Structure and Stability of the Stoichiometric Al3Fe Phase



Abstract
1. Introduction
2. Materials and Methods
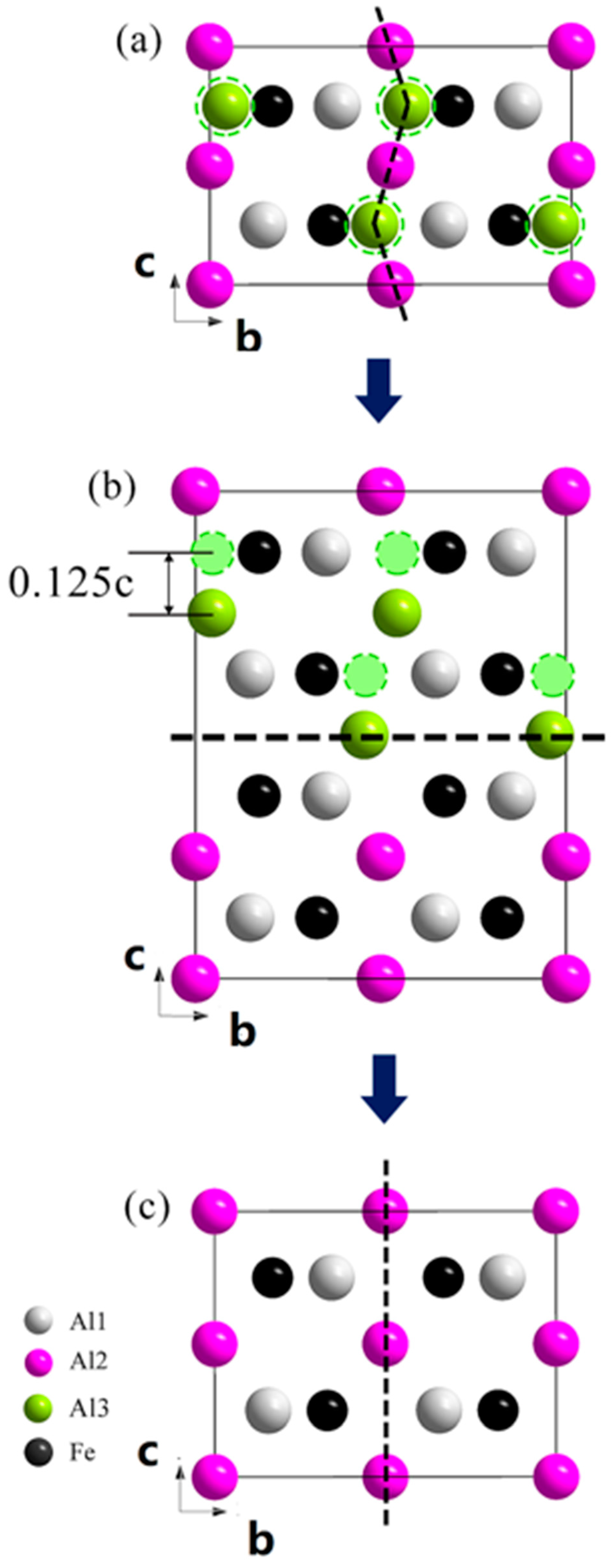
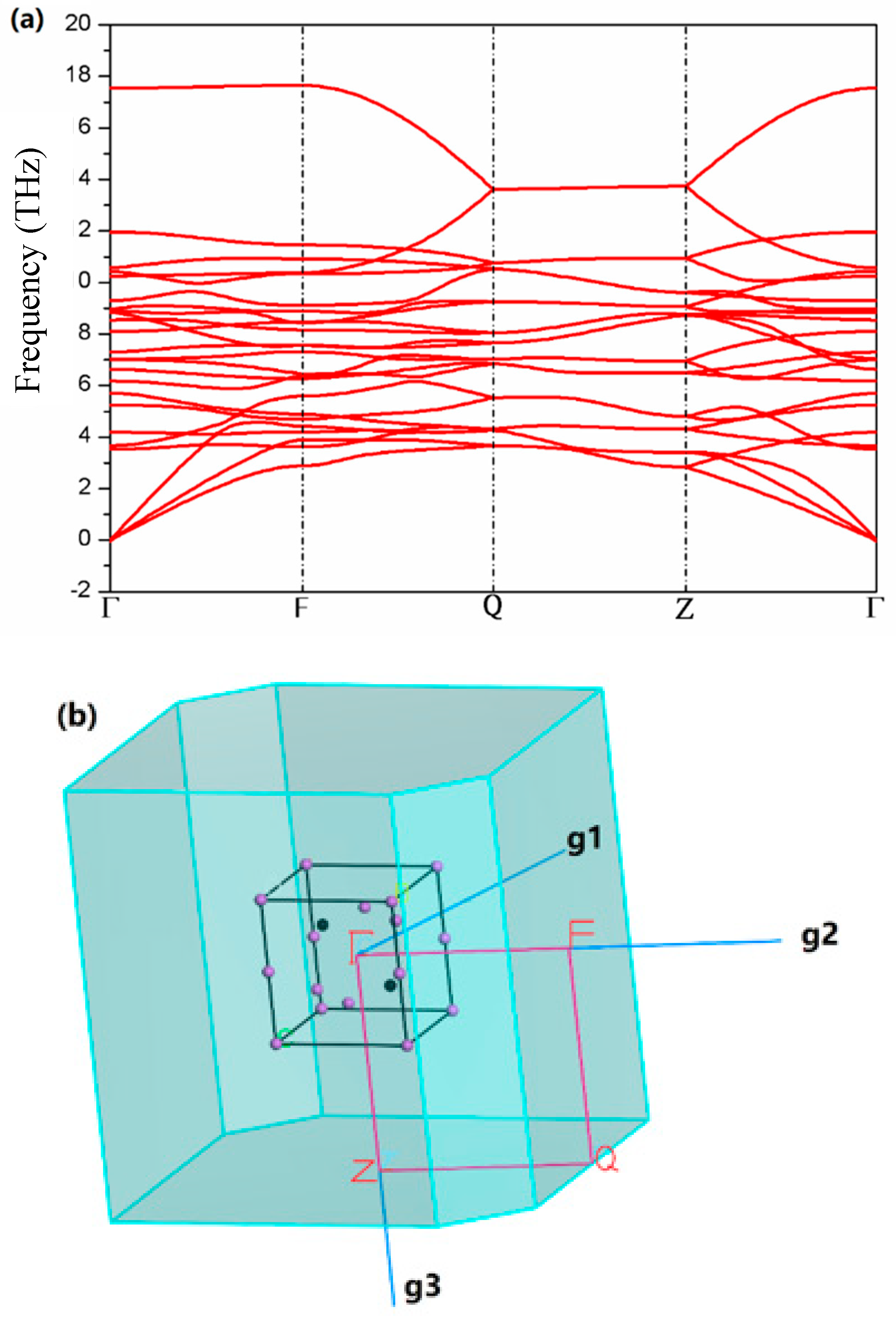
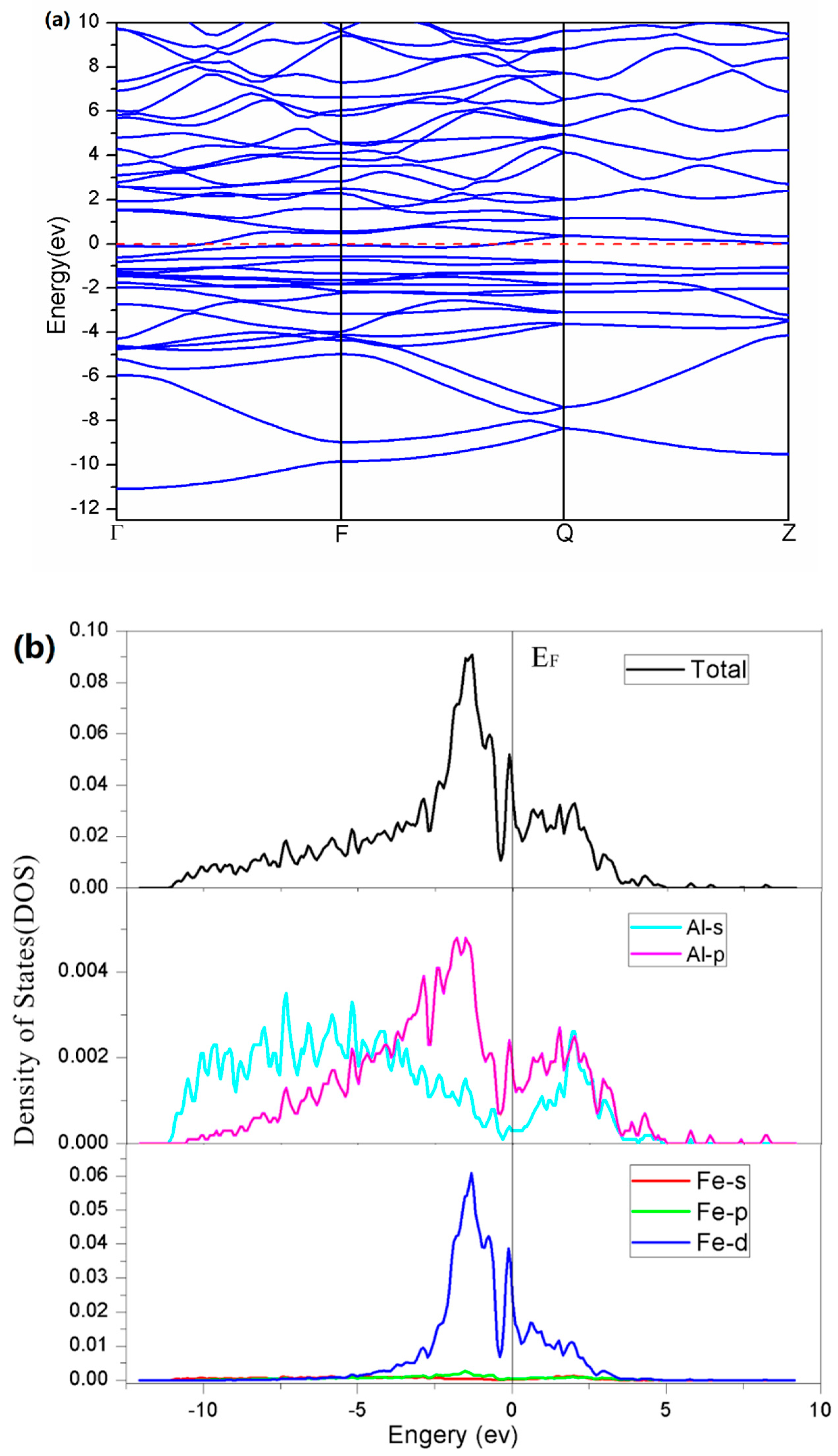
3. Results
C11 + C22 − 2C12 > 0; C11 + C33 − 2C13 > 0
C22 + C33 − 2C23 > 0; C11 + C22 + C33 + (C12 + C13 + C23) > 0
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Burkhardt, U.; Grin, Y.; Ellner, M.; Peters, K. Stucture Refinement of the Iron-Aluminium Phase with the Approximate Composition Fe2Al5. Acta Crystallogr. B 1994, 50, 313–316. [Google Scholar] [CrossRef]
- Han, K.; Ohnuma, I.; Kainuma, R. Experimental determination of phase equilibria of Al-rich portion in the Al-Fe binary system. J. Alloys Compd. 2016, 668, 97–106. [Google Scholar] [CrossRef]
- Li, X.; Scherf, A.; Heilmaier, M.; Stein, F. The Al-rich part of the Fe-Al phase diagram. J. Phase Equilib. Diff. 2016, 37, 162–173. [Google Scholar] [CrossRef]
- Denkena, B.; Stiffel, J.H.; Hasselberg, E.; Nespor, D. Chip formation and modeling of dynamic force behavior in machining polycrystalline iron–aluminum. Prod. Eng. Res. Devel. 2014, 8, 273–282. [Google Scholar] [CrossRef]
- Buršíková, V.; Homola, V.; Jirásková, Y.; Pizúrová, N.; Miháliková, I.; Friák, M.; Šob, M. Study of Local Mechanical Properties of Fe78Al22 Alloy. Key Eng. Mater. 2018, 784, 27–32. [Google Scholar] [CrossRef]
- Schuermann, E.; Kaiser, H.P. Melting Equilibria of Iron-Aluminium and Iron-Phosphorus Alloys. Arch. Eisenhuettenwes. 1980, 51, 325–327. [Google Scholar]
- Palm, M. Fe–Al materials for structural applications at high temperatures: Current research at MPIE. Int. J. Mater. Res. 2009, 100, 277–287. [Google Scholar] [CrossRef]
- Runge, J.M. The Metallurgy of Anodizing Aluminum, 1st ed.; Springer: Cham, Switzerland, 2018; pp. 1–64. [Google Scholar]
- McDonough, W.; Sun, S.S. The composition of the Earth. Chem. Geol. 1995, 120, 223–253. [Google Scholar] [CrossRef]
- Groth, P. Chemische Krystallographie, 1st ed.; Verlag von Wilhelm Engelmann: Leipzig, Deutschland, 1906; pp. 47–48. [Google Scholar]
- Osawa, A. On the equilibrium diagram of iron-aluminium system. Sci. Rep. Tohoku Univ. 1933, 22, 803–823. [Google Scholar]
- Bachmetew, E. Röntgenographische Bestimmung der FeAl3-Struktur. Z. Kristallogr. 1934, 89, 575–586. [Google Scholar]
- Bradley, A.J.; Taylor, A. An X-ray study of the iron-nickel-aluminium ternary equilibrium diagram. Proc. R. Soc. Lond. A 1938, 166, 353–375. [Google Scholar]
- Phragmen, G. On the phases occurring in alloys of aluminium with copper, magnesium, manganese, iron, and silicon. J. Inst. Metals 1950, 77, 489–551. [Google Scholar]
- Black, P.J. The structure of FeAl3.I. Acta Crystallogr. 1955, 8, 43–48. [Google Scholar] [CrossRef]
- Black, P.J. The structure of FeAl3. II. Acta Crystallogr. 1955, 8, 175–182. [Google Scholar] [CrossRef]
- Armbrüster, M.; Kovnir, K.; Friedrich, M.; Teschner, D.; Wowsnick, G.; Hahne, M.; Gille, P.; Szentmiklósi, L.; Feuerbacher, M.; Heggen, M.; et al. Al13Fe4 as a low-cost alternative for palladium in heterogeneous hydrogenation. Nat. Mater. 2012, 11, 690–693. [Google Scholar] [CrossRef]
- Sekhar, J.A.; Rajasekharan, T.; Rao, P.R.; Parthasarathy, G.; Ramkumar, S.; Gopal, E.S.R.; Lakshmi, C.S.; Mallya, R.M. Electron and x-ray diffraction studies on Al86Fe14, Al82Fe18 and Al75Fe25 quasicrystals. J. Phys. 1986, 27, 267–273. [Google Scholar] [CrossRef]
- Ma, C.; Lin, C.; Bindi, L.; Steinhardt, P.J. Hollisterite (Al3Fe), kryachkoite (Al,Cu)6 (Fe,Cu), and stolperite (AlCu): Three new minerals from the Khatyrka CV3 carbonaceous chondrite. Am. Mineral. 2017, 102, 690–693. [Google Scholar] [CrossRef]
- Xia, Z.; Liu, C.; Fan, C.Z. Al13Fe3. IUCrData 2018, 3, x180593. [Google Scholar] [CrossRef]
- Okamoto, N.L.; Okumura, J.; Higashi, M.; Inui, H. Crystal structure of ƞ′-Fe3Al8; low-temperature phase of ƞ-Fe2Al5 accompanied by an ordered arrangement of Al atoms of full occupancy in the c-axis chain sites. Acta Mater. 2017, 129, 290–299. [Google Scholar] [CrossRef]
- Becker, H.; Amirkhanyan, L.; Kortus, J.; Leineweber, A. Powder-X-ray diffraction analysis of the crystal structure of the ƞ′-Al8Fe3 (ƞ′-Al2.67Fe) phase. J. Alloys Compd. 2017, 721, 691–696. [Google Scholar] [CrossRef]
- Okamoto, N.L.; Higashi, M.; Inui, H. Crystal structure of η″-Fe3Al7+x determined by single-crystal synchrotron X-ray diffraction combined with scanning transmission electron microscopy. Sci. Technol. Adv. Mater. 2019, 20, 543–556. [Google Scholar] [CrossRef] [PubMed]
- Kresse, G.; Furthmuller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169–11181. [Google Scholar] [CrossRef] [PubMed]
- Parr, R.G. Density functional theory. Ann. Rev. Phys. Chem. 1983, 34, 631–656. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed]
- Blochl, P.E. Projector augmented-wave method. Phys. Rev. B 1994, 50, 17953–17979. [Google Scholar] [CrossRef]
- Monkhorst, H.J.; Pack, J.D. Special points for Brillouin-zone integrations. Phys. Rev. B 1976, 13, 5188–5192. [Google Scholar] [CrossRef]
- Togo, A.; Oba, F.; Tanaka, I. First-principles calculations of the ferroelastic transition between rutile-type and CaCl2-type SiO2 at high pressures. Phys. Rev. B 2008, 78, 134106. [Google Scholar] [CrossRef]
- Togo, A.; Tanaka, I. First principles phonon calculations in materials science. Scripta Mater. 2015, 108, 1–5. [Google Scholar] [CrossRef]
- Van Alboom, A.; Lemmens, B.; Breitbach, B.; De Grave, E.; Cottenier, S.; Verbeken, K. Multi-method identification and characterization of the intermetallic surface layers of hot-dip Al-coated steel: FeAl3 or Fe4Al13 and Fe2Al5 or Fe2Al5+x. Surf. Coat. Technol. 2017, 324, 419–428. [Google Scholar] [CrossRef]
- APEX3, SAINT and SADABS; Software for Data Reduction, Absorption Correction and Structure Solution; Bruker AXS Inc.: Madison, WI, USA, 2015.
- Sheldrick, G.M. SHELXT-Integrated space-group and crystal-structure determination. Acta Crystallogr. A 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. C 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Brandenburg, K.; Putz, H. Diamond; Crystal Impact GbR: Bonn, Germany, 2017. [Google Scholar]
- Mouhat, F.; Coudert, F.X. Necessary and sufficient elastic stability conditions in various crystal systems. Phys. Rev. B 2014, 90, 224104. [Google Scholar] [CrossRef]
- Pang, M.J.; Zhan, Y.Z.; Wang, H.Z. Ab initio investigation into the structural, electronic and elastic properties of AlCu2TM (TM= Ti, Zr and Hf) ternary compounds. Curr. Appl. Phys. 2012, 12, 957–962. [Google Scholar] [CrossRef]
- Huang, Y.C.; Li, Y.; Xiao, Z.B. First-Principles Study of Structure, Elastic and Electronic Properties of Precipitates Al3Fe, Al6Mn and Mg2Si in Al-Mg Alloys; Szewczyk, R., Jiang, Z.Y., Huang, Y.J., Eds.; International Workshop on Materials Science and Engineering (IWMSE): GuangZhou, China, 2016; pp. 876–883. [Google Scholar]
- Chen, X.Q.; Niu, H.Y.; Franchini, C.; Li, D.Z.; Li, Y.Y. Hardness of T -carbon: Density functional theory calculations. Phys. Rev. B. 2011, 84, 121405. [Google Scholar] [CrossRef]
- Finzel, K. ELF and Its Relatives-A Detailed Study about the Robustness of the Atomic Shell Structure in Real Space. Int. J. Quantum Chem. 2014, 114, 1546–1558. [Google Scholar] [CrossRef]
- Ptashkina, E.A.; Kabanova, E.G.; Tursina, A.I.; Yatsenko, A.V.; Kuznetsov, V.N. Synthesis and crystal structure of a new Cu3Au-type. Acta Cryst. C 2018, 74, 295–299. [Google Scholar] [CrossRef] [PubMed]
- Balagurov, A.M.; Bobrikov, I.A.; Sumnikov, S.V.; Golovin, I.S. Neutron Diffraction study of coherent atomic ordering in Fe3Al-type alloys. Acta Mater. 2018, 153, 45–52. [Google Scholar] [CrossRef]
- Freiburg, C.; Grushko, B. An Al13Fe4 phase in the Al-Cu-Fe alloy system. J. Alloys Compd. 1994, 210, 149–152. [Google Scholar] [CrossRef]
- Mihalkovič, M.; Widom, M. Structure and stability of Al2Fe and Al5Fe2: First-principles total energy and phonon calculations. Phys. Rev. B 2012, 85, 014113. [Google Scholar] [CrossRef]
- Becker, H.; Leineweber, A. Atomic channel occupation in disordered η-Al5Fe2 and in two of its low-temperatures phases, η″ and η‴. Intermetallics 2018, 93, 251–262. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Chemical Formula | Al6Fe2 |
|---|---|
| Crystal system, space group | Orthorhombic, Cmcm |
| Temperature/K | 293(2) |
| a, b, c/Å | 7.6474 (7), 6.4097 (6), 4.2114 (4) |
| α, β, γ/° | 90, 90, 90 |
| V/Å3, | 206.43 (3) |
| Z | 2 |
| Diffractometer | Bruker D8 Venture Photon 100 COMS |
| Radiation type | Mo-Kα, λ = 0.71073 (Å) |
| Scan mode | φ and ω scan |
| Absorption correction | Multi-scan |
| μ/mm−1 No. of measured, independent and observed [I > 2σ(I)] reflections | 8.09 2896, 186, 167 |
| Rint (sin θ/λ)max (Å−1) | 0.039 0.713 |
| R[F2 > 2σ(F2)], wR(F2), S No. of reflections No. of parameters Δρmax, Δρmin (e Å−3) | 0.031, 0.090, 1.26 186 17 0.70, −1.63 |
| Label | Site | x | y | z | Occ. | Ueq,Uiso |
|---|---|---|---|---|---|---|
| Al1 | 8g | 0.6875(3) | 0.8524(3) | 3/4 | 1.000 | 0.0132(6) |
| Al2 | 4c | 1/2 | 0.544(2) | 3/4 | 0.431(13) | 0.059(4) |
| Al3 | 4a | 1/2 | 1/2 | 1/2 | 0.569(13) | 0.059(4) |
| Fe4 | 4c | 1/2 | 0.17212(19) | 3/4 | 1.000 | 0.0071(5) |
| Region | Spot1 | Spot2 | Spot3 | Area1 | Area2 | Area3 |
|---|---|---|---|---|---|---|
| x(Al)/% | 70.52(±5.97) | 76.52(±5.45) | 71.42(±5.94) | 67.56(±6.21) | 69.34(±6.07) | 68.91(±6.10) |
| x(Fe)/% | 29.48(±2.25) | 23.48(±2.32) | 28.58(±2.35) | 32.44(±2.25) | 30.66(±2.26) | 31.09(±2.26) |
| Al:Fe | 2.39 | 3.26 | 2.50 | 2.08 | 2.26 | 2.22 |
| Phases | C11 | C22 | C33 | C44 | C55 | C66 | C12 | C13 | C23 | B | G | E | σ | B/G | Hv |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Al3Fe | 240 | 249 | 235 | 93 | 59 | 56 | 77 | 52 | 55 | 121 | 76 | 188 | 0.24 | 1.60 | 11.62 |
| Al13Fe3 | 150 | 143 | 139 | 61 | 59 | 54 | 8 | 16 | 25 | 59 | 59 | 133 | 0.12 | 1.00 | 18.73 |
| 1 Al13Fe4 | − | − | − | − | − | − | − | − | − | 91 | 67 | 162 | 0.20 | 1.35 | 13.36 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xia, Z.; Wen, B.; Fan, C. Structure and Stability of the Stoichiometric Al3Fe Phase. Metals 2019, 9, 1322. https://doi.org/10.3390/met9121322
Xia Z, Wen B, Fan C. Structure and Stability of the Stoichiometric Al3Fe Phase. Metals. 2019; 9(12):1322. https://doi.org/10.3390/met9121322
Chicago/Turabian StyleXia, Zheng, Bin Wen, and Changzeng Fan. 2019. "Structure and Stability of the Stoichiometric Al3Fe Phase" Metals 9, no. 12: 1322. https://doi.org/10.3390/met9121322
APA StyleXia, Z., Wen, B., & Fan, C. (2019). Structure and Stability of the Stoichiometric Al3Fe Phase. Metals, 9(12), 1322. https://doi.org/10.3390/met9121322