitzalis, C.; Jones, G.W.; Bombardieri, M.; Jones, S.A. Ectopic Lymphoid-like Structures in Infection, Cancer and Autoimmunity. Nat. Rev. Immunol. 2014, 14, 447–462. [Google Scholar] [CrossRef] [PubMed]
- Serafini, B.; Rosicarelli, B.; Franciotta, D.; Magliozzi, R.; Reynolds, R.; Cinque, P.; Andreoni, L.; Trivedi, P.; Salvetti, M.; Faggioni, A.; et al. Dysregulated Epstein-Barr Virus Infection in the Multiple Sclerosis Brain. J. Exp. Med. 2007, 204, 2899. [Google Scholar] [CrossRef]
- Cavalcante, P.; Serafini, B.; Rosicarelli, B.; Maggi, L.; Barberis, M.; Antozzi, C.; Berrih-Aknin, S.; Bernasconi, P.; Aloisi, F.; Mantegazza, R. Epstein-Barr Virus Persistence and Reactivation in Myasthenia Gravis Thymus. Ann. Neurol. 2010, 67, 726–738. [Google Scholar] [CrossRef] [PubMed]
- Lucchesi, D.; Bombardieri, M. The Role of Viruses in Autoreactive B Cell Activation within Tertiary Lymphoid Structures in Autoimmune Diseases. J. Leukoc. Biol. 2013, 94, 1191–1199. [Google Scholar] [CrossRef] [PubMed]
- Croia, C.; Serafini, B.; Bombardieri, M.; Kelly, S.; Humby, F.; Severa, M.; Rizzo, F.; Coccia, E.M.; Migliorini, P.; Aloisi, F.; et al. Epstein-Barr Virus Persistence and Infection of Autoreactive Plasma Cells in Synovial Lymphoid Structures in Rheumatoid Arthritis. Ann. Rheum. Dis. 2013, 72, 1559–1568. [Google Scholar] [CrossRef] [PubMed]
- Berrih-Aknin, S.; Ragheb, S.; Le Panse, R.; Lisak, R.P. Ectopic Germinal Centers, BAFF and Anti-B-Cell Therapy in Myasthenia Gravis. Autoimmun. Rev. 2013, 12, 885–893. [Google Scholar] [CrossRef]
- Bombardieri, M.; Pitzalis, C. Ectopic Lymphoid Neogenesis and Lymphoid Chemokines in Sjogren’s Syndrome: At the Interplay between Chronic Inflammation, Autoimmunity and Lymphomagenesis. Curr. Pharm. Biotechnol. 2012, 13, 1989–1996. [Google Scholar] [CrossRef]
- Manzo, A.; Bombardieri, M.; Humby, F.; Pitzalis, C. Secondary and Ectopic Lymphoid Tissue Responses in Rheumatoid Arthritis: From Inflammation to Autoimmunity and Tissue Damage/Remodeling. Immunol. Rev. 2010, 233, 267–285. [Google Scholar] [CrossRef]
- Jamaly, S.; Rakaee, M.; Abdi, R.; Tsokos, G.C.; Fenton, K.A. Interplay of Immune and Kidney Resident Cells in the Formation of Tertiary Lymphoid Structures in Lupus Nephritis. Autoimmun. Rev. 2021, 20, 102980. [Google Scholar] [CrossRef]
- Korpos, É.; Kadri, N.; Loismann, S.; Findeisen, C.R.; Arfuso, F.; Burke, G.W.; Richardson, S.J.; Morgan, N.G.; Bogdani, M.; Pugliese, A.; et al. Identification and Characterisation of Tertiary Lymphoid Organs in Human Type 1 Diabetes. Diabetologia 2021, 64, 1626–1641. [Google Scholar] [CrossRef] [PubMed]
- Ludewig, B.; Odermatt, B.; Landmann, S.; Hengartner, H.; Zinkernagel, R.M. Dendritic Cells Induce Autoimmune Diabetes and Maintain Disease via De Novo Formation of Local Lymphoid Tissue. J. Exp. Med. 1998, 188, 1493. [Google Scholar] [CrossRef] [PubMed]
- Jones, G.W.; Jones, S.A.; Building, T.; Jones, G.W. Ectopic Lymphoid Follicles: Inducible Centres for Generating Antigen-Specific Immune Responses within Tissues. Immunology 2016, 147, 141–151. [Google Scholar] [CrossRef] [PubMed]
- Stott, D.I.; Hiepe, F.; Hummel, M.; Steinhauser, G.; Berek, C. Antigen-Driven Clonal Proliferation of B Cells within the Target Tissue of an Autoimmune Disease. The Salivary Glands of Patients with Sjögren’s Syndrome. J. Clin. Investig. 1998, 102, 938–946. [Google Scholar] [CrossRef] [PubMed]
- Babcock, G.J.; Hochberg, D.; Thorley-Lawson, D.A. The Expression Pattern of Epstein-Barr Virus Latent Genes in Vivo Is Dependent upon the Differentiation Stage of the Infected B Cell. Immunity 2000, 13, 497–506. [Google Scholar] [CrossRef]
- Thorley-Lawson, D.A.; Gross, A. Persistence of the Epstein-Barr Virus and the Origins of Associated Lymphomas. N. Engl. J. Med. 2004, 350, 1328–1337. [Google Scholar] [CrossRef]
- Odumade, O.A.; Hogquist, K.A.; Balfour, H.H. Progress and Problems in Understanding and Managing Primary Epstein-Barr Virus Infections. Clin. Microbiol. Rev. 2011, 24, 193–209. [Google Scholar] [CrossRef]
- De Paschale, M.; Clerici, P. Serological Diagnosis of Epstein-Barr Virus Infection: Problems and Solutions. World J. Virol. 2012, 1, 31–43. [Google Scholar] [CrossRef]
- Liu, W.; Lin, Y.; Xiao, H.; Xing, S.; Chen, H.; Chi, P.; Zhang, G. Epstein-Barr Virus Infection Induces Indoleamine 2,3-Dioxygenase Expression in Human Monocyte-Derived Macrophages through P38/Mitogen-Activated Protein Kinase and NF-ΚB Pathways: Impairment in T Cell Functions. J. Virol. 2014, 88, 6660–6671. [Google Scholar] [CrossRef]
- Iwakiri, D.; Takada, K. Role of EBERs in the Pathogenesis of EBV Infection. Adv. Cancer Res. 2010, 107, 119–136. [Google Scholar] [CrossRef]
- Al Tabaa, Y.; Tuaillon, E.; Jeziorski, E.; Ouedraogo, D.E.; Bolloré, K.; Rubbo, P.A.; Foulongne, V.; Rodière, M.; Vendrell, J.P. B-Cell Polyclonal Activation and Epstein-Barr Viral Abortive Lytic Cycle Are Two Key Features in Acute Infectious Mononucleosis. J. Clin. Virol. 2011, 52, 33–37. [Google Scholar] [CrossRef] [PubMed]
- Laichalk, L.L.; Thorley-Lawson, D.A. Terminal Differentiation into Plasma Cells Initiates the Replicative Cycle of Epstein-Barr Virus in Vivo. J. Virol. 2005, 79, 1296–1307. [Google Scholar] [CrossRef] [PubMed]
- Lerner, A.M.; Beqaj, S. A Paradigm Linking Herpesvirus Immediate-Early Gene Expression Apoptosis and Myalgic Encephalomyelitis Chronic Fatigue Syndrome. Virus Adapt. Treat. 2011, 3, 19–24. [Google Scholar] [CrossRef]
- Glaser, R.; Litsky, M.L.; Padgett, D.A.; Baiocchi, R.A.; Yang, E.V.; Chen, M.; Yeh, P.E.; Green-Church, K.B.; Caligiuri, M.A.; Williams, M.V. EBV-Encoded DUTPase Induces Immune Dysregulation: Implications for the Pathophysiology of EBV-Associated Disease. Virology 2006, 346, 205–218. [Google Scholar] [CrossRef] [PubMed]
- Ariza, M.E.; Rivailler, P.; Glaser, R.; Chen, M.; Williams, M.V. Epstein-Barr Virus Encoded DUTPase Containing Exosomes Modulate Innate and Adaptive Immune Responses in Human Dendritic Cells and Peripheral Blood Mononuclear Cells. PLoS ONE 2013, 8, e69827. [Google Scholar] [CrossRef] [PubMed]
- Schroder, K.; Hertzog, P.J.; Ravasi, T.; Hume, D.A. Interferon-γ: An Overview of Signals, Mechanisms and Functions. J. Leukoc. Biol. 2004, 75, 163–189. [Google Scholar] [CrossRef]
- Grammatikos, A.P.; Tsokos, G.C. Immunodeficiency and Autoimmunity: Lessons from Systemic Lupus Erythematosus. Trends Mol. Med. 2012, 18, 101–108. [Google Scholar] [CrossRef]
- Biasi, D.; Caramaschi, P.; Ambrosetti, A.; Carletto, A.; Mocella, S.; Randon, M.; Bambara, L.M. Mucosa-Associated Lymphoid Tissue Lymphoma of the Salivary Glands Occurring in Patients Affected by Sjögren’s Syndrome: Report of 6 Cases. Acta Haematol. 2001, 105, 83–88. [Google Scholar] [CrossRef]
- Voulgarelis, M.; Moutsopoulos, H.M. Mucosa-Associated Lymphoid Tissue Lymphoma in Sjögren’s Syndrome: Risks, Management, and Prognosis. Rheum. Dis. Clin. N. Am. 2008, 34, 921–933. [Google Scholar] [CrossRef]
- Alunno, A.; Leone, M.C.; Giacomelli, R.; Gerli, R.; Carubbi, F. Lymphoma and Lymphomagenesis in Primary Sjögren’s Syndrome. Front. Med. 2018, 5, 102. [Google Scholar] [CrossRef]
- Strunk, J.E.; Schuẗtler, C.; Ziebuhr, J.; Stowasser, M.; Noḧte, M.; Bräuninger, A.; Gattenlöhner, S.; Mayer, K. Epstein-Barr Virus-Induced Secondary High-Grade Transformation of Sjögren’s Syndrome-Related Mucosa-Associated Lymphoid Tissue Lymphoma. J. Clin. Oncol. 2013, 31, e265–e268. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, R.K.; Pedersen, N.T. Primary Non-Hodgkin’s Lymphoma of the Thyroid Gland: A Population Based Study. Histopathology 1996, 28, 25–32. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, N.; Noh, J.Y.; Narimatsu, H.; Takeuchi, K.; Yamaguchi, T.; Kameyama, K.; Kobayashi, K.; Kami, M.; Kubo, A.; Kunii, Y.; et al. Clinicopathological Features of 171 Cases of Primary Thyroid Lymphoma: A Long-Term Study Involving 24 553 Patients with Hashimoto’s Disease. Br. J. Haematol. 2011, 153, 236–243. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Sun, M.; Liu, C. Secondary EBV-Positive Diffuse Large B-Cell Lymphoma of Skeletal Muscle from EBV-Positive Primary Diffuse Large B-Cell Lymphoma of Thyroid Gland: Case Report and Literature Review. Austin J. Med. Oncol. 2021, 8, 1057. Available online: https://austinpublishinggroup.com/medical-oncology/fulltext/ajmo-v8-id1057.php (accessed on 10 April 2022).
- Soldan, S.S.; Su, C.; Jason Lamontagne, R.; Grams, N.; Lu, F.; Zhang, Y.; Gesualdi, J.D.; Frase, D.M.; Tolvinski, L.E.; Martin, K.; et al. Epigenetic Plasticity Enables CNS-Trafficking of EBV-Infected B Lymphocytes. PLoS Pathog. 2021, 17, e1009618. [Google Scholar] [CrossRef]
- Bender, G.P.; Schapiro, R.T. Primary CNS Lymphoma Presenting as Multiple Sclerosis. Minn. Med. 1989, 72, 157–160. Available online: https://pubmed.ncbi.nlm.nih.gov/2784533/ (accessed on 10 March 2021).
- Salavoura, K.; Kolialexi, A.; Tsangaris, G.; Mavrou, A. Development of Cancer in Patients with Primary Immunodeficiencies. Anticancer Res. 2008, 28, 1263–1269. Available online: https://pubmed.ncbi.nlm.nih.gov/18505064/ (accessed on 8 January 2021).
- Verhoeven, D.; Stoppelenburg, A.J.; Meyer-Wentrup, F.; Boes, M. Increased Risk of Hematologic Malignancies in Primary Immunodeficiency Disorders: Opportunities for Immunotherapy. Clin. Immunol. 2018, 190, 22–31. [Google Scholar] [CrossRef]
- Jones, K.; Nourse, J.P.; Morrison, L.; Nguyen-Van, D.; Moss, D.J.; Burrows, S.R.; Gandhi, M.K. Expansion of EBNA1-Specific Effector T Cells in Posttransplantation Lymphoproliferative Disorders. Blood 2010, 116, 2245–2252. [Google Scholar] [CrossRef]
- Heller, K.N.; Arrey, F.; Steinherz, P.; Portlock, C.; Chadburn, A.; Kelly, K.; Münz, C. Patients with Epstein Barr Virus-Positive Lymphomas Have Decreased CD4+ T-Cell Responses to the Viral Nuclear Antigen 1. Int. J. Cancer 2008, 123, 2824–2831. [Google Scholar] [CrossRef]
- Piriou, E.; Van Dort, K.; Nanlohy, N.M.; Van Oers, M.H.J.; Miedema, F.; Van Baarle, D. Loss of EBNA1-Specific Memory CD4+ and CD8+ T Cells in HIV-Infected Patients Progressing to AIDS-Related Non-Hodgkin Lymphoma. Blood 2005, 106, 3166–3174. [Google Scholar] [CrossRef] [PubMed]
- Gasser, O.; Bihl, F.K.; Wolbers, M.; Loggi, E.; Steffen, I.; Hirsch, H.H.; Günthard, H.F.; Walker, B.D.; Brander, C.; Battegay, M.; et al. HIV Patients Developing Primary CNS Lymphoma Lack EBV-Specific CD4+ T Cell Function Irrespective of Absolute CD4+ T Cell Counts. PLoS Med. 2007, 4, e96. [Google Scholar] [CrossRef]
- Moormann, A.M.; Heller, K.N.; Chelimo, K.; Embury, P.; Ploutz-Snyder, R.; Otieno, J.A.; Oduor, M.; Münz, C.; Rochford, R. Children with Endemic Burkitt Lymphoma Are Deficient in EBNA1-Specific IFN-γ T Cell Responses. Int. J. Cancer 2009, 124, 1721–1726. [Google Scholar] [CrossRef] [PubMed]
- Gallois, A.; Silva, I.; Osman, I.; Bhardwaj, N. Reversal of Natural Killer Cell Exhaustion by TIM-3 Blockade. Oncoimmunology 2015, 3, e946365. [Google Scholar] [CrossRef] [PubMed]
- Kahan, S.M.; Wherry, E.J.; Zajac, A.J. T Cell Exhaustion during Persistent Viral Infections. Virology 2015, 479, 180–193. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Fueyo, A.; Markmann, J.F. Immune Exhaustion and Transplantation. Am. J. Transplant. 2016, 16, 1953–1957. [Google Scholar] [CrossRef]
- Wiesmayr, S.; Webber, S.A.; Macedo, C.; Popescu, I.; Smith, L.; Luce, J.; Metes, D. Decreased NKp46 and NKG2D and Elevated PD-1 Are Associated with Altered NK-Cell Function in Pediatric Transplant Patients with PTLD. Eur. J. Immunol. 2012, 42, 541–550. [Google Scholar] [CrossRef]
- Ruiz-Pablos, M.; Paiva, B.; Montero-Mateo, R.; Garcia, N.; Zabaleta, A. Epstein-Barr Virus and the Origin of Myalgic Encephalomyelitis or Chronic Fatigue Syndrome. Front. Immunol. 2021, 12, 4637. [Google Scholar] [CrossRef]
- Song, H.; Park, H.; Kim, J.; Park, G.; Kim, Y.S.; Kim, S.M.; Kim, D.; Seo, S.K.; Lee, H.K.; Cho, D.H.; et al. IDO Metabolite Produced by EBV-Transformed B Cells Inhibits Surface Expression of NKG2D in NK Cells via the c-Jun N-Terminal Kinase (JNK) Pathway. Immunol. Lett. 2011, 136, 187–193. [Google Scholar] [CrossRef]
- Thompson, M.P.; Kurzrock, R. Epstein-Barr Virus and Cancer. Clin. Cancer Res. 2004, 10, 803–821. [Google Scholar] [CrossRef]
- Mangalam, A.K.; Taneja, V.; David, C.S. HLA Class II Molecules Influence Susceptibility vs. Protection in Inflammatory Diseases by Determining the Cytokine Profile. J. Immunol. 2013, 190, 513. [Google Scholar] [CrossRef] [PubMed]
- Fukayama, M.; Abe, H.; Kunita, A.; Shinozaki-Ushiku, A.; Matsusaka, K.; Ushiku, T.; Kaneda, A. Thirty Years of Epstein-Barr Virus-Associated Gastric Carcinoma. Virchows Arch. 2020, 476, 353–365. [Google Scholar] [CrossRef] [PubMed]
- Ghasemi, F.; Tessier, T.M.; Gameiro, S.F.; Maciver, A.H.; Cecchini, M.J.; Mymryk, J.S. High MHC-II Expression in Epstein–Barr Virus-Associated Gastric Cancers Suggests That Tumor Cells Serve an Important Role in Antigen Presentation. Sci. Rep. 2020, 10, 14786. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.C.; Hattori, T.; Kushima, R.; Terata, N.; Kodama, M. Expression of Hla-Class II Antigen in Gastric Carcinomas: Its Relationship to Histopathological Grade, Lymphocyte Infiltration and Five-Year Survival Rate. Acta Oncol. 2009, 33, 187–190. [Google Scholar] [CrossRef]
- Rihane, F.E.; Hassou, N.; Nadifi, S.; Ennaji, M.M. Status of Helicobacter pylori Coinfection with Epstein–Barr Virus in Gastric Cancer. In Emerging and Reemerging Viral Pathogens; Volume 1: Fundamental and Basic Virology Aspects of Human, Animal and Plant Pathogens; Academic Press: Cambridge, MA, USA, 2020; Volume 1, pp. 571–585. [Google Scholar] [CrossRef]
- Stanland, L.J.; Luftig, M.A. The Role of EBV-Induced Hypermethylation in Gastric Cancer Tumorigenesis. Viruses 2020, 12, 1222. [Google Scholar] [CrossRef]
- Gold, J.E.; Okyay, R.A.; Licht, W.E.; Hurley, D.J. Investigation of Long COVID Prevalence and Its Relationship to Epstein-Barr Virus Reactivation. Pathogens 2021, 10, 763. [Google Scholar] [CrossRef]
- Espinosa Rodríguez, P.; Martínez Aguilar, A.; Ripoll Muñoz, M.P.; Rodríguez Navarro, M. COVID Persistente: ¿es En Realidad Una Encefalomielitis Miálgica? Revisión Bibliográfica y Consideraciones. Semergen 2022, 48, 63. [Google Scholar] [CrossRef]
- Wong, T.L.; Weitzer, D.J. Long COVID and Myalgic Encephalomyelitis/Chronic Fatigue Syndrome (ME/CFS)—A Systemic Review and Comparison of Clinical Presentation and Symptomatology. Medicina 2021, 57, 418. [Google Scholar] [CrossRef]
- Kronzer, V.L.; Bridges, S.L.; Davis, J.M. Why Women Have More Autoimmune Diseases than Men: An Evolutionary Perspective. Evol. Appl. 2021, 14, 629. [Google Scholar] [CrossRef]
- Bakken, I.J.; Tveito, K.; Gunnes, N.; Ghaderi, S.; Stoltenberg, C.; Trogstad, L.; Håberg, S.E.; Magnus, P. Two Age Peaks in the Incidence of Chronic Fatigue Syndrome/Myalgic Encephalomyelitis: A Population-Based Registry Study from Norway 2008-2012. BMC Med. 2014, 12, 167. [Google Scholar] [CrossRef]
- Bai, F.; Tomasoni, D.; Falcinella, C.; Barbanotti, D.; Castoldi, R.; Mulè, G.; Augello, M.; Mondatore, D.; Allegrini, M.; Cona, A.; et al. Female Gender Is Associated with Long COVID Syndrome: A Prospective Cohort Study. Clin. Microbiol. Infect. 2022, 28, 611.e9–611.e16. [Google Scholar] [CrossRef] [PubMed]
- Fernández-De-las-peñas, C.; Martín-Guerrero, J.D.; Pellicer-Valero, Ó.J.; Navarro-Pardo, E.; Gómez-Mayordomo, V.; Cuadrado, M.L.; Arias-Navalón, J.A.; Cigarán-Méndez, M.; Hernández-Barrera, V.; Arendt-Nielsen, L. Female Sex Is a Risk Factor Associated with Long-Term Post-COVID Related-Symptoms but Not with COVID-19 Symptoms: The LONG-COVID-EXP-CM Multicenter Study. J. Clin. Med. 2022, 11, 413. [Google Scholar] [CrossRef] [PubMed]
- Faro, M.; Sàez-Francás, N.; Castro-Marrero, J.; Aliste, L.; Fernández de Sevilla, T.; Alegre, J. Diferencias de Género En Pacientes Con Síndrome de Fatiga Crónica. Reumatol. Clínica 2016, 12, 72–77. [Google Scholar] [CrossRef] [PubMed]
- Angum, F.; Khan, T.; Kaler, J.; Siddiqui, L.; Hussain, A. The Prevalence of Autoimmune Disorders in Women: A Narrative Review. Cureus 2020, 12, e8094. [Google Scholar] [CrossRef]
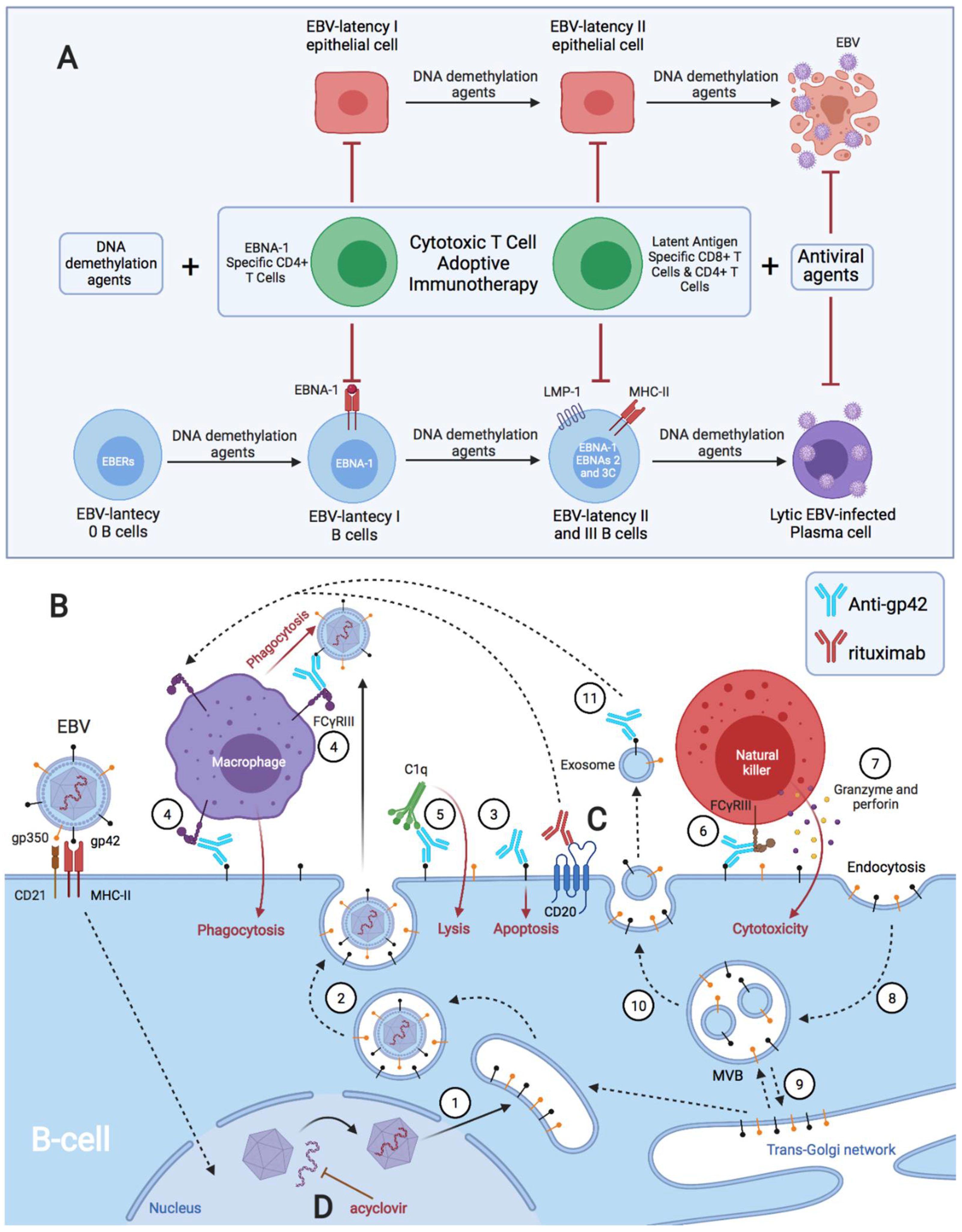
- Heslop, H.E. How I Treat EBV Lymphoproliferation. Blood 2009, 114, 4002–4008. [Google Scholar] [CrossRef]
- Pender, M.P. Preventing and Curing Multiple Sclerosis by Controlling Epstein-Barr Virus Infection. Autoimmun. Rev. 2009, 8, 563–568. [Google Scholar] [CrossRef]
- Savoldo, B.; Rooney, C.M.; Quiros-Tejeira, R.E.; Caldwell, Y.; Wagner, H.; Lee, T.; Finegold, M.J.; Dotti, G.; Heslop, H.E.; Goss, J.A. Cellular Immunity to Epstein-Barr Virus in Liver Transplant Recipients Treated with Rituximab for Post-Transplant Lymphoproliferative Disease. Am. J. Transplant. 2005, 5, 566–572. [Google Scholar] [CrossRef]
- Suzan, F.; Ammor, M.; Ribrag, V. Fatal Reactivation of Cytomegalovirus Infection after Use of Rituximab for a Post-Transplantation Lymphoproliferative Disorder. N. Engl. J. Med. 2001, 345, 1000. [Google Scholar] [CrossRef]
- Dalton, T.; Doubrovina, E.; Pankov, D.; Reynolds, R.; Scholze, H.; Selvakumar, A.; Vizconde, T.; Savalia, B.; Dyomin, V.; Weigel, C.; et al. Epigenetic Reprogramming Sensitizes Immunologically Silent EBV+ Lymphomas to Virus-Directed Immunotherapy. Blood 2020, 135, 1870–1881. [Google Scholar] [CrossRef]
- Turrini, R.; Merlo, A.; Dolcetti, R.; Zanovello, P.; Rosato, A. Differential Down-Modulation of HLA Class i and II Molecule Expression on Human Tumor Cell Lines upon in Vivo Transfer. Cancer Immunol. Immunother. 2011, 60, 1639–1645. [Google Scholar] [CrossRef][Green Version]
- Merlo, A.; Turrini, R.; Bobisse, S.; Zamarchi, R.; Alaggio, R.; Dolcetti, R.; Mautner, J.; Zanovello, P.; Amadori, A.; Rosato, A. Virus-Specific Cytotoxic CD4+ T Cells for the Treatment of EBV-Related Tumors. J. Immunol. 2010, 184, 5895–5902. [Google Scholar] [CrossRef] [PubMed]
- Nishikawa, J.; Iizasa, H.; Yoshiyama, H.; Nakamura, M.; Saito, M.; Sasaki, S.; Shimokuri, K.; Yanagihara, M.; Sakai, K.; Suehiro, Y.; et al. The Role of Epigenetic Regulation in Epstein-Barr Virus-Associated Gastric Cancer. Int. J. Mol. Sci. 2017, 18, 1606. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, M.; Nishikawa, J.; Saito, M.; Sakai, K.; Sasaki, S.; Hashimoto, S.; Okamoto, T.; Suehiro, Y.; Yamasaki, T.; Sakaida, I. Decitabine Inhibits Tumor Cell Proliferation and Up-Regulates e-Cadherin Expression in Epstein-Barr Virus-Associated Gastric Cancer. J. Med. Virol. 2017, 89, 508–517. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| EBV-Associated Diseases | |
|---|---|
| DR2-DQ6 (DRB1*1501, DQA1*0102, DQB1*0602) | Positive correlation with:
|
| DR3-DQ2 (DRB1*0301, DQA1*0501, DQB1*0201) | Positive correlation with: |
| DR4-DQ8 (DRB1*04, DQA1*03, DQB1*0302) | Positive correlation with:
|
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ruiz-Pablos, M. CD4+ Cytotoxic T Cells Involved in the Development of EBV-Associated Diseases. Pathogens 2022, 11, 831. https://doi.org/10.3390/pathogens11080831
Ruiz-Pablos M. CD4+ Cytotoxic T Cells Involved in the Development of EBV-Associated Diseases. Pathogens. 2022; 11(8):831. https://doi.org/10.3390/pathogens11080831
Chicago/Turabian StyleRuiz-Pablos, Manuel. 2022. "CD4+ Cytotoxic T Cells Involved in the Development of EBV-Associated Diseases" Pathogens 11, no. 8: 831. https://doi.org/10.3390/pathogens11080831
APA StyleRuiz-Pablos, M. (2022). CD4+ Cytotoxic T Cells Involved in the Development of EBV-Associated Diseases. Pathogens, 11(8), 831. https://doi.org/10.3390/pathogens11080831