
Impact of HCV Infection on Hepatocyte Polarity and Plasticity


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. HCV Proteins
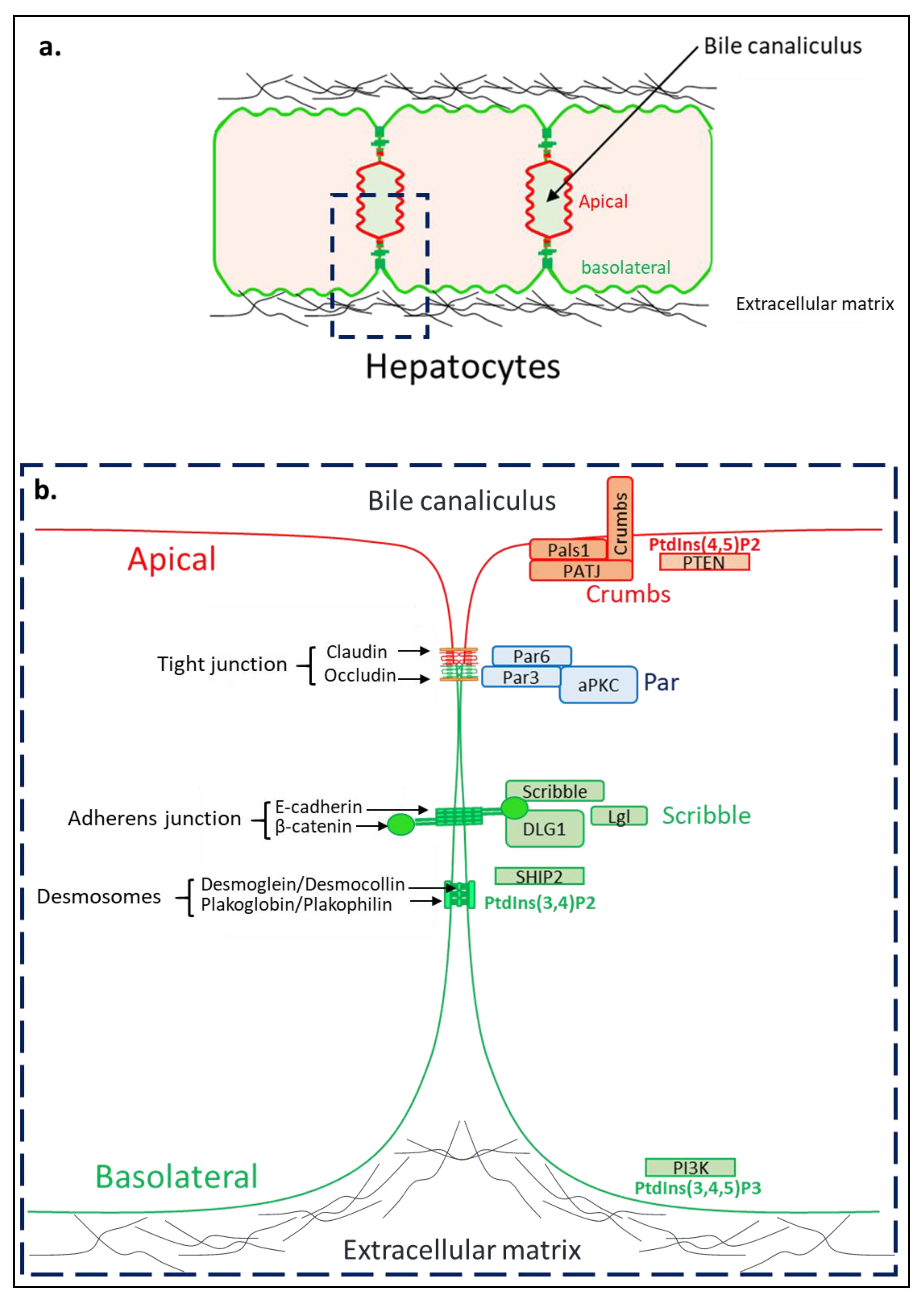
3. Hepatocyte Polarity
4. HCV Infection and Cell–Cell Junctions
5. HCV Infection and Intracellular Trafficking
6. HCV Infection and ECM
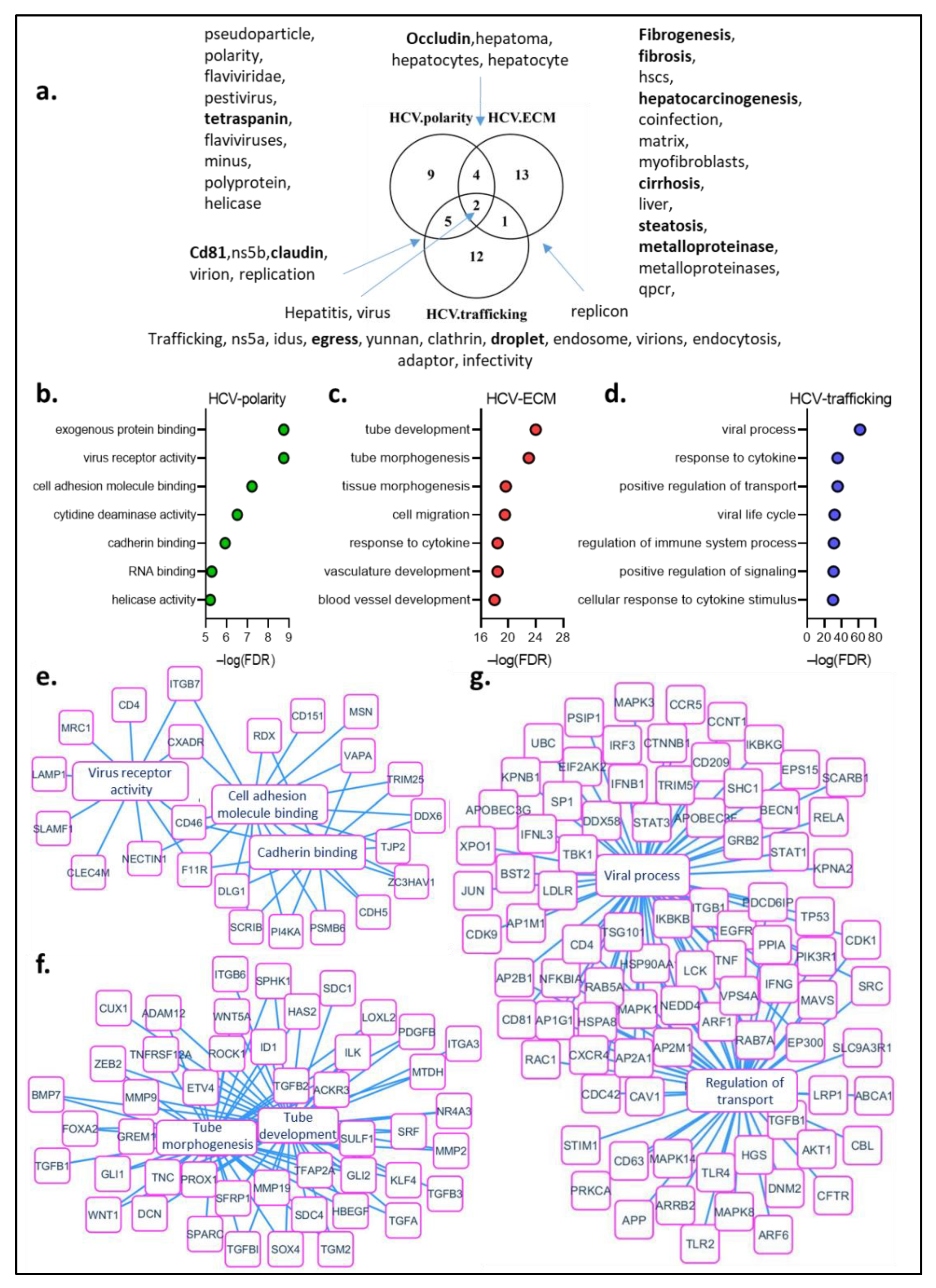
7. Text-Mining Analysis of HCV Infection and Hepatocyte Polarity
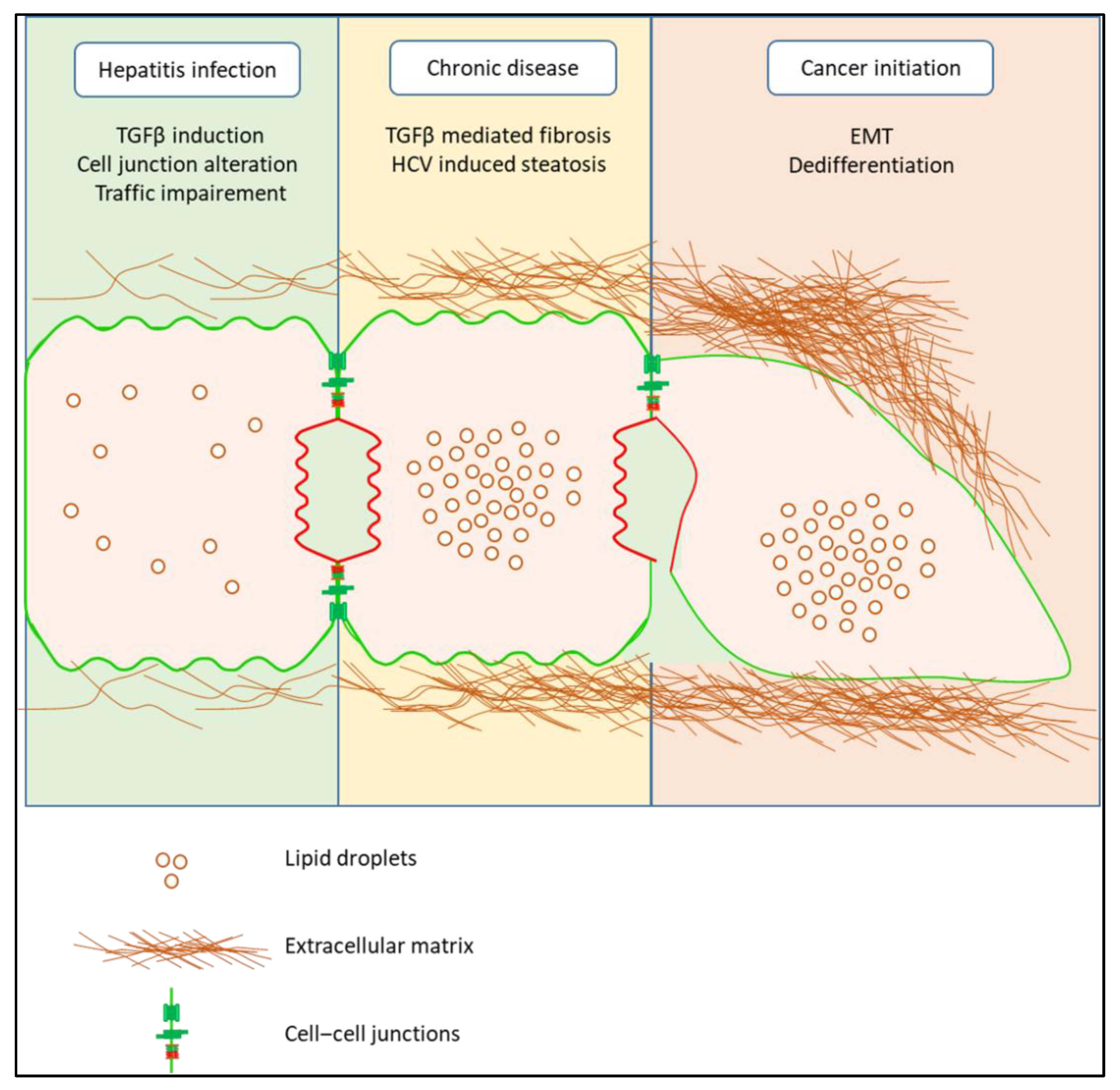
8. HCV Infection and EMT
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ABC | ATP binding cassette |
| CD81 | Cluster of differentiation |
| DAA | Direct acting antivirals |
| DDC | 3,5-diethoxycarbonyl-1,4-dihydrocollidine |
| DMV | Double membrane vesicle |
| ECM | Extracellular matrix |
| EMT | Epithelial–mesenchymal transition |
| FAK | Focal adhesion kinase |
| HCC | Hepatocellular carcinoma |
| HCV | Hepatitis C virus |
| HNF4α | Hepatocyte nuclear factor 4 alpha |
| HSC | Hepatic stellate cells |
| LD | Lipid droplet |
| MRP2 | Multidrug resistance-associated protein 2 |
| NS | Nonstructural protein |
| PI3K | phosphoinositide 3-kinase |
| PtdIns | Phosphatidylinositol |
| PTEN | Phosphatase and TENsin homolog |
| ROCK | Rho-associated protein kinase |
| SHIP2 | SH2-containing 5′-inositol phosphatase 2 |
| SVR | Sustained virological response |
| TGFβ | Transforming growth factor beta |
| VEGF | Vascular endothelial growth factor |
| WHO | World Health Organization |
References
- Feinstone, S.M.; Kapikian, A.Z.; Purcell, R.H.; Alter, H.J.; Holland, P.V. Transfusion-Associated Hepatitis Not Due to Viral Hepatitis Type A or B. N. Engl. J. Med. 1975, 292, 767–770. [Google Scholar] [CrossRef] [PubMed]
- Choo, Q.-L.; Kuo, G.; Weiner, A.J.; Overby, L.R.; Bradley, D.W.; Houghton, M. Isolation of a cDNA cLone Derived from a Blood-Borne Non-A, Non-B Viral Hepatitis Genome. Science 1989, 244, 359–362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clouston, A.D.; Powell, E.; Walsh, M.J.; Richardson, M.M.; Demetris, A.J.; Jonsson, J.R. Fibrosis correlates with a ductular reaction in hepatitis C: Roles of impaired replication, progenitor cells and steatosis. Hepatology 2005, 41, 809–818. [Google Scholar] [CrossRef] [PubMed]
- Colombo, M.; Choo, Q.; Del Ninno, E.; Dioguardi, N.; Kuo, G.; Donato, M.; Tommasini, M.; Houghton, M. Prevalence of Antibodies to Hepatitis C Virus in Italian Patients with Hepatocellular Carcinoma. Lancet 1989, 334, 1006–1008. [Google Scholar] [CrossRef]
- Kew, M.; Houghton, M.; Choo, Q.; Kuo, G. Hepatitis C virus antibodies in southern African blacks with hepatocellular carcinoma. Lancet 1990, 335, 873–874. [Google Scholar] [CrossRef]
- Lonardo, A.; Adinolfi, L.E.; Loria, P.; Carulli, N.; Ruggiero, G.; Day, C.P. Steatosis and hepatitis C virus: Mechanisms and significance for hepatic and extrahepatic disease. Gastroenterology 2004, 126, 586–597. [Google Scholar] [CrossRef]
- Polo, M.L.; Laufer, N. Extrahepatic manifestations of HCV: The role of direct acting antivirals. Expert Rev. Anti-Infect. Ther. 2017, 15, 737–746. [Google Scholar] [CrossRef]
- Cacoub, P.; Poynard, T.; Ghillani, P.; Charlotte, F.; Olivi, M.; Piette, J.C.; Opolon, P. Extrahepatic manifestations of chronic hepatitis C. Arthr. Rheum. Off. J. Am. Coll. Rheumatol. 1999, 42, 2204–2212. [Google Scholar] [CrossRef]
- Carrat, F.; Fontaine, H.; Dorival, C.; Simony, M.; Diallo, A.; Hezode, C.; De Ledinghen, V.; Larrey, D.; Haour, G.; Bronowicki, J.-P.; et al. Clinical outcomes in patients with chronic hepatitis C after direct-acting antiviral treatment: A prospective cohort study. Lancet 2019, 393, 1453–1464. [Google Scholar] [CrossRef]
- Hamdane, N.; Jühling, F.; Crouchet, E.; El Saghire, H.; Thumann, C.; Oudot, M.A.; Bandiera, S.; Saviano, A.; Ponsolles, C.; Roca Suarez, A.A.R.; et al. HCV-Induced Epigenetic Changes Associated with Liver Cancer Risk Persist after Sustained Virologic Response. Gastroenterology 2019, 156, 2313–2329.e7. [Google Scholar] [CrossRef] [Green Version]
- Polyak, S.J.; Crispe, I.N.; Baumert, T.F. Liver Abnormalities after Elimination of HCV Infection: Persistent Epigenetic and Immunological Perturbations Post-Cure. Pathogens 2021, 10, 44. [Google Scholar] [CrossRef] [PubMed]
- Perez, S.; Kaspi, A.; Domovitz, T.; Davidovich, A.; Lavi-Itzkovitz, A.; Meirson, T.; Alison Holmes, J.; Dai, C.-Y.; Huang, C.-F.; Chung, R.T.; et al. Hepatitis C virus leaves an epigenetic signature post cure of infection by direct-acting antivirals. PLoS Genet. 2019, 15, e1008181. [Google Scholar] [CrossRef] [PubMed]
- Hoshida, Y.; Villanueva, A.; Kobayashi, M.; Peix, J.; Chiang, D.Y.; Camargo, A.; Gupta, S.; Moore, J.; Wrobel, M.J.; Lerner, J.; et al. Gene Expression in Fixed Tissues and Outcome in Hepatocellular Carcinoma. N. Engl. J. Med. 2008, 359, 1995–2004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoshida, Y.; Villanueva, A.; SanGiovanni, A.; Sole, M.; Hur, C.; Andersson, K.L.; Chung, R.T.; Gould, J.; Kojima, K.; Gupta, S.; et al. Prognostic Gene Expression Signature for Patients with Hepatitis C–Related Early-Stage Cirrhosis. Gastroenterology 2013, 144, 1024–1030. [Google Scholar] [CrossRef] [Green Version]
- Nieto, M.A.; Huang, R.Y.-J.; Jackson, R.A.; Thiery, J.P. EMT: 2016. Cell 2016, 166, 21–45. [Google Scholar] [CrossRef] [Green Version]
- Thiery, J.P. Epithelial–mesenchymal transitions in tumour progression. Nat. Cancer 2002, 2, 442–454. [Google Scholar] [CrossRef]
- Jung, H.-Y.; Fattet, L.; Tsai, J.H.; Kajimoto, T.; Chang, Q.; Newton, A.C.; Yang, J. Apical–basal polarity inhibits epithelial–mesenchymal transition and tumour metastasis by PAR-complex-mediated SNAI1 degradation. Nat. Cell Biol. 2019, 21, 359–371. [Google Scholar] [CrossRef]
- Persa, O.-D.; Niessen, C.M. Epithelial polarity limits EMT. Nat. Cell Biol. 2019, 21, 299–300. [Google Scholar] [CrossRef]
- Smith, D.B.; Bukh, J.; Kuiken, C.; Muerhoff, A.S.; Rice, C.M.; Stapleton, J.T.; Simmonds, P. Expanded classification of hepatitis C virus into 7 genotypes and 67 subtypes: Updated criteria and genotype assignment web resource. Hepatology 2013, 59, 318–327. [Google Scholar] [CrossRef] [Green Version]
- Miyanari, Y.; Atsuzawa, K.; Usuda, N.; Watashi, K.; Hishiki, T.; Zayas, M.; Bartenschlager, R.; Wakita, T.; Hijikata, M.; Shimotohno, K. The lipid droplet is an important organelle for hepatitis C virus production. Nat. Cell Biol. 2007, 9, 1089–1097. [Google Scholar] [CrossRef]
- Shavinskaya, A.; Boulant, S.; Penin, F.; McLauchlan, J.; Bartenschlager, R. The Lipid Droplet Binding Domain of Hepatitis C Virus Core Protein Is a Major Determinant for Efficient Virus Assembly. J. Biol. Chem. 2007, 282, 37158–37169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Romero-Brey, I.; Berger, C.; Kallis, S.; Kolovou, A.; Paul, D.; Lohmann, V.; Bartenschlager, R. NS5A Domain 1 and Polyprotein Cleavage Kinetics Are Critical for Induction of Double-Membrane Vesicles Associated with Hepatitis C Virus Replication. mBio 2015, 6, e00759-15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gosert, R.; Egger, D.; Lohmann, V.; Bartenschlager, R.; Blum, H.E.; Bienz, K.; Moradpour, D. Identification of the Hepatitis C Virus RNA Replication Complex in Huh-7 Cells Harboring Subgenomic Replicons. J. Virol. 2003, 77, 5487–5492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paul, D.; Hoppe, S.; Saher, G.; Krijnse-Locker, J.; Bartenschlager, R. Morphological and Biochemical Charac-terization of the Membranous Hepatitis C Virus Replication Compartment. J. Virol. 2013, 87, 10612–10627. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bryant, D.M.; Mostov, K.E. From cells to organs: Building polarized tissue. Nat. Rev. Mol. Cell Biol. 2008, 9, 887–901. [Google Scholar] [CrossRef]
- Overeem, A.; Bryant, D.M.; van Ijzendoorn, S.C. Mechanisms of apical–basal axis orientation and epithelial lumen positioning. Trends Cell Biol. 2015, 25, 476–485. [Google Scholar] [CrossRef]
- Wen, W.; Zhang, M. Protein Complex Assemblies in Epithelial Cell Polarity and Asymmetric Cell Division. J. Mol. Biol. 2018, 430, 3504–3520. [Google Scholar] [CrossRef]
- Awad, A.; Sar, S.; Barré, R.; Cariven, C.; Marin, M.; Salles, J.P.; Erneux, C.; Samuel, D.; Gassama-Diagne, A. SHIP2 regulates epithelial cell polarity through its lipid product, which binds to Dlg1, a pathway subverted by hepatitis C virus core protein. Mol. Biol. Cell 2013, 24, 2171–2185. [Google Scholar] [CrossRef]
- Gassama-Diagne, A.; Yu, W.; Ter Beest, M.; Martin-Belmonte, F.; Kierbel, A.; Engel, J.N.; Mostov, K. Phosphatidylinositol-3,4,5-trisphosphate regulates the formation of the basolateral plasma membrane in epithelial cells. Nat. Cell Biol. 2006, 8, 963–970. [Google Scholar] [CrossRef]
- Martin-Belmonte, F.; Gassama, A.; Datta, A.; Yu, W.; Rescher, U.; Gerke, V.; Mostov, K. PTEN-Mediated Apical Segregation of Phosphoinositides Controls Epithelial Morphogenesis through Cdc. Cell 2007, 128, 383–397. [Google Scholar] [CrossRef] [Green Version]
- Treyer, A.; Müsch, A. Hepatocyte Polarity. Compr. Physiol. 2013, 3, 243–287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cohen, D.; Fernandez, D.; Lazaro-Dieguez, F.; Müsch, A. The serine/threonine kinase Par1b regulates epithelial lumen polarity via IRSp53-mediated cell–ECM signaling. J. Cell Biol. 2011, 192, 525–540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lazaro-Dieguez, F.; Cohen, D.; Fernandez, D.; Hodgson, L.; Van Ijzendoorn, S.C.D.; Muesch, A. Par1b links lumen polarity with LGN–NuMA positioning for distinct epithelial cell division phenotypes. J. Cell Biol. 2013, 203, 251–264. [Google Scholar] [CrossRef] [PubMed]
- Slim, C.L.; Lazaro-Dieguez, F.; Bijlard, M.; Toussaint, M.J.M.; De Bruin, A.; Du, Q.; Müsch, A.; Van Ijzendoorn, S.C.D. Par1b Induces Asymmetric Inheritance of Plasma Membrane Domains via LGN-Dependent Mitotic Spindle Orientation in Proliferating Hepatocytes. PLoS Biol. 2013, 11, e1001739. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Slim, C.L.; Van Ijzendoorn, S.C.; Lázaro-Diéguez, F.; Müsch, A. The special case of hepatocytes. BioArchitecture 2014, 4, 47–52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kipp, H.; Arias, I.M. Trafficking of Canalicular ABC Transporters in Hepatocytes. Annu. Rev. Physiol. 2002, 64, 595–608. [Google Scholar] [CrossRef]
- Kipp, H.; Pichetshote, N.; Arias, I.M. Transporters on Demand: Intrahepatic Pools of Canalicular Atp Binding Cassette Transporters In Rat Liver. J. Biol. Chem. 2001, 276, 7218–7224. [Google Scholar] [CrossRef] [Green Version]
- Mee, C.; Grove, J.; Harris, H.J.; Hu, K.; Balfe, P.; McKeating, J. Effect of Cell Polarization on Hepatitis C Virus Entry. J. Virol. 2008, 82, 461–470. [Google Scholar] [CrossRef] [Green Version]
- Mee, C.; Harris, H.J.; Farquhar, M.J.; Wilson, G.; Reynolds, G.; Davis, C.; van Ijzendoorn, S.C.D.; Balfe, P.; McKeating, J. Polarization Restricts Hepatitis C Virus Entry into HepG2 Hepatoma Cells. J. Virol. 2009, 83, 6211–6221. [Google Scholar] [CrossRef] [Green Version]
- Mailly, L.; Xiao, F.; Lupberger, J.; Wilson, G.K.; Aubert, P.; Duong, H.T.F.; Calabrese, D.; Leboeuf, C.; Fofana, I.; Thumann, C.; et al. Clearance of persistent hepatitis C virus infection in humanized mice using a claudin-1-targeting monoclonal antibody. Nat. Biotechnol. 2015, 33, 549–554. [Google Scholar] [CrossRef] [Green Version]
- Douam, F.; Lavillette, D.; Cosset, F.-L. Chapter Three—The Mechanism of HCV Entry into Host Cells. In Progress in Molecular Biology and Translational Science; Klasse, P.J., Ed.; The Molecular Basis of Viral Infection; Elsevier: Waltham, MA, USA, 2015; Volume 129, pp. 63–107. [Google Scholar]
- Baktash, Y.; Madhav, A.; Coller, K.E.; Randall, G. Single Particle Imaging of Polarized Hepatoma Organoids upon Hepatitis C Virus Infection Reveals an Ordered and Sequential Entry Process. Cell Host Microbe 2018, 23, 382–394.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- So, C.-W.; Randall, G. Three-Dimensional Cell Culture Systems for Studying Hepatitis C Virus. Viruses 2021, 13, 211. [Google Scholar] [CrossRef] [PubMed]
- Belouzard, S.; Danneels, A.; Fénéant, L.; Séron, K.; Rouillé, Y.; Dubuisson, J. Entry and Release of Hepatitis C Virus in Polarized Human Hepatocytes. J. Virol. 2017, 91, e00478-17, Correction in J. Virol. 2018, 92, e00309-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Evans, M.; Von Hahn, T.; Tscherne, D.M.; Syder, A.J.; Panis, M.; Wölk, B.; Hatziioannou, T.; McKeating, J.; Bieniasz, P.D.; Rice, C.M. Claudin-1 is a hepatitis C virus co-receptor required for a late step in entry. Nature 2007, 446, 801–805. [Google Scholar] [CrossRef]
- Liu, S.; Kuo, W.; Yang, W.; Liu, W.; Gibson, G.A.; Dorko, K.; Watkins, S.C.; Strom, S.C.; Wang, T. The second extracellular loop dictates Occludin-mediated HCV entry. Virology 2010, 407, 160–170. [Google Scholar] [CrossRef] [Green Version]
- Ploss, A.; Evans, M.; Gaysinskaya, V.A.; Panis, M.; You, H.; De Jong, Y.P.; Rice, C.M. Human occludin is a hepatitis C virus entry factor required for infection of mouse cells. Nature 2009, 457, 882–886. [Google Scholar] [CrossRef] [Green Version]
- Zeisel, M.B.; Felmlee, D.J.; Baumert, T.F. Hepatitis C VirusEntry. Curr. Top. Microbiol. Immunol. 2013, 369, 87–112. [Google Scholar] [CrossRef] [Green Version]
- Arora, P.; Kim, E.-O.; Jung, J.K.; Jang, K.L. Hepatitis C virus core protein downregulates E-cadherin expression via activation of DNA methyltransferase 1 and 3b. Cancer Lett. 2008, 261, 244–252. [Google Scholar] [CrossRef]
- Tiwari, I.; Yoon, M.-H.; Park, B.-J.; Jang, K.L. Hepatitis C virus core protein induces epithelial–mesenchymal transition in human hepatocytes by upregulating E12/E47 levels. Cancer Lett. 2015, 362, 131–138. [Google Scholar] [CrossRef]
- Pérez-Moreno, M.A.; Locascio, A.; Rodrigo, I.; Dhondt, G.; Portillo, F.; Nieto, M.A.; Cano, A. A New Role for E12/E47 in the Repression of E-cadherin Expression and Epithelial-Mesenchymal Transitions. J. Biol. Chem. 2001, 276, 27424–27431. [Google Scholar] [CrossRef] [Green Version]
- Hu, B.; Li, S.; Zhang, Z.; Xie, S.; Hu, Y.; Huang, X.; Zheng, Y. HCV NS4B targets Scribble for proteasome-mediated degradation to facilitate cell transformation. Tumor Biol. 2016, 37, 12387–12396. [Google Scholar] [CrossRef] [PubMed]
- Awad, A.; Gassama-Diagne, A. PI3K/SHIP2/PTEN pathway in cell polarity and hepatitis C virus pathogenesis. World J. Hepatol. 2017, 9, 18–29. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Sodroski, C.; Lowey, B.; Schweitzer, C.J.; Cha, H.; Zhang, F.; Liang, T.J. Hepatitis C virus depends on E-cadherin as an entry factor and regulates its expression in epithelial-to-mesenchymal transition. Proc. Natl. Acad. Sci. USA 2016, 113, 7620–7625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colpitts, C.; Lupberger, J.; Baumert, T.F. Multifaceted role of E-cadherin in hepatitis C virus infection and pathogenesis. Proc. Natl. Acad. Sci. USA 2016, 113, 7298–7300. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; Yang, W.; Shen, L.; Turner, J.R.; Coyne, C.B.; Wang, T. Tight Junction Proteins Claudin-1 and Occludin Control Hepatitis C Virus Entry and Are Downregulated during Infection To Prevent Superinfection. J. Virol. 2009, 83, 2011–2014. [Google Scholar] [CrossRef] [Green Version]
- Mee, C.J.; Farquhar, M.J.; Harris, H.J.; Hu, K.; Ramma, W.; Ahmed, A.; Maurel, P.; Bicknell, R.; Balfe, P.; McKeating, J.A. Hepatitis C Virus Infection Reduces Hepatocellular Polarity in a Vascular Endothelial Growth Factor–Dependent Manner. Gastroenterology 2010, 138, 1134–1142. [Google Scholar] [CrossRef] [Green Version]
- Hall, A. The Cellular Functions of Small GTP-Binding Proteins. Science 1990, 249, 635–640. [Google Scholar] [CrossRef]
- Amano, M.; Nakayama, M.; Kaibuchi, K. Rho-kinase/ROCK: A key regulator of the cytoskeleton and cell polarity. Cytoskeleton 2010, 67, 545–554. [Google Scholar] [CrossRef] [Green Version]
- Mostowy, S.; Cossart, P. Septins: The fourth component of the cytoskeleton. Nat. Rev. Mol. Cell Biol. 2012, 13, 183–194. [Google Scholar] [CrossRef]
- Pol, A.; Gross, S.P.; Parton, R.G. Biogenesis of the multifunctional lipid droplet: Lipids, proteins, and sites. J. Cell Biol. 2014, 204, 635–646. [Google Scholar] [CrossRef] [Green Version]
- Egger, D.; Wolk, B.; Gosert, R.; Bianchi, L.; Blum, H.E.; Moradpour, D.; Bienz, K. Expression of Hepatitis C Virus Proteins Induces Distinct Membrane Alterations Including a Candidate Viral Replication Complex. J. Virol. 2002, 76, 5974–5984. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boulant, S.; Douglas, M.W.; Moody, L.; Budkowska, A.; Targett-Adams, P.; McLauchlan, J. Hepatitis C Virus Core Protein Induces Lipid Droplet Redistribution in a Microtubule- and Dynein-Dependent Manner. Traffic 2008, 9, 1268–1282. [Google Scholar] [CrossRef] [PubMed]
- Akil, A.; Peng, J.; Omrane, M.; Gondeau, C.; Desterke, C.; Marin, M.; Tronchère, H.; Taveneau, C.; Sar, S.; Briolotti, P.; et al. Septin 9 induces lipid droplets growth by a phosphatidylinositol-5-phosphate and microtubule-dependent mechanism hijacked by HCV. Nat. Commun. 2016, 7, 12203. [Google Scholar] [CrossRef] [PubMed]
- Ozeki, S.; Cheng, J.; Tauchi-Sato, K.; Hatano, N.; Taniguchi, H.; Fujimoto, T. Rab18 localizes to lipid droplets and induces their close apposition to the endoplasmic reticulum-derived membrane. J. Cell Sci. 2005, 118, 2601–2611. [Google Scholar] [CrossRef] [Green Version]
- Dansako, H.; Hiramoto, H.; Ikeda, M.; Wakita, T.; Kato, N. Rab18 is required for viral assembly of hepatitis C virus through trafficking of the core protein to lipid droplets. Virology 2014, 462–463, 166–174. [Google Scholar] [CrossRef] [Green Version]
- Salloum, S.; Wang, H.; Ferguson, C.; Parton, R.G.; Tai, A.W. Rab18 Binds to Hepatitis C Virus NS5A and Promotes Interaction between Sites of Viral Replication and Lipid Droplets. PLoS Pathog. 2013, 9, e1003513. [Google Scholar] [CrossRef]
- Bissell, D.M.; Guzelian, P.S. Phenotypic Stability of Adult Rat Hepatocytes In Primary Monolayer Culture. Ann. N. Y. Acad. Sci. 1980, 349, 85–98. [Google Scholar] [CrossRef]
- Moghe, P.V.; Berthiaume, F.; Ezzell, R.M.; Toner, M.; Tompkins, R.G.; Yarmush, M.L. Culture matrix configuration and composition in the maintenance of hepatocyte polarity and function. Biomaterials 1996, 17, 373–385. [Google Scholar] [CrossRef]
- Zeigerer, A.; Wuttke, A.; Marsico, G.; Seifert, S.; Kalaidzidis, Y.; Zerial, M. Functional properties of hepatocytes in vitro are correlated with cell polarity maintenance. Exp. Cell Res. 2016, 350, 242–252. [Google Scholar] [CrossRef]
- Herrema, H.; Czajkowska, D.; Théard, D.; Van Der Wouden, J.M.; Kalicharan, D.; Zolghadr, B.; Hoekstra, D.; Van Ijzendoorn, S.C. Rho Kinase, Myosin-II, and p42/44 MAPK Control Extracellular Matrix-mediated Apical Bile Canalicular Lumen Morphogenesis in HepG2 Cells. Mol. Biol. Cell 2006, 17, 3291–3303. [Google Scholar] [CrossRef] [Green Version]
- Mccaughan, G. Fibrosis progression in chronic hepatitis C virus infection. Gut 2004, 53, 318–321. [Google Scholar] [CrossRef] [PubMed]
- Desai, S.S.; Tung, J.C.; Zhou, V.X.; Grenert, J.P.; Malato, Y.; Rezvani, M.; Español-Suñer, R.; Willenbring, H.; Weaver, V.M.; Chang, T.T. Physiological ranges of matrix rigidity modulate primary mouse hepatocyte function in part through hepatocyte nuclear factor 4 alpha. Hepatology 2016, 64, 261–275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deegan, D.B.; Zimmerman, C.; Skardal, A.; Atala, A.; Shupe, T.D. Stiffness of hyaluronic acid gels containing liver extracellular matrix supports human hepatocyte function and alters cell morphology. J. Mech. Behav. Biomed. Mater. 2015, 55, 87–103. [Google Scholar] [CrossRef] [PubMed]
- Martin-Belmonte, F.; Perez-Moreno, M. Epithelial cell polarity, stem cells and cancer. Nat. Cancer 2011, 12, 23–38. [Google Scholar] [CrossRef] [PubMed]
- Kaufhold, S.; Bonavida, B. Central role of Snail1 in the regulation of EMT and resistance in cancer: A target for therapeutic intervention. J. Exp. Clin. Cancer Res. 2014, 33, 62. [Google Scholar] [CrossRef]
- Lamouille, S.; Xu, J.; Derynck, R. Molecular mechanisms of epithelial–mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2014, 15, 178–196. [Google Scholar] [CrossRef] [Green Version]
- Wan, S.; Meyer, A.-S.; Weiler, S.M.E.; Rupp, C.; Tóth, M.; Sticht, C.; Singer, S.; Thomann, S.; Roessler, S.; Schorpp-Kistner, M.; et al. Cytoplasmic localization of the cell polarity factor scribble supports liver tumor formation and tumor cell invasiveness. Hepatology 2017, 67, 1842–1856. [Google Scholar] [CrossRef] [Green Version]
- Asselah, T.; Rubbia-Brandt, L.; Marcellin, P.; Negro, F. Steatosis in Chronic Hepatitis C: Why Does It Really Matter? Gut 2006, 55, 123–130. [Google Scholar] [CrossRef] [Green Version]
- Piodi, A.; Chouteau, P.; Lerat, H.; Hezode, C.; Pawlotsky, J. Morphological changes in intracellular lipid droplets induced by different hepatitis C virus genotype core sequences and relationship with steatosis. Hepatology 2008, 48, 16–27. [Google Scholar] [CrossRef] [Green Version]
- Abramczyk, H.; Surmacki, J.; Kopeć, M.; Olejnik, A.K.; Lubecka-Pietruszewska, K.; Fabianowska-Majewska, K. The role of lipid droplets and adipocytes in cancer. Raman imaging of cell cultures: MCF10A, MCF7, and MDA-MB-231 compared to adipocytes in cancerous human breast tissue. Analyst 2015, 140, 2224–2235. [Google Scholar] [CrossRef]
- De Gonzalo-Calvo, D.; López-Vilaró, L.; Nasarre, L.; Perez-Olabarria, M.; Vázquez, T.; Escuin, D.; Badimon, L.; Barnadas, A.; Lerma, E.; Llorente-Cortés, V. Intratumor cholesteryl ester accumulation is associated with human breast cancer proliferation and aggressive potential: A molecular and clinicopathological study. BMC Cancer 2015, 15, 460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yue, S.; Li, J.; Lee, S.-Y.; Lee, H.J.; Shao, T.; Song, B.; Cheng, L.; Masterson, T.A.; Liu, X.; Ratliff, T.L.; et al. Cholesteryl Ester Accumulation Induced by PTEN Loss and PI3K/AKT Activation Underlies Human Prostate Cancer Aggressiveness. Cell Metab. 2014, 19, 393–406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corbet, C.; Bastien, E.; De Jesus, J.P.S.; Dierge, E.; Martherus, R.; Linden, C.V.; Doix, B.; Degavre, C.; Guilbaud, C.; Petit, L.; et al. TGFβ2-induced formation of lipid droplets supports acidosis-driven EMT and the metastatic spreading of cancer cells. Nat. Commun. 2020, 11, 454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benzoubir, N.; Lejamtel, C.; Battaglia, S.; Testoni, B.; Benassi, B.; Gondeau, C.; Perrin-Cocon, L.; Desterke, C.; Thiers, V.; Samuel, D.; et al. HCV core-mediated activation of latent TGF-β via thrombospondin drives the crosstalk between hepatocytes and stromal environment. J. Hepatol. 2013, 59, 1160–1168. [Google Scholar] [CrossRef] [PubMed]
- Seki, E.; De Minicis, S.; Österreicher, C.H.; Kluwe, J.; Osawa, Y.; Brenner, D.; Schwabe, R.F. TLR4 enhances TGF-β signaling and hepatic fibrosis. Nat. Med. 2007, 13, 1324–1332. [Google Scholar] [CrossRef]
- Drabsch, Y.; ten Dijke, P. TGF-β signalling and its role in cancer progression and metastasis. Cancer Metast. Rev. 2012, 31, 553–568. [Google Scholar] [CrossRef]
- Meulmeester, E.; Ten Dijke, P. The dynamic roles of TGF-β in cancer. J. Pathol. 2011, 223, 206–219. [Google Scholar] [CrossRef]
- Godoy, P.; Hengstler, J.G.; Ilkavets, I.; Meyer, C.; Bachmann, A.; Müller, A.; Tuschl, G.; Mueller, S.O.; Dooley, S. Extracellular matrix modulates sensitivity of hepatocytes to fibroblastoid dedifferentiation and transforming growth factor β-induced apoptosis. Hepatology 2009, 49, 2031–2043. [Google Scholar] [CrossRef]
- Meyer, C.; Liebe, R.; Breitkopf-Heinlein, K.; Liu, Y.; Müller, A.; Rakoczy, P.; Thomas, M.; Weng, H.; Bachmann, A.; Ebert, M.; et al. Hepatocyte fate upon TGF-β challenge is determined by the matrix environment. Differentiation 2015, 89, 105–116. [Google Scholar] [CrossRef]
- Battaglia, S.; Benzoubir, N.; Nobilet, S.; Charneau, P.; Samuel, D.; Zignego, A.L.; Atfi, A.; Brechot, C.; Bourgeade, M.-F. Liver Cancer-Derived Hepatitis C Virus Core Proteins Shift TGF-Beta Responses from Tumor Suppression to Epithelial-Mesenchymal Transition. PLoS ONE 2009, 4, e4355. [Google Scholar] [CrossRef] [Green Version]
- Nie, D.; Shan, X.; Nie, L.; Duan, Y.; Chen, Z.; Yang, Y.; Li, Z.; Tian, L.; Gao, Q.; Shan, Y.; et al. Hepatitis C virus core protein interacts with Snail and histone deacetylases to promote the metastasis of hepatocellular carcinoma. Oncogene 2015, 35, 3626–3635. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Agnetti, J.; Desterke, C.; Gassama-Diagne, A. Impact of HCV Infection on Hepatocyte Polarity and Plasticity. Pathogens 2022, 11, 337. https://doi.org/10.3390/pathogens11030337
Agnetti J, Desterke C, Gassama-Diagne A. Impact of HCV Infection on Hepatocyte Polarity and Plasticity. Pathogens. 2022; 11(3):337. https://doi.org/10.3390/pathogens11030337
Chicago/Turabian StyleAgnetti, Jean, Christophe Desterke, and Ama Gassama-Diagne. 2022. "Impact of HCV Infection on Hepatocyte Polarity and Plasticity" Pathogens 11, no. 3: 337. https://doi.org/10.3390/pathogens11030337
APA StyleAgnetti, J., Desterke, C., & Gassama-Diagne, A. (2022). Impact of HCV Infection on Hepatocyte Polarity and Plasticity. Pathogens, 11(3), 337. https://doi.org/10.3390/pathogens11030337