
Helicobacter pylori-Induced Inflammation: Possible Factors Modulating the Risk of Gastric Cancer


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
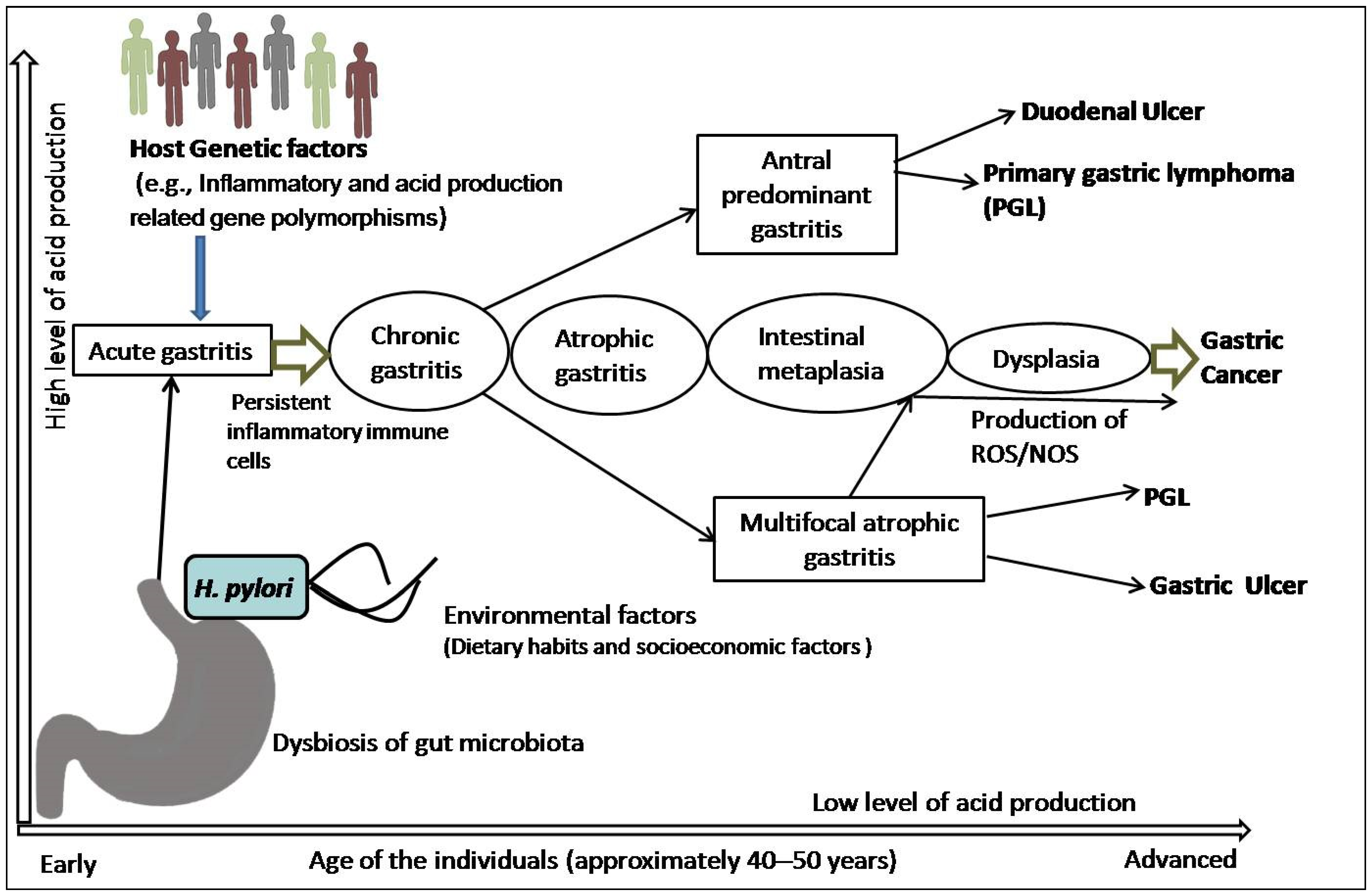
2. Mechanism of Chronic Inflammation and Multi-Step Sequel of Gastric Carcinogenesis
3. H. pylori-Triggered Inflammation and Gastric Precursor Lesions
3.1. The Link between H. pylori-Induced Inflammation and Gastric Carcinogenesis
3.2. The Link between H. pylori-Triggered AG, IM, and GC
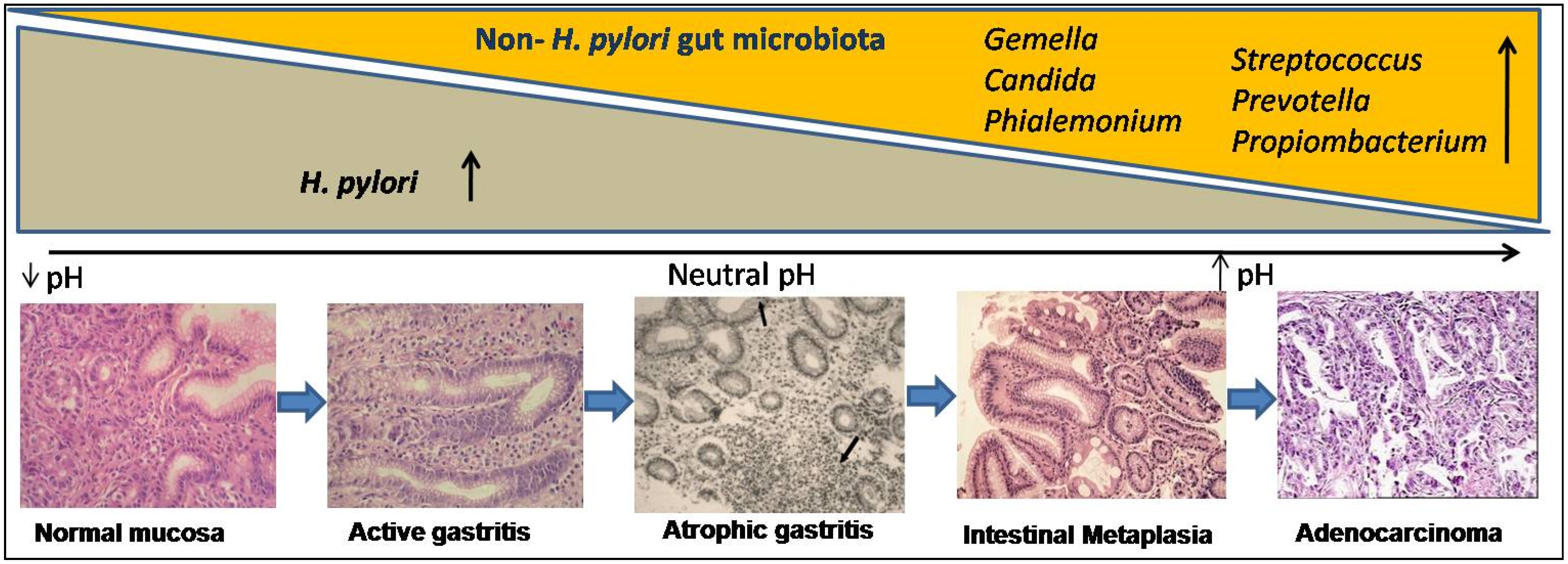
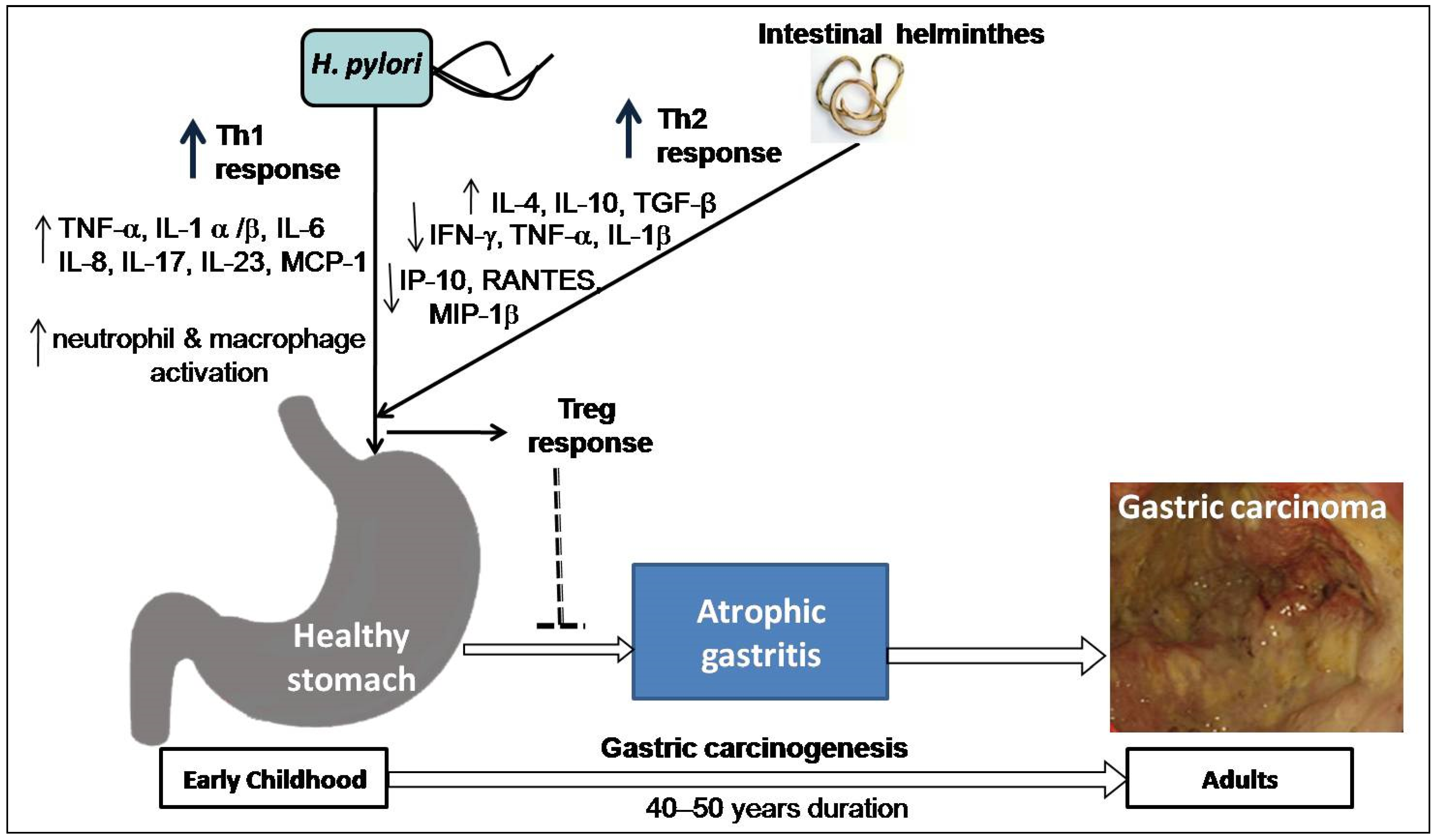
4. Risk Modulation of GC by Co-Existence of Gut Microbiota and Infestation with Intestinal Helminths
4.1. Gut Microbiota Other Than H. pylori and Their Role in Cascade of Gastric Cancer
4.2. Intestinal Helminth Infection Down-Regulates the Effect of H. pylori in Gastric Carcinogenesis
5. Relationship between Dietary Behavior of the People and Risk of GC
6. Inflammation-Related Genetic Polymorphisms Together with Other Factors as Initiators of Precancerous Lesions and GC
6.1. Cytokine Genes Influence Individual Response to Carcinogenic Exposures
6.2. Inflammatory Gene Polymorphism Alters Acid Production
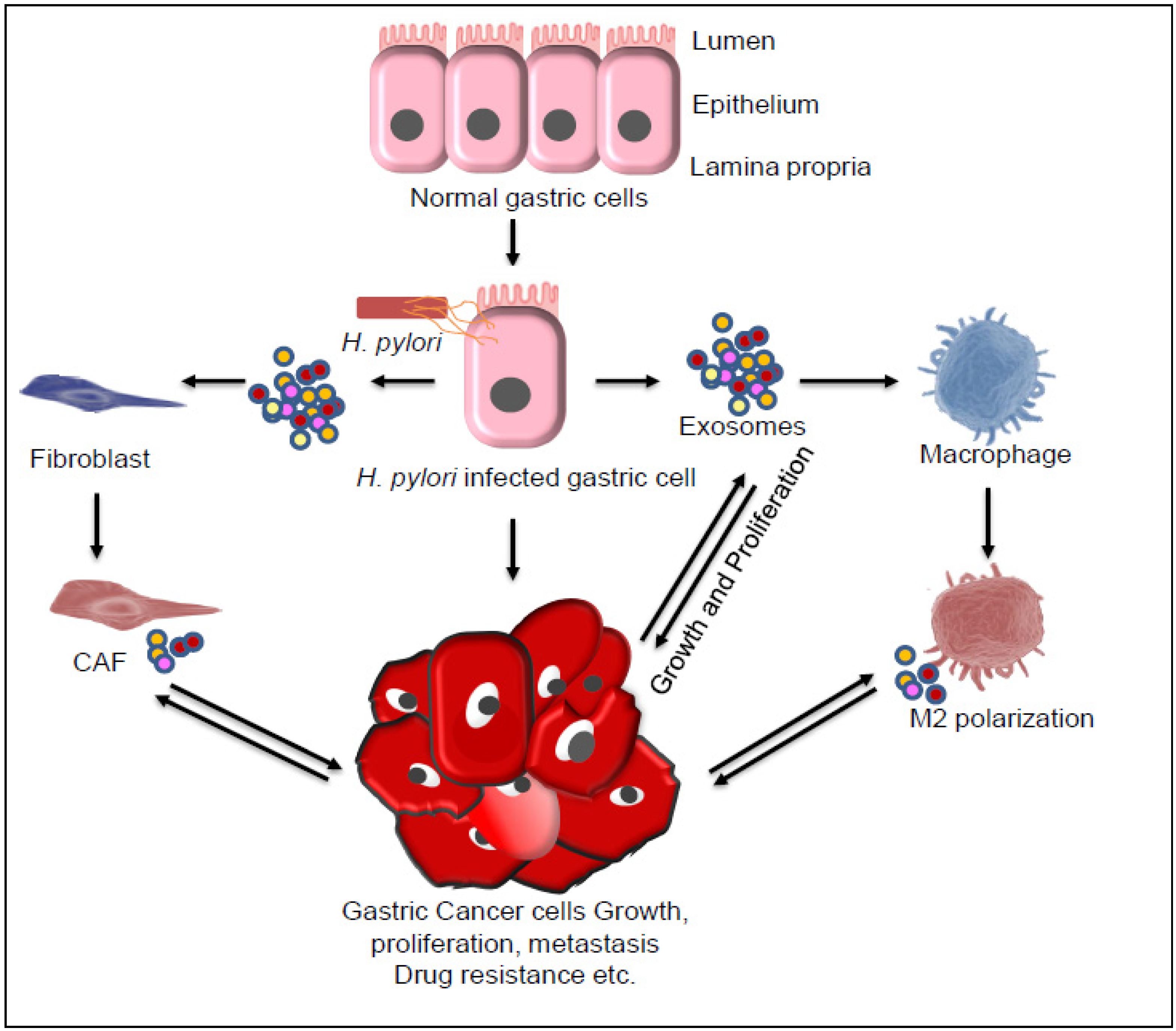
7. Role of Exosomes in Gastric Cancer
8. Conclusions and Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Berardi, R.; Scartozzi, M.; Romagnoli, E.; Antognoli, S.; Cascinu, S. Gastric cancer treatment: A systematic review. Oncol. Rep. 2004, 11, 911–916. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Plummer, M.; Franceschi, S.; Vignat, J. Global burden of gastric cancer attributable to Helicobacter pylori. Int. J. Cancer 2015, 136, 487–490. [Google Scholar] [CrossRef]
- Uemura, N.; Okamoto, S.; Yamamoto, S.; Matsumura, N.; Yamaguchi, S.; Yamakido, M.; Taniyama, K.; Sasaki, N.; Schlemper, R.J. Helicobacter pylori Infection and the Development of Gastric Cancer. N. Engl. J. Med. 2001, 345, 784–789. [Google Scholar] [CrossRef]
- Hooi, J.K.; Lai, W.Y.; Ng, W.K.; Suen, M.M.; Underwood, F.E.; Tanyingoh, D.; Malfertheiner, P.; Graham, D.Y.; Wong, V.W.; Wu, J.C.; et al. Global Prevalence of Helicobacter pylori Infection: Systematic Review and Meta-Analysis. Gastroenterology 2017, 153, 420–429. [Google Scholar] [CrossRef] [PubMed]
- Kidd, M.; Lastovica, A.J.; Atherton, J.C. Heterogeneity in the Helicobacter pylori vacA and cagA genes: Association with gastroduodenal disease in South Africa? Gut 1999, 45, 499–502. [Google Scholar] [CrossRef] [PubMed]
- Correa, P. Human gastric carcinogenesis: A multistep and multifactorial process—First American Cancer Society Award Lecture on Cancer Epidemiology and Prevention. Cancer Res. 1992, 52, 6735–6740. [Google Scholar] [PubMed]
- Sugano, K. Premalignant conditions of gastric cancer. J. Gastroenterol. Hepatol. 2013, 28, 906–911. [Google Scholar] [CrossRef]
- Ghoshal, U.C.; Chaturvedi, R.; Correa, P. The enigma of Helicobacter pylori infection and gastric cancer. Indian J. Gastroenterol. 2010, 29, 95–100. [Google Scholar] [CrossRef]
- Correa, P.; Piazuelo, B.M.; Wilson, K. Pathology of Gastric Intestinal Metaplasia: Clinical Implications. Am. J. Gastroenterol. 2010, 105, 493–498. [Google Scholar] [CrossRef]
- Wroblewski, L.E.; Peek, R.M.; Wilson, K. Helicobacter pylori and Gastric Cancer: Factors That Modulate Disease Risk. Clin. Microbiol. Rev. 2010, 23, 713–739. [Google Scholar] [CrossRef]
- Singh, K.; Ghoshal, U.C. Causal role of Helicobacter pylori infection in gastric cancer: An Asian enigma. World J. Gastroenterol. 2006, 12, 1346–1351. [Google Scholar] [CrossRef]
- Arnold, M.; Park, J.Y.; Camargo, M.C.; Lunet, N.; Forman, D.; Soerjomataram, I. Is gastric cancer becoming a rare disease? A global assessment of predicted incidence trends to 2035. Gut 2020, 69, 823–829. [Google Scholar] [CrossRef] [PubMed]
- Heer, E.V.; Harper, A.S.; Sung, H.; Jemal, A.; Fidler-Benaoudia, M.M. Emerging cancer incidence trends in Canada: The growing burden of young adult cancers. Cancer 2020, 126, 4553–4562. [Google Scholar] [CrossRef]
- Terao, S.; Suzuki, S.; Yaita, H.; Kurahara, K.; Shunto, J.; Furuta, T.; Maruyama, Y.; Ito, M.; Kamada, T.; Aoki, R.; et al. Multicenter study of autoimmune gastritis in Japan: Clinical and endoscopic characteristics. Dig. Endosc. 2020, 32, 364–372. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Kumari, N.; Mittal, R.D.; Mohindra, S.; Ghoshal, U.C. Association between pro-(IL-8) and anti-inflammatory (IL-10) cytokine variants and their serum levels and H. pylori-related gastric carcinogenesis in northern India. Meta Gene 2015, 6, 9–16. [Google Scholar] [CrossRef]
- Shukla, S.K.; Prasad, K.N.; Tripathi, A.; Singh, A.; Saxena, A.; Ghoshal, U.C.; Krishnani, N.; Husain, N. Epstein-Barr virus DNA load and its association with Helicobacter pylori infection in gastroduodenal diseases. Braz. J. Infect. Dis. 2012, 15, 583–590. [Google Scholar] [CrossRef]
- Valenzuela, M.A.; Canales, J.; Corvalan, A.; Quest, A.F.G. Helicobacter pylori-induced inflammation and epigenetic changes during gastric carcinogenesis. World J. Gastroenterol. 2015, 21, 12742–12756. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Meng, W.; Wang, B.; Qiao, L. Helicobacter pylori-induced gastric inflammation and gastric cancer. Cancer Lett. 2014, 345, 196–202. [Google Scholar] [CrossRef]
- Balkwill, F.; Mantovani, A. Inflammation and cancer: Back to Virchow? Lancet 2001, 357, 539–545. [Google Scholar] [CrossRef]
- Correa, P. Gastric cancer: Overview. Gastroenterol. Clin. N. Am. 2013, 42, 211–217. [Google Scholar] [CrossRef]
- Mishra, K.K.; Srivastava, S.; Aayyagari, A.; Ghosh, K. Development of an animal model of Helicobacter pylori (Indian strain) infection. Indian J. Gastroenterol. 2019, 38, 167–172. [Google Scholar] [CrossRef]
- Coussens, L.M.; Werb, Z. Inflammation and cancer. Nature 2002, 420, 860–867. [Google Scholar] [CrossRef]
- Correa, P.; Schneider, B.G. Etiology of gastric cancer: What is new? Cancer Epidemiol. Biomark. Prev. 2005, 14, 1865–1868. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Correa, P.; Haenszel, W.; Cuello, C.; Zavala, D.; Fontham, E.; Zarama, G.; Tannenbaum, S.; Collazos, T.; Ruiz, B. Gastric precancerous process in a high risk population: Cohort follow-up. Cancer Res. 1990, 50, 4737–4740. [Google Scholar]
- Correa, P.; Piazuelo, M. Natural history of Helicobacter pylori infection. Dig. Liver Dis. 2008, 40, 490–496. [Google Scholar] [CrossRef] [PubMed]
- Díaz, P.; Valderrama, M.V.; Bravo, J.; Quest, A.F.G. Helicobacter pylori and Gastric Cancer: Adaptive Cellular Mechanisms Involved in Disease Progression. Front. Microbiol. 2018, 9, 5. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. International Agency for Research on Cancer: Schistosomes LFaHp. IARC Monogr. Eval. Carcinog. Risks Hum. 1994, 61, 9–243. [Google Scholar]
- Nomura, A.; Stemmermann, G.N.; Chyou, P.-H.; Perez, G.P.; Blaser, M.J. Helicobacter pylori Infection and the Risk for Duodenal and Gastric Ulceration. Ann. Intern. Med. 1994, 120, 977–981. [Google Scholar] [CrossRef] [PubMed]
- Houghton, J.; Wang, T.C. Helicobacter pylori and Gastric Cancer: A New Paradigm for Inflammation-Associated Epithelial Cancers. Gastroenterology 2005, 128, 1567–1578. [Google Scholar] [CrossRef]
- Correa, P. Helicobacter pylori and gastric cancer: State of the art. Cancer Epidemiol. Biomark. Prev. 1996, 5, 477–481. [Google Scholar]
- Atherton, J.C.H. Pylori virulence factors. Br Med. Bull. 1998, 54, 105–120. [Google Scholar] [CrossRef] [PubMed]
- Graham, D.Y.; Yamaoka, Y. Disease-specific Helicobacter pylori Virulence Factors: The Unfulfilled Promise. Helicobacter 2000, 5, 3–9. [Google Scholar] [CrossRef]
- Bodger, K.; Crabtree, J.E. Helicobacter pylori and gastric inflammation. Br Med. Bull. 1998, 54, 139–150. [Google Scholar] [CrossRef]
- Crabtree, J.E. Role of cytokines in pathogenesis of Helicobacter pylori-induced mucosal damage. Dig. Dis. Sci. 1998, 43, 46S–55S. [Google Scholar]
- Harris, P.R.; Smythies, L.E.; Smith, P.D.; Dubois, A. Inflammatory Cytokine mRNA Expression during Early and Persistent Helicobacter pylori Infection in Nonhuman Primates. J. Infect. Dis. 2000, 181, 783–786. [Google Scholar] [CrossRef][Green Version]
- Tomita, T.; Jackson, A.M.; Hida, N. Expression of Interleukin-18, a Th1 cytokine, in human gastric mucosa is increased in Helicobacter pylori infection. J. Infect. Dis. 2001, 183, 620–627. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Wang, J.; Brooks, E.G.; Bamford, K.B. Negative selection of T cells by Helicobacter pylori as a model for bacterial strain selection by immune evasion. J. Immunol. 2001, 167, 926–934. [Google Scholar] [CrossRef] [PubMed]
- Smythies, L.E.; Waites, K.B.; Lindsey, J.R.; Harris, P.R.; Ghiara, P.; Smith, P.D. Helicobacter pylori-Induced Mucosal Inflammation Is Th1 Mediated and Exacerbated in IL-4, But Not IFN-γ, Gene-Deficient Mice. J. Immunol. 2000, 165, 1022–1029. [Google Scholar] [CrossRef] [PubMed]
- Amedei, A.; Munari, F.; DELLA Bella, C.; Niccolai, E.; Benagiano, M.; Bencini, L.; Cianchi, F.; Farsi, M.; Emmi, G.; Zanotti, G.; et al. Helicobacter pylori secreted peptidyl prolyl cis, trans-isomerase drives Th17 inflammation in gastric adenocarcinoma. Intern. Emerg. Med. 2012, 9, 303–309. [Google Scholar] [CrossRef]
- Dixon, M.F.; Genta, R.M.; Yardley, J.H. Classification and grading of gastritis. The updated Sydney System. International Workshop on the Histopathology of Gastritis, Houston 1994. Am. J. Surg. Pathol. 1996, 20, 1161–1181. [Google Scholar] [CrossRef] [PubMed]
- Genta, R.M.; Lew, G.M.; Graham, D.Y. Changes in the gastric mucosa following eradication of Helicobacter pylori. Mod. Pathol. 1993, 6, 271–289. [Google Scholar]
- Fallone, C.A.; Chiba, N.; van Zanten, S.V.; Fischbach, L.; Gisbert, J.P.; Hunt, R.H.; Jones, N.L.; Render, C.; Leontiadis, G.I.; Moayyedi, P. The Toronto Consensus for the Treatment of Helicobacter pylori Infection in Adults. Gastroenterology 2016, 151, 51–69.e14. [Google Scholar] [CrossRef] [PubMed]
- Jha, S.K.; Mishra, M.K.; Saharawat, K.; Jha, P.; Purkayastha, S.; Ranjan, R. Comparison of concomitant therapy versus standard triple-drug therapy for eradication of Helicobacter pylori infection: A prospective open-label randomized controlled trial. Indian J. Gastroenterol. 2019, 38, 325–331. [Google Scholar] [CrossRef] [PubMed]
- Sipponen, P.; Maaroos, H.I. Chronic gastritis. Scand. J. Gastroenterol. 2015, 50, 657–667. [Google Scholar] [CrossRef]
- Sipponen, P.; Riihela, M.; Hyvarinen, H. Chronic nonatropic (’superficial’) gastritis increases the risk of gastric carcinoma. A case-control study. Scand. J. Gastroenterol. 1994, 29, 336–340. [Google Scholar] [CrossRef]
- Correa, P.; Haenszel, W.; Cuello, C. A model for gastric cancer epidemiology. Lancet 1975, 2, 58–60. [Google Scholar] [CrossRef]
- Rugge, M.; Capelle, L.G.; Cappellesso, R.; Nitti, D.; Kuipers, E.J. Precancerous lesions in the stomach: From biology to clinical patient management. Best Pr. Res. Clin. Gastroenterol. 2013, 27, 205–223. [Google Scholar] [CrossRef]
- Kuipers, E.J.; Lee, A.; Klinkenberg-Knol, E.C. Review article: The development of atrophic gastritis--Helicobacter pylori and the effects of acid suppressive therapy. Aliment Pharmacol. Ther. 1995, 9, 331–340. [Google Scholar] [CrossRef]
- Kuipers, E.J.; Uyterlinde, A.M.; Peña, A.S.; Hazenberg, H.J.; Bloemena, E.; Lindeman, J.; Klinkenberg-Knol, E.C.; Meuwissen, S.G. Increase of Helicobacter pylori-associated corpus gastritis during acid suppressive therapy: Implications for long-term safety. Am. J. Gastroenterol. 1995, 90, 1401–1406. [Google Scholar]
- Crabtree, J.; Taylor, J.; Heatley, R.; Shallcross, T.; Rathbone, B.; Wyatt, J.; Tompkins, D. Mucosal IgA recognition of Helicobacter pylori 120 kDa protein, peptic ulceration, and gastric pathology. Lancet 1991, 338, 332–335. [Google Scholar] [CrossRef]
- Kuipers, E.J.; Pérez-Pérez, G.I.; Meuwissen, S.G.M.; Blaser, M.J. Helicobacter pylori and Atrophic Gastritis: Importance of the cagA Status. J. Natl. Cancer Inst. 1995, 87, 1777–1780. [Google Scholar] [CrossRef] [PubMed]
- Parsonnet, J.; Friedman, G.D.; Orentreich, N.; Vogelman, H. Risk for gastric cancer in people with CagA positive or CagA negative Helicobacter pylori infection. Gut 1997, 40, 297–301. [Google Scholar] [CrossRef] [PubMed]
- Ghoshal, U.C.; Tiwari, S.; Dhingra, S.; Pandey, R.; Ghoshal, U.; Tripathi, S.; Singh, H.; Gupta, V.K.; Nagpal, A.K.; Naik, S. Frequency of Helicobacter pylori and CagA Antibody in Patients with Gastric Neoplasms and Controls: The Indian Enigma. Dig. Dis. Sci. 2008, 53, 1215–1222. [Google Scholar] [CrossRef]
- Sipponen, P.; Kimura, K. Intestinal metaplasia, atrophic gastritis and stomach cancer: Trends over time. Eur. J. Gastroenterol. Hepatol. 1994, 6 (Suppl. 1), S79–S83. [Google Scholar] [PubMed]
- Sipponen, P.; Kekki, M.; Haapakoski, J.; Ihamäki, T.; Siurala, M. Gastric cancer risk in chronic atrophic gastritis: Statistical calculations of cross-sectional data. Int. J. Cancer 1985, 35, 173–177. [Google Scholar] [CrossRef] [PubMed]
- Semino-Mora, C.; Doi, S.Q.; Marty, A.; Simko, V.; Carlstedt, I.; Dubois, A. Intracellular and Interstitial Expression ofHelicobacterpyloriVirulence Genes in Gastric Precancerous Intestinal Metaplasia and Adenocarcinoma. J. Infect. Dis. 2003, 187, 1165–1177. [Google Scholar] [CrossRef] [PubMed]
- Ghoshal, U.C.; Kumar, S.; Krishnani, N.; Kumari, N.; Chourasia, D.; Tripathi, S. Serological assessment of gastric intestinal metaplasia and atrophy using pepsinogen-I, pepsinogen-II and gastrin-17 levels in a low incidence area of gastric cancer endemic for H. pylori infection. Trop. Gastroenterol. 2012, 32, 292–298. [Google Scholar]
- Weis, V.; Goldenring, J.R. Current understanding of SPEM and its standing in the preneoplastic process. Gastric Cancer 2009, 12, 189–197. [Google Scholar] [CrossRef]
- Stemmermann, G.N. Intestinal metaplasia of the stomach. A status report. Cancer 1994, 74, 556–564. [Google Scholar] [CrossRef]
- Jass, J.R.; Filipe, M.I. Sulphomucins and precancerous lesions of the human stomach. Histopathology 1980, 4, 271–279. [Google Scholar] [CrossRef]
- De Vries, A.C.; van Grieken, N.C.; Looman, C.W. Gastric cancer risk in patients with premalignant gastric lesions: A nationwide cohort study in the Netherlands. Gastroenterology 2008, 134, 945–952. [Google Scholar] [CrossRef]
- Filipe, M.I.; Muñoz, N.; Matko, I.; Kato, I.; Pompe-Kirn, V.; Jutersek, A.; Teuchmann, S.; Benz, M.; Prijon, T. Intestinal metaplasia types and the risk of gastric cancer: A cohort study in Slovenia. Int. J. Cancer 1994, 57, 324–329. [Google Scholar] [CrossRef] [PubMed]
- Safatle-Ribeiro, A.V.; Ribeiro, U.; Clarke, M.R.; Sakai, P.; Ishioka, S.; Garrido, A.B.; Gama-Rodrigues, J.; Safatle, N.F.; Reynolds, J.C. Relationship between persistence of Helicobacter pylori and dysplasia, intestinal metaplasia, atrophy, inflammation, and cell proliferation following partial gastrectomy. Dig. Dis. Sci. 1999, 44, 243–252. [Google Scholar] [CrossRef] [PubMed]
- Pulipati, P.; Sarkar, P.; Jakkampudi, A. The Indian gut microbiota-Is it unique? Indian. J. Gastroenterol. 2020, 39, 133–140. [Google Scholar] [CrossRef] [PubMed]
- Ghoshal, U.C.; Goel, A.; Quigley, E.M.M. Gut microbiota abnormalities, small intestinal bacterial overgrowth, and non-alcoholic fatty liver disease: An emerging paradigm. Indian J. Gastroenterol. 2020, 39, 9–21. [Google Scholar] [CrossRef]
- Schulz, C.; Schütte, K.; Malfertheiner, P. Helicobacter pylori and Other Gastric Microbiota in Gastroduodenal Pathologies. Dig. Dis. 2016, 34, 210–216. [Google Scholar] [CrossRef]
- Schulz, C.; Schütte, K.; Mayerle, J.; Malfertheiner, P. The role of the gastric bacterial microbiome in gastric cancer: Helicobacter pylori and beyond. Ther. Adv. Gastroenterol. 2019, 12, 1756284819894062. [Google Scholar] [CrossRef]
- Tu, S.; Bhagat, G.; Cui, G. Overexpression of interleukin-1beta induces gastric inflammation and cancer and mobilizes myeloid-derived suppressor cells in mice. Cancer Cell. 2008, 14, 408–419. [Google Scholar] [CrossRef]
- Huang, F.Y.; Chan, A.O.; Rashid, A. Interleukin-1beta increases the risk of gastric cancer through induction of aberrant DNA methylation in a mouse model. Oncol. Lett. 2016, 11, 2919–2924. [Google Scholar] [CrossRef]
- Lofgren, J.L.; Whary, M.T.; Ge, Z.; Muthupalani, S.; Taylor, N.S.; Mobley, M.; Potter, A.; Varro, A.; Eibach, D.; Suerbaum, S.; et al. Lack of Commensal Flora in Helicobacter pylori–Infected INS-GAS Mice Reduces Gastritis and Delays Intraepithelial Neoplasia. Gastroenterology 2011, 140, 210–220.e4. [Google Scholar] [CrossRef]
- Wang, T.C.; Dangler, C.A.; Chen, D.; Goldenring, J.R.; Koh, T.; Raychowdhury, R.; Coffey, R.J.; Ito, S.; Varro, A.; Dockray, G.J.; et al. Synergistic interaction between hypergastrinemia and Helicobacter infection in a mouse model of gastric cancer. Gastroenterology 2000, 118, 36–47. [Google Scholar] [CrossRef]
- Ferreira, R.M.; Pereira-Marques, J.; Pinto-Ribeiro, I.; Costa, J.L.; Carneiro, F.; Machado, J.C.; Figueiredo, C. Gastric microbial community profiling reveals a dysbiotic cancer-associated microbiota. Gut 2017, 67, 226–236. [Google Scholar] [CrossRef]
- Guo, Y.; Zhang, Y.; Gerhard, M.; Gao, J.-J.; Mejias-Luque, R.; Zhang, L.; Vieth, M.; Ma, J.-L.; Bajbouj, M.; Suchanek, S.; et al. Effect of Helicobacter pylori on gastrointestinal microbiota: A population-based study in Linqu, a high-risk area of gastric cancer. Gut 2019, 69, 1598–1607. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Zhou, X.; Liu, X.; Ling, Z.; Ji, F. Role of the Gastric Microbiome in Gastric Cancer: From Carcinogenesis to Treatment. Front. Microbiol. 2021, 12, 641322. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Shao, L.; Liu, X. Alterations of gastric mucosal microbiota across different stomach microhabitats in a cohort of 276 patients with gastric cancer. E Bio. Med. 2019, 40, 336–348. [Google Scholar] [CrossRef] [PubMed]
- Sung, J.J.Y.; Coker, O.O.; Chu, E.; Szeto, C.H.; Luk, S.T.Y.; Lau, H.; Yu, J. Gastric microbes associated with gastric inflammation, atrophy and intestinal metaplasia 1 year after Helicobacter pylori eradication. Gut 2020, 69, 1572–1581. [Google Scholar] [CrossRef] [PubMed]
- Barra, W.F.; Sarquis, D.P.; Khayat, A.S. Gastric Cancer Microbiome. Pathobiology 2021, 88, 156–169. [Google Scholar] [CrossRef]
- Bravo, L.E.; van Doom, L.J.; Realpe, J.L. Virulence-associated genotypes of Helicobacter pylori: Do they explain the African enigma? Am. J. Gastroenterol. 2002, 97, 2839–2842. [Google Scholar] [CrossRef]
- Fox, J.G.; Beck, P.; Dangler, C.A.; Whary, M.T.; Wang, T.C.; Shi, H.N.; Nagler-Anderson, C. Concurrent enteric helminth infection modulates inflammation and gastric immune responses and reduces helicobacter-induced gastric atrophy. Nat. Med. 2000, 6, 536–542. [Google Scholar] [CrossRef]
- Fuenmayor-Boscán, A.; Hernández-Rincón, I.; Arismendi-Morillo, G.; Mengual, E.; Rivero, Z.; Romero, G.; Lizarzábal, M.; Álvarez-Mon, M. Changes in the severity of gastric mucosal inflammation associated with Helicobacter pylori in humans coinfected with intestinal helminths. Indian J. Gastroenterol. 2020, 39, 186–195. [Google Scholar] [CrossRef]
- Hussain, Z.; El-Omar, E.; Lee, Y.Y. Dual infective burden of Helicobacter pylori and intestinal parasites: Good or bad news for the host? Indian J. Gastroenterol. 2020, 39, 111–116. [Google Scholar] [CrossRef]
- Du, Y.; Agnew, A.; Ye, X.-P.; Robinson, P.A.; Forman, D.; Crabtree, J.E. Helicobacter pylori and Schistosoma japonicum co-infection in a Chinese population: Helminth infection alters humoral responses to H. pylori and serum pepsinogen I/II ratio. Microbes Infect. 2006, 8, 52–60. [Google Scholar] [CrossRef]
- Whary, M.T.; Sundina, N.; Bravo, L.E.; Correa, P.; Quiñones, F.; Caro, F.; Fox, J.G. Intestinal Helminthiasis in Colombian Children Promotes a Th2 Response to Helicobacter pylori: Possible Implications for Gastric Carcinogenesis. Cancer Epidemiol. Biomark. Prev. 2005, 14, 1464–1469. [Google Scholar] [CrossRef] [PubMed]
- McGinty, J.W.; Ting, H.-A.; Billipp, T.E.; Nadjsombati, M.S.; Khan, D.M.; Barrett, N.A.; Liang, H.-E.; Matsumoto, I.; von Moltke, J. Tuft-Cell-Derived Leukotrienes Drive Rapid Anti-helminth Immunity in the Small Intestine but Are Dispensable for Anti-protist Immunity. Immunity 2020, 52, 528–541.e7. [Google Scholar] [CrossRef]
- Von Moltke, J.; Ji, M.; Liang, H.E. Tuft-cell-derived IL-25 regulates an intestinal ILC2-epithelial response circuit. Nature 2016, 529, 221–225. [Google Scholar] [CrossRef]
- Yousefi, B.; Mohammadlou, M.; Abdollahi, M.; Farrokhi, A.S.; Karbalaei, M.; Keikha, M.; Kokhaei, P.; Valizadeh, S.; Rezaiemanesh, A.; Arabkari, V.; et al. Epigenetic changes in gastric cancer induction by Helicobacter pylori. J. Cell. Physiol. 2019, 234, 21770–21784. [Google Scholar] [CrossRef] [PubMed]
- Minarovits, J. Microbe-induced epigenetic alterations in host cells: The coming era of patho-epigenetics of microbial infections. Acta Microbiol. Immunol. Hung. 2009, 56, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Ghoshal, U.C.; Tripathi, S.; Ghoshal, U. The Indian enigma of frequent H. pylori infection but infrequent gastric cancer: Is the magic key in Indian diet, host’s genetic make up, or friendly bug? Am. J. Gastroenterol. 2007, 102, 2113–2114. [Google Scholar] [CrossRef] [PubMed]
- Parsonnet, J.; Friedman, G.D.; Vandersteen, D.P.; Chang, Y.; Vogelman, J.H.; Orentreich, N.; Sibley, R.K. Helicobacter pyloriInfection and the Risk of Gastric Carcinoma. N. Engl. J. Med. 1991, 325, 1127–1131. [Google Scholar] [CrossRef] [PubMed]
- Jakszyn, P.; Gonzalez, C.A. Nitrosamine and related food intake and gastric and oesophageal cancer risk: A systematic review of the epidemiological evidence. World J. Gastroenterol. 2006, 12, 4296–4303. [Google Scholar] [CrossRef]
- Butt, J.; Varga, M.G.; Wang, T.; Tsugane, S.; Shimazu, T.; Zheng, W.; Abnet, C.C.; Yoo, K.-Y.; Park, S.K.; Kim, J.; et al. Smoking, Helicobacter Pylori Serology, and Gastric Cancer Risk in Prospective Studies from China, Japan, and Korea. Cancer Prev. Res. 2019, 12, 667–674. [Google Scholar] [CrossRef]
- Kumar, S.; Metz, D.C.; Ellenberg, S.; Kaplan, D.E.; Goldberg, D.S. Risk Factors and Incidence of Gastric Cancer After Detection of Helicobacter pylori Infection: A Large Cohort Study. Gastroenterology 2020, 158, 527–536.e7. [Google Scholar] [CrossRef]
- Van Hecke, T.; van Camp, J.; De Smet, S. Oxidation During Digestion of Meat: Interactions with the Diet and Helicobacter pylori Gastritis, and Implications on Human Health. Compr. Rev. Food Sci. Food Saf. 2017, 16, 214–233. [Google Scholar] [CrossRef] [PubMed]
- La Torre, G.; Chiaradia, G.; Gianfagna, F. Smoking status and gastric cancer risk: An updated meta-analysis of case-control studies published in the past ten years. Tumori 2009, 95, 13–22. [Google Scholar] [CrossRef]
- Ladeiras-Lopes, R.; Pereira, A.K.; Nogueira, A.; Pinheiro-Torres, T.; Pinto, I.; Santos-Pereira, R.; Lunet, N. Smoking and gastric cancer: Systematic review and meta-analysis of cohort studies. Cancer Causes Control. 2008, 19, 689–701. [Google Scholar] [CrossRef] [PubMed]
- D’Elia, L.; Rossi, G.; Ippolito, R.; Cappuccio, F.P.; Strazzullo, P. Habitual salt intake and risk of gastric cancer: A meta-analysis of prospective studies. Clin. Nutr. 2012, 31, 489–498. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Liu, L.; Wang, X.; Wang, J.; Yan, Z.; Cheng, J.; Gong, G.; Li, G. Body Mass Index and Risk of Gastric Cancer: A Meta-analysis of a Population with More Than Ten Million from 24 Prospective Studies. Cancer Epidemiol. Biomark. Prev. 2013, 22, 1395–1408. [Google Scholar] [CrossRef]
- López-Carrillo, L.; López-Cervantes, M.; Ramírez-Espitia, A.; Rueda, C.; Fernández-Ortega, C.; Orozco-Rivadeneyra, S. Alcohol consumption and gastric cancer in Mexico. Cad. Saude Publica 1998, 14, S25–S32. [Google Scholar] [CrossRef][Green Version]
- Clinton, S.K.; Giovannucci, E.L.; Hursting, S.D. The World Cancer Research Fund/American Institute for Cancer Research Third Expert Report on Diet, Nutrition, Physical Activity, and Cancer: Impact and Future Directions. J. Nutr. 2019, 150, 663–671. [Google Scholar] [CrossRef]
- González, C.A.; Pera, G.; Agudo, A.; Bueno-De-Mesquita, H.B.; Ceroti, M.; Boeing, H.; Schulz, M.; Del Giudice, G.; Plebani, M.; Carneiro, F.; et al. Fruit and vegetable intake and the risk of stomach and oesophagus adenocarcinoma in the European Prospective Investigation into Cancer and Nutrition (EPIC–EURGAST). Int. J. Cancer 2006, 118, 2559–2566. [Google Scholar] [CrossRef] [PubMed]
- Serafini, M.; Bellocco, R.; Wolk, A.; Ekström, A.M. Total antioxidant potential of fruit and vegetables and risk of gastric cancer. Gastroenterology 2002, 123, 985–991. [Google Scholar] [CrossRef]
- Foryst-Ludwig, A.; Neumann, M.; Schneider-Brachert, W. Curcumin blocks NF-kappaB and the motogenic response in Helicobacter pylori-infected epithelial cells. Biochem. Biophys. Res. Commun. 2004, 316, 1065–1072. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.P.; Ahuja, V.; Ghoshal, U.C.; Makharia, G.; Dutta, U.; Zargar, S.A.; Venkataraman, J.; Dutta, A.K.; Mukhopadhyay, A.K.; Singh, A.; et al. Management of Helicobacter pylori infection: The Bhubaneswar Consensus Report of the Indian Society of Gastroenterology. Indian J. Gastroenterol. 2021, 1–25. [Google Scholar] [CrossRef]
- Okada, F.; Izutsu, R.; Goto, K.; Osaki, M. Inflammation-Related Carcinogenesis: Lessons from Animal Models to Clinical Aspects. Cancers 2021, 13, 921. [Google Scholar] [CrossRef] [PubMed]
- Machado, J.C.; Figueiredo, C.; Canedo, P.; Pharoah, P.; Carvalho, R.; Nabais, S.; Alves, C.C.; Campos, M.L.; Van Doorn, L.-J.; Caldas, C.; et al. A proinflammatory genetic profile increases the risk for chronic atrophic gastritis and gastric carcinoma. Gastroenterology 2003, 125, 364–371. [Google Scholar] [CrossRef]
- Graham, D.Y. Helicobacter pylori Update: Gastric Cancer, Reliable Therapy, and Possible Benefits. Gastroenterology 2015, 148, 719–731.e3. [Google Scholar] [CrossRef] [PubMed]
- Forman, D.; Newell, D.G.; Fullerton, F.; Yarnell, J.W.; Stacey, A.R.; Wald, N.; Sitas, F. Association between infection with Helicobacter pylori and risk of gastric cancer: Evidence from a prospective investigation. BMJ 1991, 302, 1302–1305. [Google Scholar] [CrossRef] [PubMed]
- Sugimoto, M.; Furuta, T.; Yamaoka, Y. Influence of inflammatory cytokine polymorphisms on eradication rates ofHelicobacter pylori. J. Gastroenterol. Hepatol. 2009, 24, 1725–1732. [Google Scholar] [CrossRef][Green Version]
- White, J.R.; Winter, J.A.; Robinson, K. Differential inflammatory response to Helicobacter pylori infection: Etiology and clinical outcomes. J. Inflamm. Res. 2015, 8, 137–147. [Google Scholar]
- Morse, H.; Olomolaiye, O.; Wood, N.; Keen, L.; Bidwell, J. Induced Heteroduplex Genotyping of Tnf-A, Il-1β, Il-6 And Il-10 Polymorphisms Associated With Transcriptional Regulation. Cytokine 1999, 11, 789–795. [Google Scholar] [CrossRef] [PubMed]
- Bidwell, J.; Keen, L.; Gallagher, G.; Kimberly, R.; Huizinga, T.; McDermott, M.; Oksenberg, J.; McNicholl, J.; Pociot, F.; Hardt, C.; et al. Cytokine gene polymorphism in human disease: Online databases. Genes Immun. 1999, 1, 3–19. [Google Scholar] [CrossRef] [PubMed]
- Yuzhalin, A. The role of interleukin DNA polymorphisms in gastric cancer. Hum. Immunol. 2011, 72, 1128–1136. [Google Scholar] [CrossRef]
- Sugimoto, M.; Yamaoka, Y.; Furuta, T. Influence of interleukin polymorphisms on development of gastric cancer and peptic ulcer. World J. Gastroenterol. 2010, 16, 1188–1200. [Google Scholar] [CrossRef]
- Smoot, D.T.; Elliott, T.B.; Verspaget, H.W.; Jones, D.; Allen, C.R.; Vernon, K.G.; Bremner, T.; Kidd, L.C.R.; Kim, K.S.; Groupman, J.D.; et al. Influence of Helicobacter pylori on reactive oxygen-induced gastric epithelial cell injury. Carcinogenesis 2000, 21, 2091–2095. [Google Scholar] [CrossRef] [PubMed]
- Hwang, I.R.; Kodama, T.; Kikuchi, S. Effect of interleukin 1 polymorphisms on gastric mucosal interleukin 1beta production in Helicobacter pylori infection. Gastroenterology 2002, 123, 1793–1803. [Google Scholar] [CrossRef]
- Wolfe, M.; Nompleggi, D. Cytokine inhibition of gastric acid secretion—A little goes a long way. Gastroenterology 1992, 102, 2177–2178. [Google Scholar] [CrossRef]
- El-Omar, E.; Carrington, M.; Chow, W.-H.; McColl, K.E.L.; Bream, J.; Young, H.A.; Herrera, J.; Lissowska, J.; Yuan, C.-C.; Rothman, N.; et al. Interleukin-1 polymorphisms associated with increased risk of gastric cancer. Nature 2000, 404, 398–402. [Google Scholar] [CrossRef]
- Kahroba, H.; Hejazi, M.S.; Samadi, N. Exosomes: From carcinogenesis and metastasis to diagnosis and treatment of gastric cancer. Cell. Mol. Life Sci. 2019, 76, 1747–1758. [Google Scholar] [CrossRef]
- Patel, G.K.; Khan, M.A.; Bhardwaj, A.; Srivastava, S.K.; Zubair, H.; Patton, M.C.; Singh, S.; Khushman, M.; Singh, A. Exosomes confer chemoresistance to pancreatic cancer cells by promoting ROS detoxification and miR-155-mediated suppression of key gemcitabine-metabolising enzyme, DCK. Br. J. Cancer 2017, 116, 609–619. [Google Scholar] [CrossRef]
- Che, Y.; Geng, B.; Xu, Y.; Miao, X.; Chen, L.; Mu, X.; Pan, J.; Zhang, C.; Zhao, T.; Wang, C.; et al. Helicobacter pylori -induced exosomal MET educates tumour-associated macrophages to promote gastric cancer progression. J. Cell. Mol. Med. 2018, 22, 5708–5719. [Google Scholar] [CrossRef]
- Shimoda, A.; Ueda, K.; Nishiumi, S.; Murata-Kamiya, N.; Mukai, S.-A.; Sawada, S.-I.; Azuma, T.; Hatakeyama, M.; Akiyoshi, K. Exosomes as nanocarriers for systemic delivery of the Helicobacter pylori virulence factor CagA. Sci. Rep. 2016, 6, 18346. [Google Scholar] [CrossRef]
- Xu, X.; Cheng, J.; Luo, S.; Gong, X.; Huang, D.; Xu, J.; Qian, Y.; Wan, X.; Zhou, H. Deoxycholic acid-stimulated macrophage-derived exosomes promote spasmolytic polypeptide-expressing metaplasia in the stomach. Biochem. Biophys. Res. Commun. 2020, 524, 649–655. [Google Scholar] [CrossRef] [PubMed]
- Zheng, P.; Chen, L.; Yuan, X.; Luo, Q.; Liu, Y.; Xie, G.; Ma, Y.; Shen, L. Exosomal transfer of tumor-associated macrophage-derived miR-21 confers cisplatin resistance in gastric cancer cells. J. Exp. Clin. Cancer Res. 2017, 36, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Qu, J.-L.; Qu, X.-J.; Zhao, M.-F.; Teng, Y.-E.; Zhang, Y.; Hou, K.-Z.; Jiang, Y.-H.; Yang, X.-H.; Liu, Y.-P. Gastric cancer exosomes promote tumour cell proliferation through PI3K/Akt and MAPK/ERK activation. Dig. Liver Dis. 2009, 41, 875–880. [Google Scholar] [CrossRef]
- Wu, L.; Zhang, X.; Zhang, B. Exosomes derived from gastric cancer cells activate NF-kappa B pathway in macrophages to promote cancer progression. Tumour Biol. 2016, 37, 12169–12180. [Google Scholar] [CrossRef] [PubMed]
- Pan, L.; Liang, W.; Fu, M.; Huang, Z.-H.; Li, X.; Zhang, W.; Zhang, P.; Qian, H.; Jiang, P.-C.; Xu, W.-R.; et al. Exosomes-mediated transfer of long noncoding RNA ZFAS1 promotes gastric cancer progression. J. Cancer Res. Clin. Oncol. 2017, 143, 991–1004. [Google Scholar] [CrossRef] [PubMed]
- Wei, S.; Peng, L.; Yang, J. Exosomal transfer of miR-15b-3p enhances tumorigenesis and malignant transformation through the DYNLT1/Caspase-3/Caspase-9 signaling pathway in gastric cancer. J. Exp. Clin. Cancer Res. 2020, 39, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Gu, J.; Li, X.; Zhao, L. The role of PKM2 nuclear translocation in the constant activation of the NF-kappa B signaling pathway in cancer-associated fibroblasts. Cell Death Dis. 2021, 12, 1–13. [Google Scholar] [CrossRef]
- Zhang, H.; Deng, T.; Liu, R.; Ning, T.; Yang, H.; Liu, D.; Zhang, Q.; Lin, D.; Ge, S.; Bai, M.; et al. CAF secreted miR-522 suppresses ferroptosis and promotes acquired chemo-resistance in gastric cancer. Mol. Cancer 2020, 19, 1–17. [Google Scholar] [CrossRef]
- Zhang, X.; Shi, H.; Yuan, X.; Jiang, P.; Qian, H.; Xu, W. Tumor-derived exosomes induce N2 polarization of neutrophils to promote gastric cancer cell migration. Mol. Cancer 2018, 17, 1–16. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kumar, S.; Patel, G.K.; Ghoshal, U.C. Helicobacter pylori-Induced Inflammation: Possible Factors Modulating the Risk of Gastric Cancer. Pathogens 2021, 10, 1099. https://doi.org/10.3390/pathogens10091099
Kumar S, Patel GK, Ghoshal UC. Helicobacter pylori-Induced Inflammation: Possible Factors Modulating the Risk of Gastric Cancer. Pathogens. 2021; 10(9):1099. https://doi.org/10.3390/pathogens10091099
Chicago/Turabian StyleKumar, Sushil, Girijesh Kumar Patel, and Uday C. Ghoshal. 2021. "Helicobacter pylori-Induced Inflammation: Possible Factors Modulating the Risk of Gastric Cancer" Pathogens 10, no. 9: 1099. https://doi.org/10.3390/pathogens10091099
APA StyleKumar, S., Patel, G. K., & Ghoshal, U. C. (2021). Helicobacter pylori-Induced Inflammation: Possible Factors Modulating the Risk of Gastric Cancer. Pathogens, 10(9), 1099. https://doi.org/10.3390/pathogens10091099