
First-Principles Study on the Structural Stability and Segregation Behavior of γ-Fe/Cr2N Interface with Alloying Additives M (M = Mn, V, Ti, Mo, and Ni)


Abstract
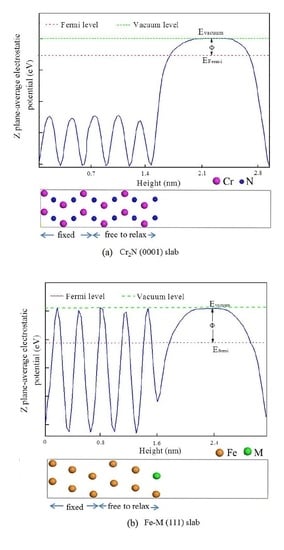
:
1. Introduction
2. Calculation Method
3. Results and Discussion
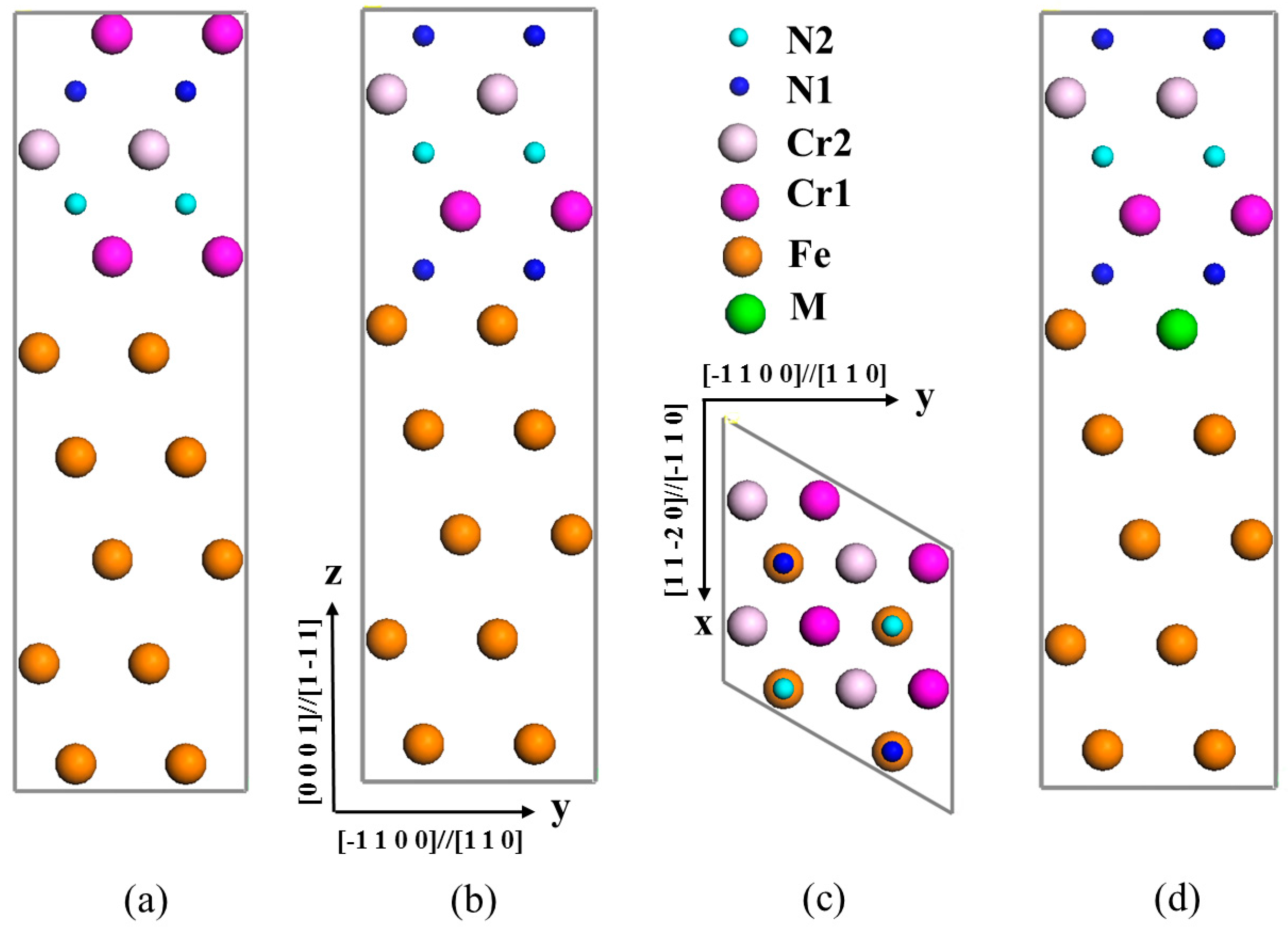
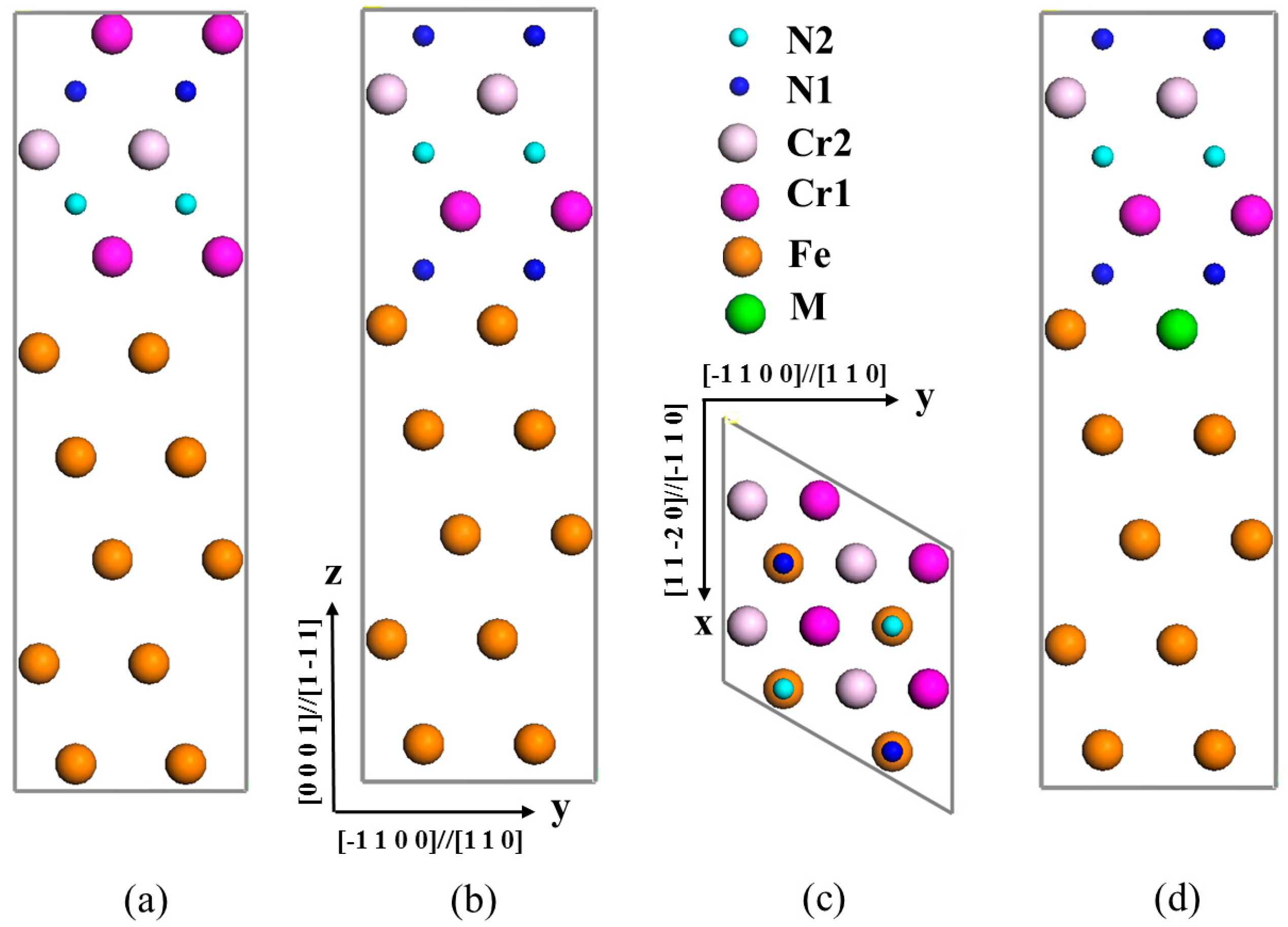
3.1. Interface Structure of γ-Fe/Cr2N
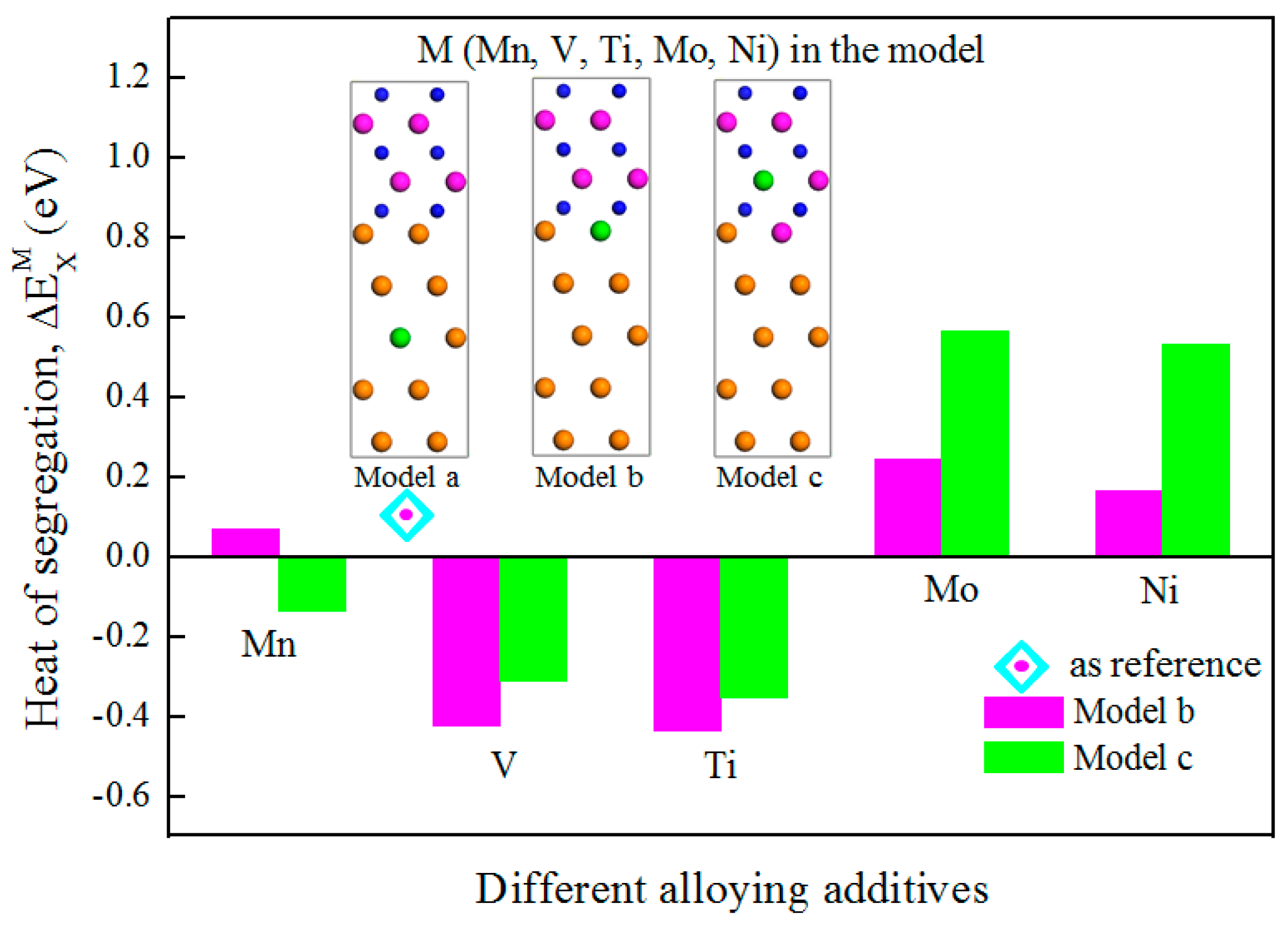
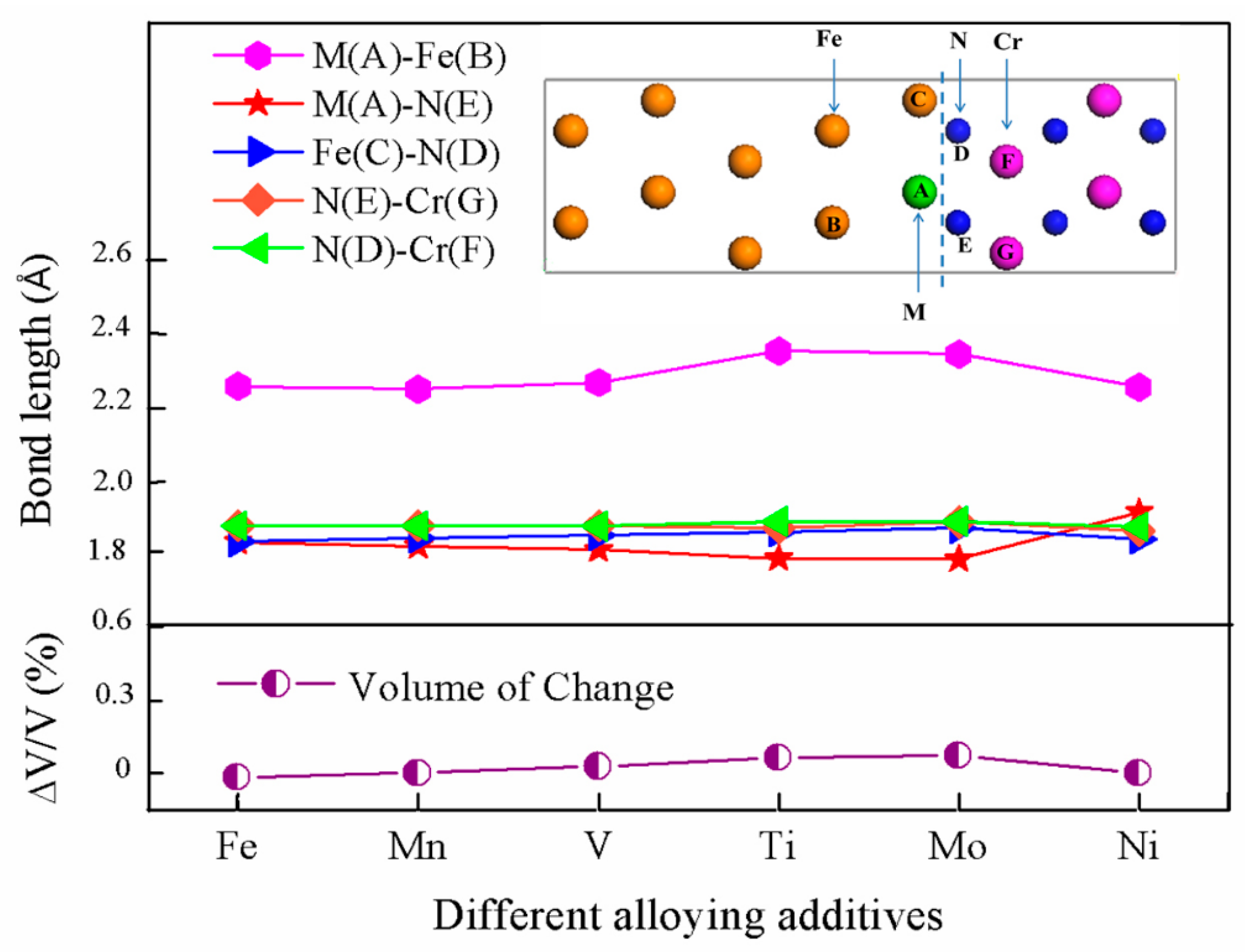
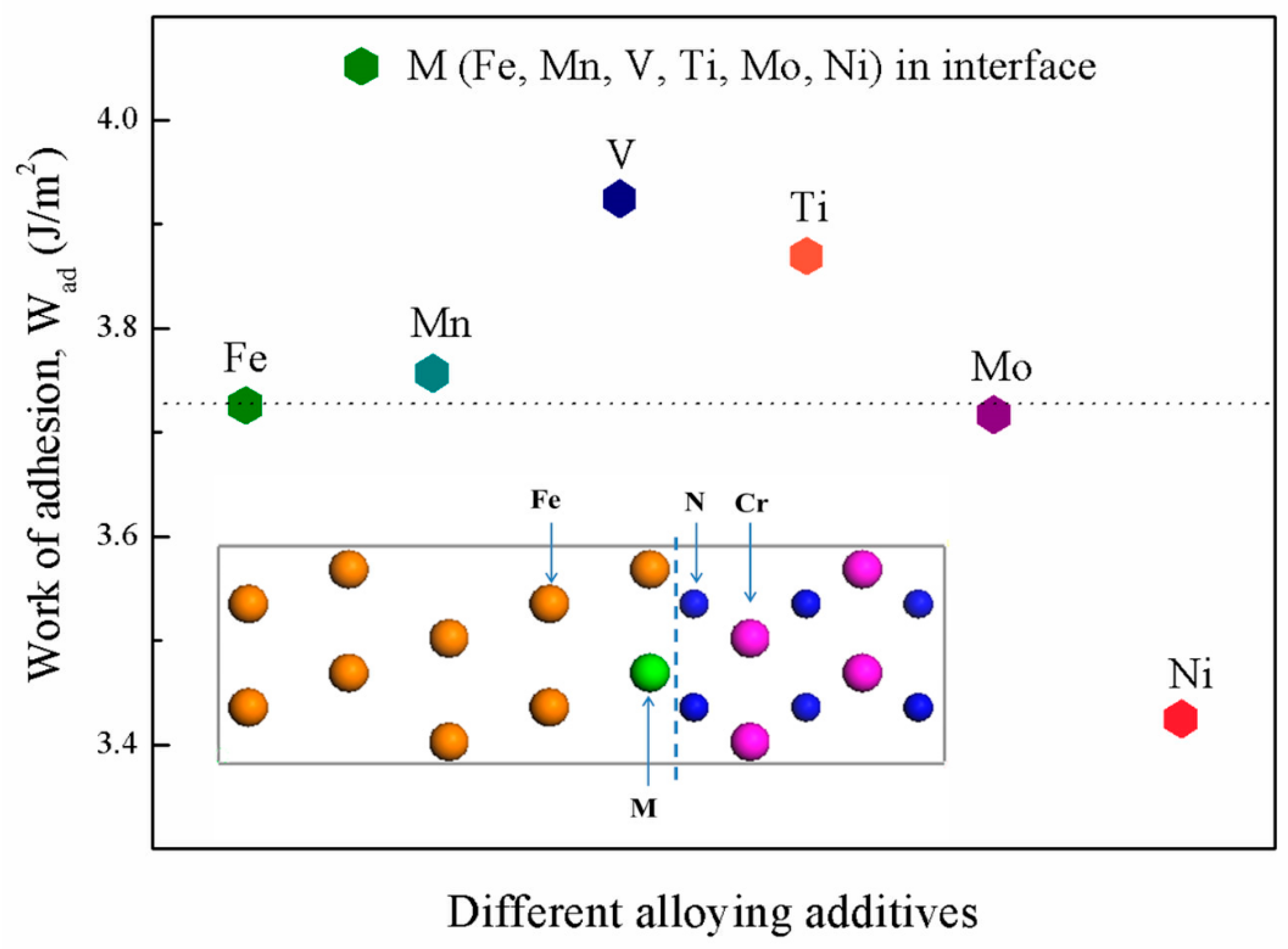
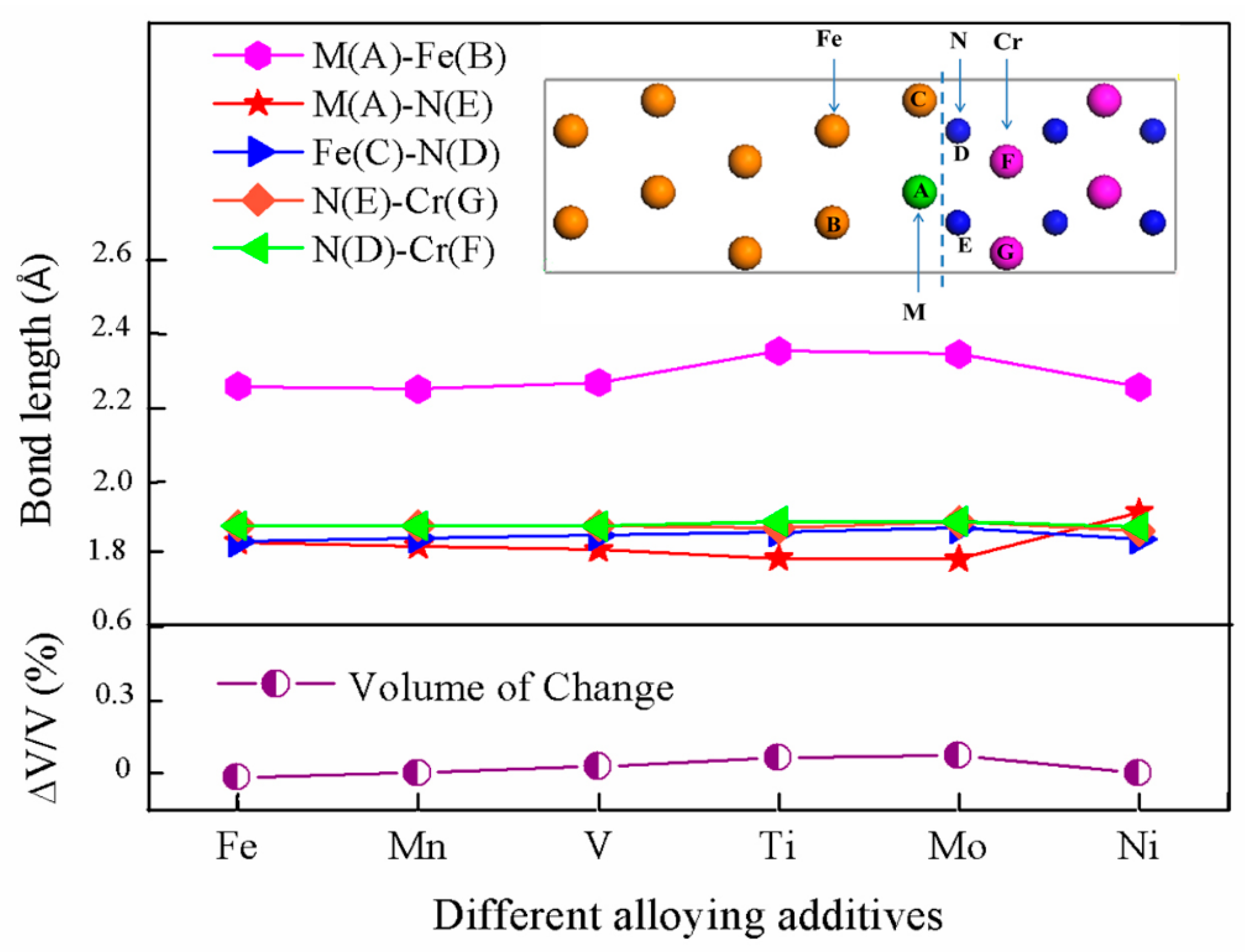
3.2. Segregation Behavior
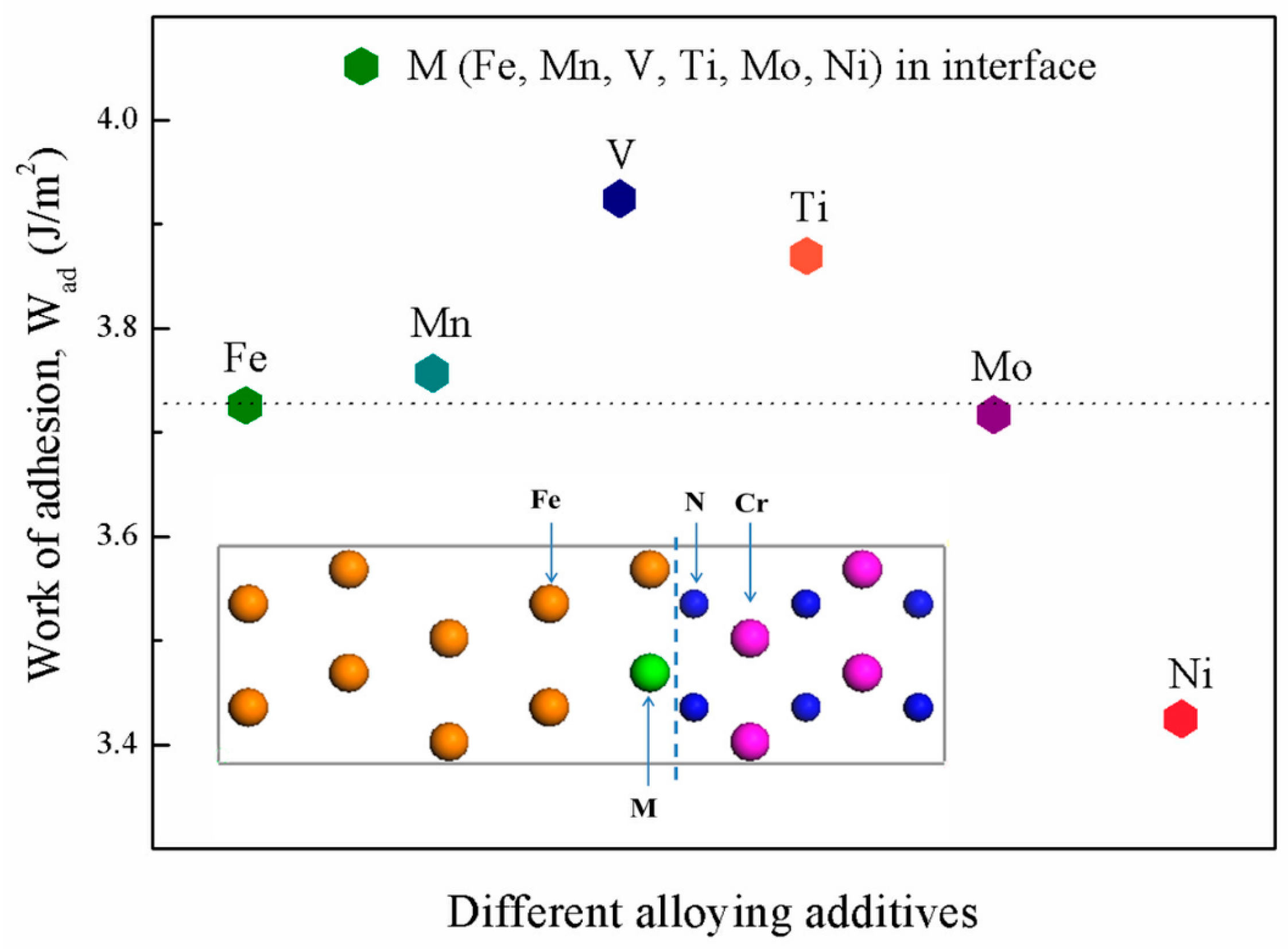
3.3. Interfacial Adhesive Behavior
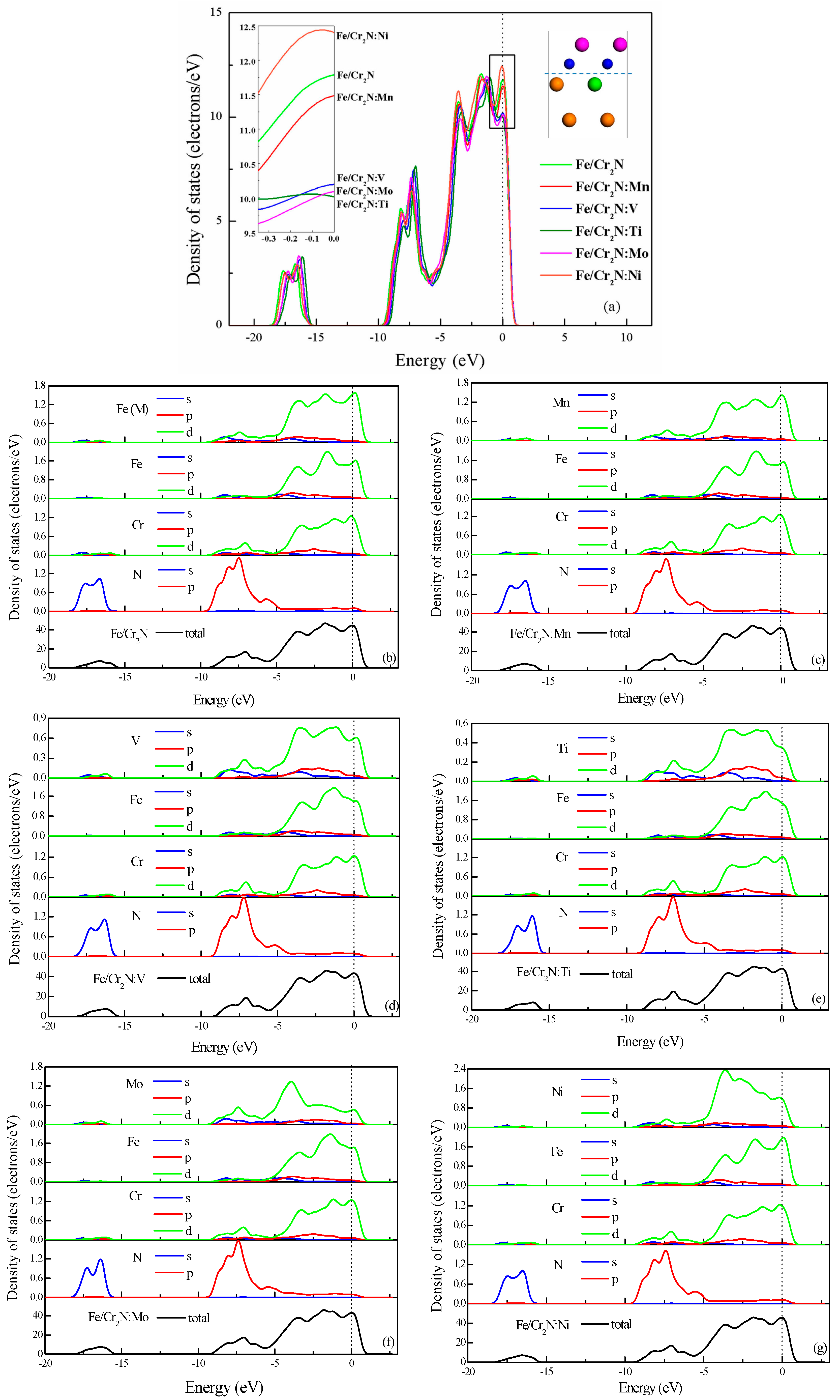
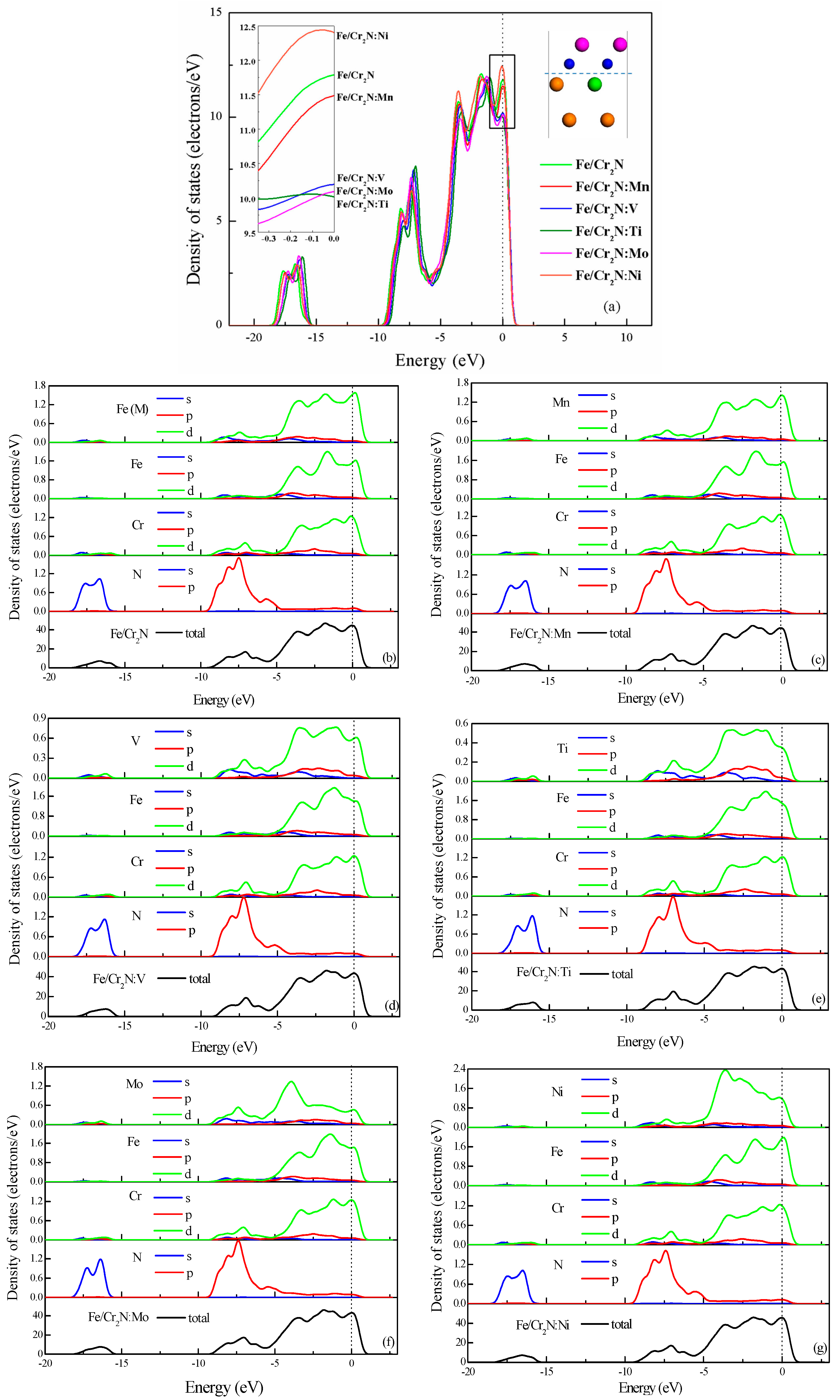
3.4. Electronic Structure
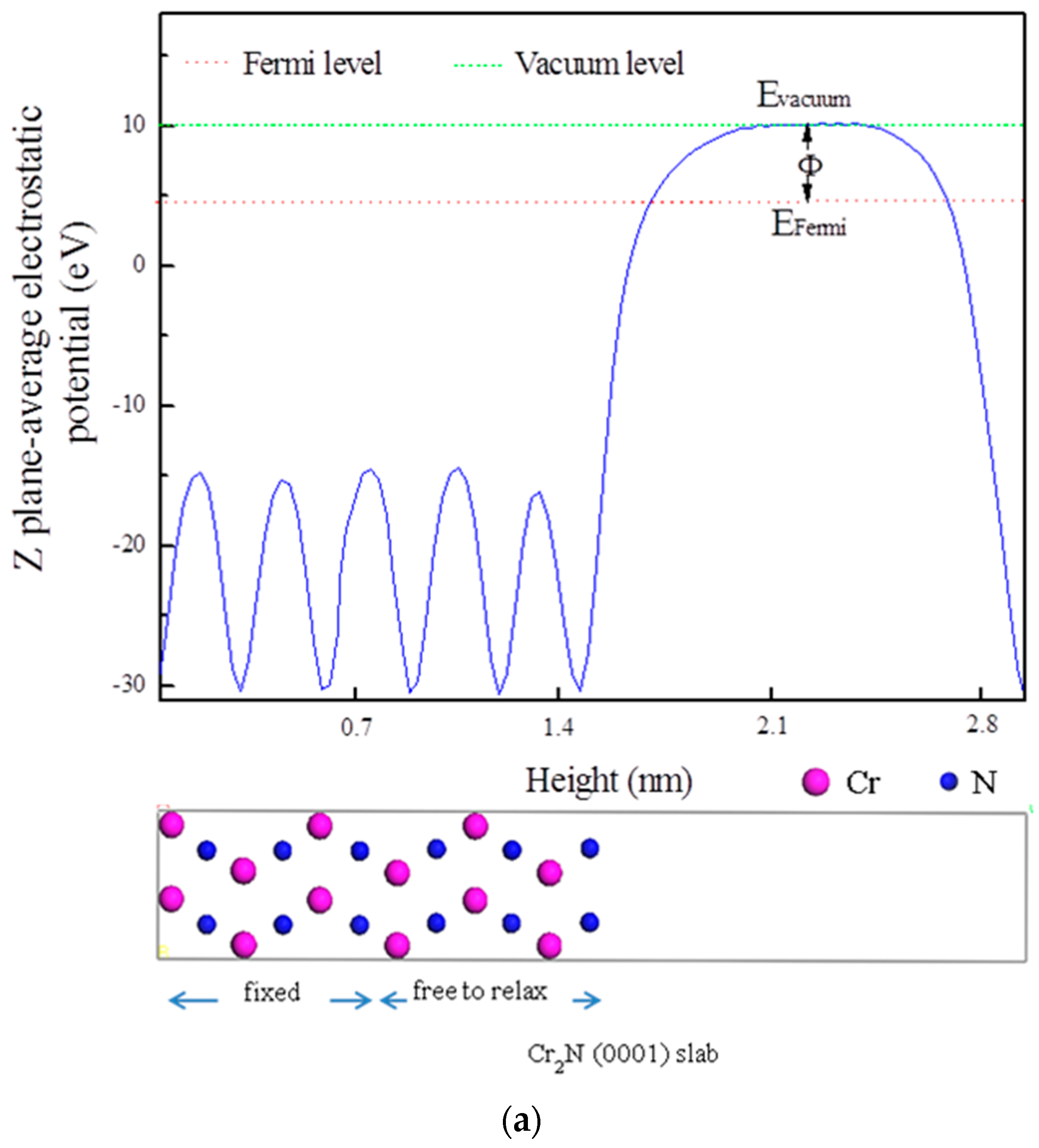
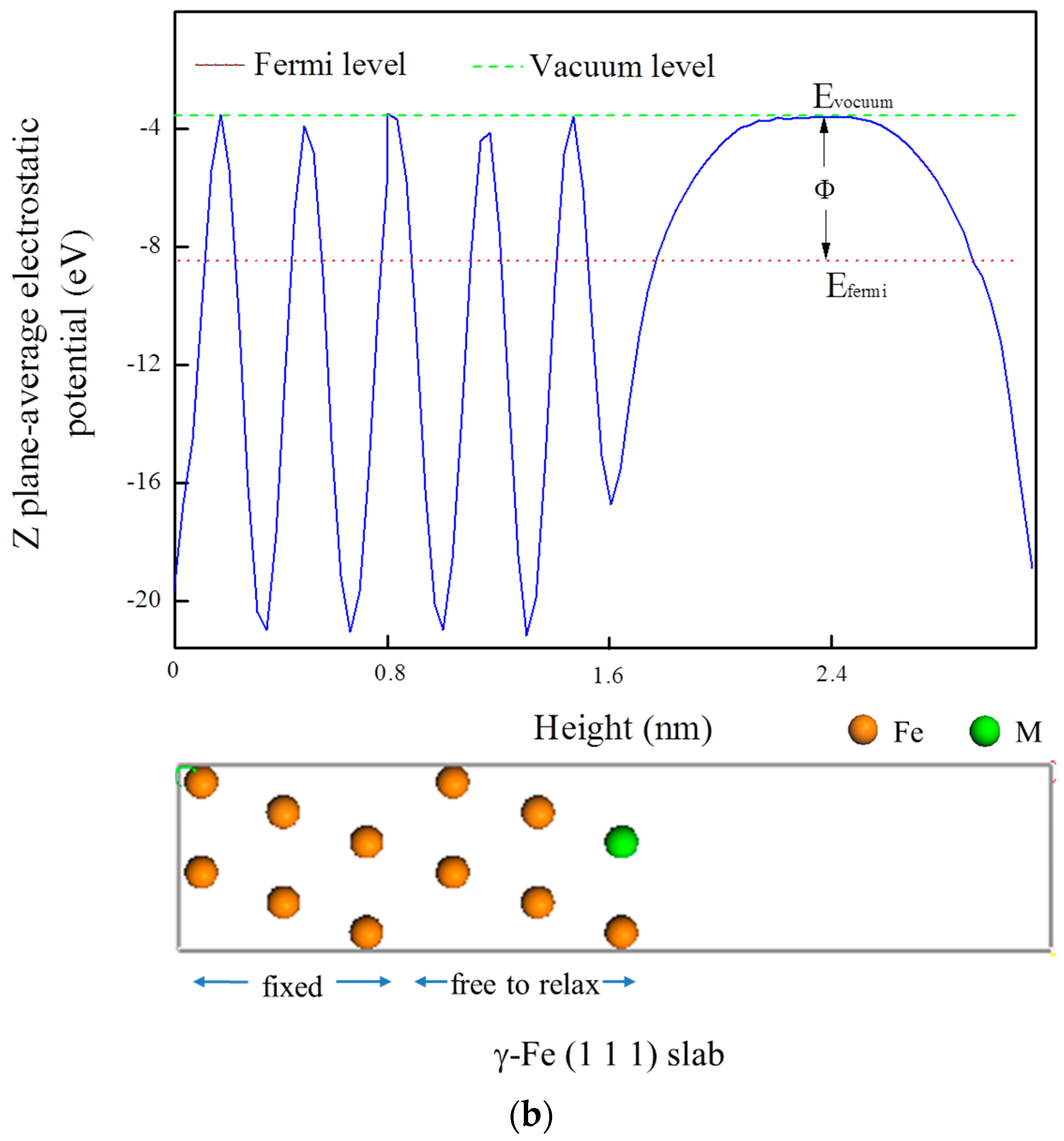
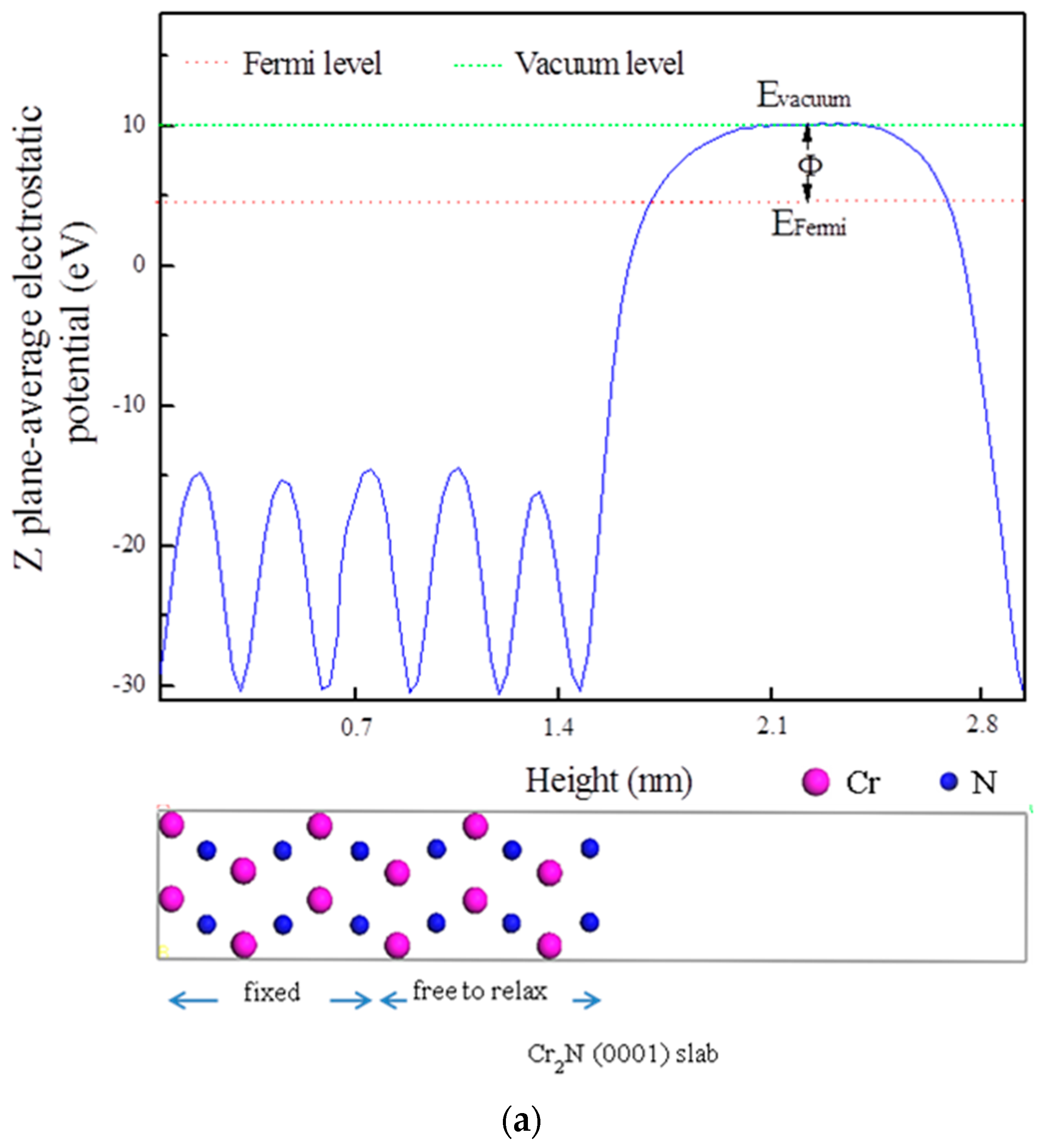
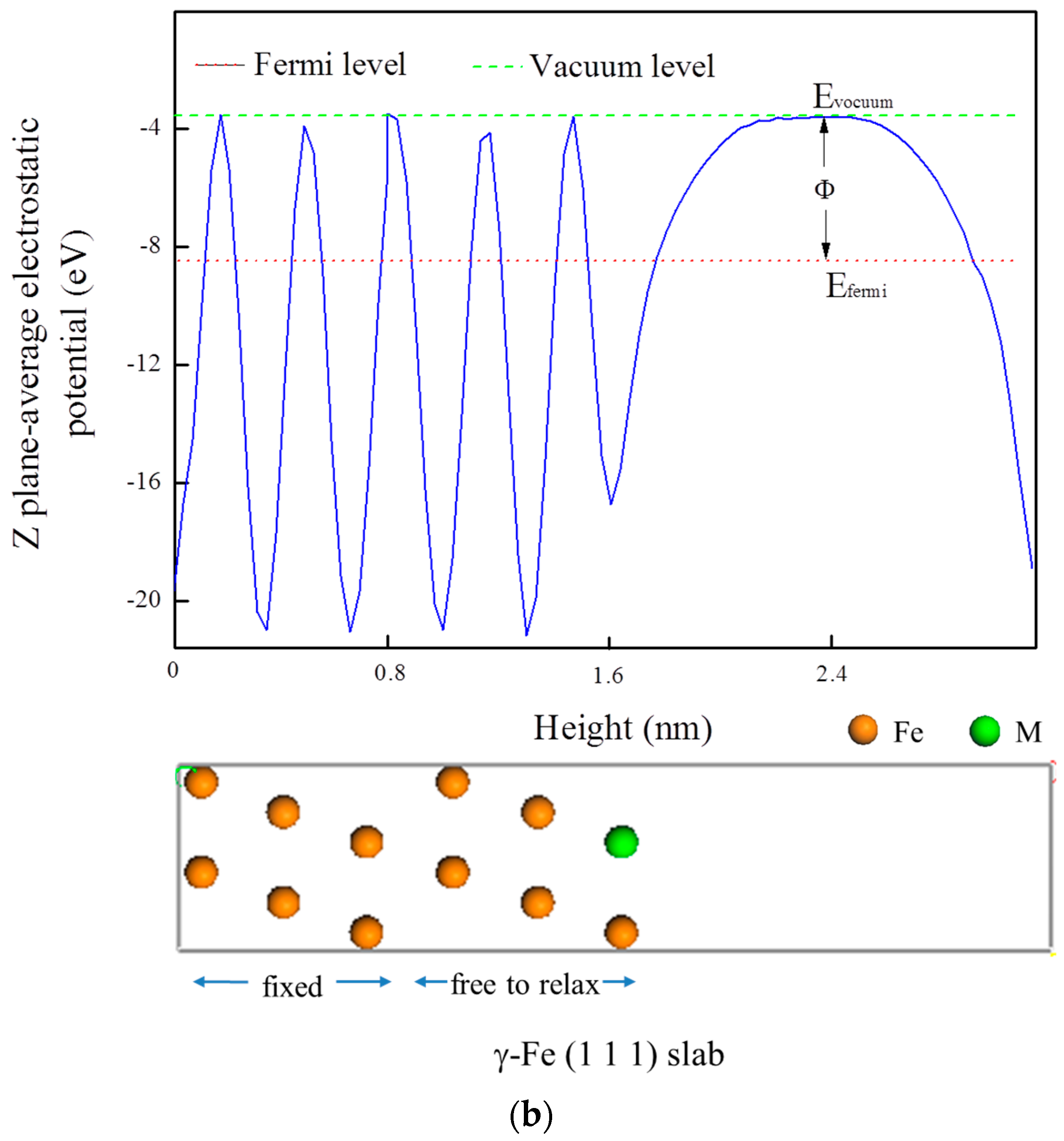
3.5. Electrochemical Properties
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Gavriljuk, V.G.; Berns, H. High Nitrogen Steels; Springer-Verlag: Berlin, Germany, 1999. [Google Scholar]
- Simmons, J.W. Influence of nitride (Cr2N) precipitation on the plastic flow behavior of high-nitrogen austenitic stainless steel. Scr. Metall. Mater. 1995, 32, 265–270. [Google Scholar] [CrossRef]
- Shi, F.; Wang, L.; Cui, W.; Liu, C. Precipitation Kinetics of Cr2N in High Nitrogen Austenitic Stainless Steel. J. Iron Steel Res. Int. 2008, 15, 72–77. [Google Scholar] [CrossRef]
- Lee, T.H.; Oh, C.S.; Lee, C.G.; Kim, S.J.; Takaki, S. Precipitation of σ-Phase in High-Nitrogen Austenitic 18Cr-18Mn-2Mo-0.9N Stainless Steel During Isothermal Aging. Scr. Mater. 2004, 50, 1325–1328. [Google Scholar] [CrossRef]
- Xu, W.; Martin, D.S.; del Castillo, P.E.J.R.D.; van der Zwaag, S. Modelling and characterization of chi-phase grain boundary precipitation during aging of Fe-Cr-Ni-Mo stainless steel. Mater. Sci. Eng. A 2007, 467, 24–32. [Google Scholar] [CrossRef]
- Kartik, B.; Veerababu, R.; Sundararaman, M.; Satyanarayana, D.V.V. Effect of high temperature ageing on microstructure and mechanical properties of a nickel-free high nitrogen austenitic stainless steel. Mater. Sci. Eng. A 2015, 642, 288–296. [Google Scholar] [CrossRef]
- Cobelli, P.; Kerschenbauer, C.; Speidel, M.O. Nitrides in high nitrogen stainless steels. Trans. Indian Inst. Met. 2002, 55, 473–480. [Google Scholar]
- Ha, H.Y.; Kwon, H.S. Effects of Cr2N on the pitting corrosion of high nitrogen stainless steels. Electrochim. Acta 2007, 52, 2175–2180. [Google Scholar] [CrossRef]
- Toro, A.; Misiolek, W.Z.; Tschiptschin, A.P. Correlations between microstructure and surface properties in a high nitrogen martensitic stainless steel. Acta Mater. 2003, 51, 3363–3374. [Google Scholar] [CrossRef]
- Lee, T.H.; Kim, S.J.; Takaki, S. Time-temperature-precipitation characteristics of high-nitrogen austenitic Fe-18Cr-18Mn-2Mo-0.9N Steel. Metall. Mater. Trans. A 2006, 37, 3445–3454. [Google Scholar] [CrossRef]
- Paulauskas, I.E.; Brady, M.P.; Meyer, H.M.; Buchanan, R.A.; Walker, L.R. Corrosion behavior of CrN, Cr2N and pi phase surfaces on nitrided Ni-50Cr for proton exchange membrane fuel cell bipolar plates. Corros. Sci. 2006, 48, 3157–3171. [Google Scholar] [CrossRef]
- Knutsen, R.D.; Lang, C.I.; Basson, J.A. Discontinuous cellular precipitation in a Cr-Mn-N steel with niobium and vanadium additions. Acta Mater. 2004, 52, 2407–2417. [Google Scholar] [CrossRef]
- Hirata, S.; Ikegami, Y. Effect of molybdenum on mechanical properties of high-nitrogen containing Cr-Mn-Ni austenitic stainless steels. Trans. Indian Inst. Met. 2002, 55, 349–353. [Google Scholar]
- Sauvage, X.; Wilde, G.; Divinski, S.V.; Horita, Z.; Valiev, R.Z. Grain boundaries in ultrafine grained materials processed by severe plastic deformation and related phenomena. Mater. Sci. Eng. A 2012, 540, 1–12. [Google Scholar] [CrossRef]
- Kohn, W.; Sham, L.J. Self-Consistent Equations Including Exchange and Correlation Effects. Phys. Rev. A 1965, 140, 1133–1138. [Google Scholar] [CrossRef]
- Segall, M.D.; Lindan, P.J.D.; Probert, M.J.; Pickard, C.J.; Hasnip, P.J.; Clark, S.J. First-principles simulation: Ideas, illustrations and the CASTEP code. J. Phys. Condens. Matter. 2002, 14, 2717–2744. [Google Scholar] [CrossRef]
- Perdew, J.P.; Zunger, A. Self-interaction correction to density-functional approximations for many-electron systems. Phys. Rev. B 1981, 23, 5048–5079. [Google Scholar] [CrossRef]
- Ceperley, D.M.; Alder, B.J. Ground State of the Electron Gas by a Stochastic Method. Phys. Rev. Lett. 1980, 45, 566–569. [Google Scholar] [CrossRef]
- Monkhorst, H.; Pack, J.D. Special points for Brillouin-zone integrations. Phys. Rev. B 1976, 13, 5188–5192. [Google Scholar] [CrossRef]
- Lee, T.H.; Kim, S.J.; Shin, E.; Takaki, S. On the crystal structure of Cr2N precipitates in high-nitrogen austenitic stainless steel. III. Neutron diffraction study on the ordered Cr2N superstructure. Acta Crystallogr. Sect. B Struct. Sci. 2006, 62, 979–986. [Google Scholar] [CrossRef] [PubMed]
- Basinski, Z.S.; Hume-Rothery, W.; Sotton, A.L. The Lattice Expansion of Iron. Math. Phys. Sci. 1955, 229, 459–467. [Google Scholar] [CrossRef]
- Straumanis, M.E.; Kim, D.C. Lattice constants, thermal expansion coefficients, densities and perfection of structure of pure iron and iron loaded with hydrogen. Trans. Metall. Soc. AIME 1965, 233, 1519–1525. [Google Scholar]
- Haglund, J. Theory of bonding in transition-metal carbides and nitrides. Phys. Rev. B 1993, 48, 11685–11691. [Google Scholar] [CrossRef]
- Ma, S.; Liu, Y.; Ye, J.; Zhang, H.; Pang, J. Theoretical study on the elastic, electronic and thermodynamic properties of trigonal-type Cr2N under high pressures. Comput. Mater. Sci. 2014, 95, 620–625. [Google Scholar] [CrossRef]
- Kim, S.J.; Marquart, T.; Franzen, H.F. Structure refinement for Cr2N. J. Less Common Met. 1990, 158, 9–10. [Google Scholar] [CrossRef]
- Rayaprolu, D.B.; Hendry, A. Cellular precipitation in a nitrogen alloyed stainless steel. Mater. Sci. Technol. 1989, 5, 328–332. [Google Scholar] [CrossRef]
- Ramirez, A.J.; Lippold, J.C.; Brandi, S.D. The Relationship between Chromium Nitride and Secondary Austenite Precipitation in Duplex Stainless Steels. Metall. Mater. Trans. A 2003, 34, 1575–1597. [Google Scholar] [CrossRef]
- Jiang, Q.; Lu, H.M.; Zhao, M. Modelling of surface energies of elemental crystals. J. Phys. Condens. Matter 2004, 16, 521–530. [Google Scholar] [CrossRef]
- Hashibon, A.; Elsässer, C.; Rühle, M. Structure at abrupt copper-alumina interfaces: An ab initio study. Acta Mater. 2005, 53, 5323–5332. [Google Scholar] [CrossRef]
- Finnis, M.W. The theory of metal-ceramic interfaces. J. Phys. Condens. Matter 1996, 8, 5811–5836. [Google Scholar] [CrossRef]
- Wang, H.; Gao, X.; Yang, J.; Jia, Y.; Gong, J. First-Principles Study of Cr2N/γ-Fe Interface in High Nitrogen Steel. Mater. Tran. 2015, 56, 1047–1051. [Google Scholar] [CrossRef]
- Nan, D.; Zhang, C.; Liu, H.; Fan, G.; Fang, X.; Han, P. Effects of different alloying additives X (X = Si, Al, V, Ti, Mo, W, Nb, Y) on the adhesive behavior of Fe/Cr2O3 interfaces: A first-principles study. Comput. Mater. Sci. 2015, 109, 293–299. [Google Scholar]
- Xie, Y.P.; Zhao, S.J. First principles study of Al and Ni segregation to the α-Fe/Cu(100) coherent interface and their effects on the interfacial cohesion. Comp. Mater. Sci. 2012, 63, 329–335. [Google Scholar] [CrossRef]
- Ande, C.K.; Sluiter, M.H.F. First-priciples prediction of partitioning of alloying elements between cementite and ferrite. Acta Mater. 2010, 58, 6276–6281. [Google Scholar] [CrossRef]
- Ozfidan, I.; Chen, K.Y.; Fu, M. Effects of Additives and Impurity on the Adhesive Behavior of the NiAl(110)/Al2O3(0001) Interface: An ab initio Study. Metall. Mater. Trans. A 2011, 42, 4126–4136. [Google Scholar] [CrossRef]
- Cantele, G. First-principles study of n- and p-doped silicon nanoclusters. Phys. Rev. B 2005, 72, 113303. [Google Scholar] [CrossRef]
- Hu, R.; Cheng, G.M.; Zhang, J.Q.; Li, J.S.; Zhang, T.B.; Fu, H.Z. First principles investigation on the stability and elastic properties of Ni2Cr1−xMx (M = Nb, Mo, Ta, and W) superlattices. Intermetallics 2013, 33, 60–66. [Google Scholar] [CrossRef]
- Segall, M.D.; Shah, R.; Pickard, C.J.; Payne, M.C. Population analysis of plane-wave electronic structure calculations of bulk material. Phys. Rev. B 1996, 54, 16317–16320. [Google Scholar] [CrossRef]
- International Union of Pure and Applied Chemistry. Compendium of Chemical Terminology Gold Book; International Union of Pure and Applied Chemistry: Research Triangle Park, NC, USA, 2014; p. 327. [Google Scholar]
- Qin, Y.F.; Wang, S.Q. Ab-Initio Study of the Role of Mg2Si and Al2CuMg Phases in Electrochemical Corrosion of Al Alloys. J. Electrochem. Soc. 2015, 162, 503–508. [Google Scholar] [CrossRef]
- Wang, J.; Wang, S.Q. Correlation between Galvanic Corrosion and Electronic Work Function of Al Alloy Surfaces. Acta Phys. Chim. Sin. 2014, 30, 551–558. [Google Scholar]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compositions | This Work (Å) | Others (Å) | Exp. (Å) |
|---|---|---|---|
| γ-Fe | a = b = c = 3.428 | a = b = c = 3.430 [23] | a = b = c = 3.647 [21] a = b = c = 3.654 [22] |
| Cr2N | a = b = 4.799; c = 4.452 | a = b = 4.773; c = 4.406 [24] | a = b = 4.752 c = 4.429 [25] |
| Atom | Total Electron (e) | Transfer Charge (e) | Atom | Total Electron (e) | Transfer Charge (e) |
|---|---|---|---|---|---|
| Fe | 7.91 | 0.09 | N | 5.55 | −0.55 |
| Mn | 6.37 | 0.63 | N | 5.55 | −0.55 |
| V | 12.23 | 0.77 | N | 5.58 | −0.58 |
| Ti | 10.99 | 1.01 | N | 5.58 | −0.58 |
| Mo | 13.07 | 0.93 | N | 5.58 | −0.58 |
| Ni | 9.97 | 0.03 | N | 5.51 | −0.51 |
| Atom | Total Electron (e) | Transfer Charge (e) | Atom | Total Electron (e) | Transfer Charge (e) | Atom | Total Electron (e) | Transfer Charge (e) |
|---|---|---|---|---|---|---|---|---|
| Fe | 8.01 | −0.01 | Fe | 8.03 | −0.03 | Fe | 8.06 | −0.06 |
| Fe | 8.01 | −0.01 | Fe | 8.04 | −0.04 | Fe | 8.03 | −0.03 |
| Fe | 8.04 | −0.04 | Fe | 7.99 | 0.01 | Fe | 8.00 | 0.00 |
| Fe | 8.04 | −0.04 | Fe | 7.99 | 0.01 | Fe | 8.00 | 0.00 |
| Fe | 7.97 | 0.03 | Fe | 7.98 | 0.02 | Fe | 7.98 | 0.02 |
| Fe | 7.97 | 0.03 | Fe | 7.97 | 0.03 | Fe | 7.98 | 0.02 |
| Fe | 7.96 | 0.04 | Fe | 7.96 | 0.04 | Fe | 7.96 | 0.04 |
| Fe | 7.96 | 0.04 | Fe | 7.96 | 0.04 | Fe | 7.95 | 0.05 |
| Fe | 7.83 | 0.17 | Fe | 7.83 | 0.17 | Fe | 7.83 | 0.17 |
| Fe | 7.83 | 0.17 | Fe | 7.83 | 0.17 | Fe | 7.83 | 0.17 |
| Fe | 7.89 | 0.11 | Fe | 7.87 | 0.13 | Fe | 7.88 | 0.12 |
| Fe | 7.89 | 0.11 | Fe | 7.88 | 0.12 | Fe | 7.90 | 0.10 |
| Fe | 7.90 | 0.10 | Fe | 7.89 | 0.11 | Fe | 7.91 | 0.09 |
| Fe | 7.90 | 0.10 | Fe | 7.89 | 0.11 | Fe | 7.91 | 0.09 |
| Fe | 7.94 | 0.06 | Fe | 7.92 | 0.08 | Fe | 7.93 | 0.07 |
| Fe | 7.94 | 0.06 | Fe | 7.92 | 0.08 | Fe | 7.95 | 0.05 |
| Fe | 7.91 | 0.09 | Fe | 7.91 | 0.09 | Fe | 8.08 | −0.08 |
| Fe | 7.91 | 0.09 | Fe | 7.91 | 0.09 | Fe | 8.08 | −0.08 |
| Fe | 7.91 | 0.09 | Fe | 7.91 | 0.09 | Fe | 8.08 | −0.08 |
| Fe | 7.91 | 0.09 | Ni | 9.97 | 0.03 | Ti | 10.99 | 1.01 |
| N | 5.55 | −0.55 | N | 5.51 | −0.51 | N | 5.58 | −0.58 |
| N | 5.55 | −0.55 | N | 5.52 | −0.52 | N | 5.55 | −0.55 |
| Cr | 13.89 | 0.11 | Cr | 13.89 | 0.11 | Cr | 13.89 | 0.11 |
| Cr | 13.85 | 0.15 | Cr | 13.88 | 0.12 | Cr | 13.88 | 0.12 |
| Cr | 13.69 | 0.31 | Cr | 13.70 | 0.30 | Cr | 13.70 | 0.30 |
| Cr | 13.75 | 0.25 | Cr | 13.76 | 0.24 | Cr | 13.76 | 0.24 |
| N | 5.56 | −0.56 | N | 5.56 | −0.56 | N | 5.56 | −0.56 |
| N | 5.56 | −0.56 | N | 5.56 | −0.56 | N | 5.56 | −0.56 |
| Cr | 13.89 | 0.11 | Cr | 13.89 | 0.11 | Cr | 13.89 | 0.11 |
| Cr | 13.85 | 0.15 | Cr | 13.88 | 0.12 | Cr | 13.88 | 0.12 |
| Cr | 13.69 | 0.31 | Cr | 13.67 | 0.33 | Cr | 13.67 | 0.33 |
| Cr | 13.75 | 0.25 | Cr | 13.68 | 0.32 | Cr | 13.68 | 0.32 |
| N | 5.54 | −0.54 | N | 5.54 | −0.54 | N | 5.54 | −0.54 |
| N | 5.53 | −0.53 | N | 5.53 | −0.53 | N | 5.53 | −0.53 |
| Cr | 13.89 | 0.11 | Cr | 13.89 | 0.11 | Cr | 13.89 | 0.11 |
| Cr | 13.85 | 0.15 | Cr | 13.88 | 0.12 | Cr | 13.88 | 0.12 |
| Cr | 13.71 | 0.29 | Cr | 13.71 | 0.29 | Cr | 13.71 | 0.29 |
| Cr | 13.73 | 0.27 | Cr | 13.73 | 0.27 | Cr | 13.73 | 0.27 |
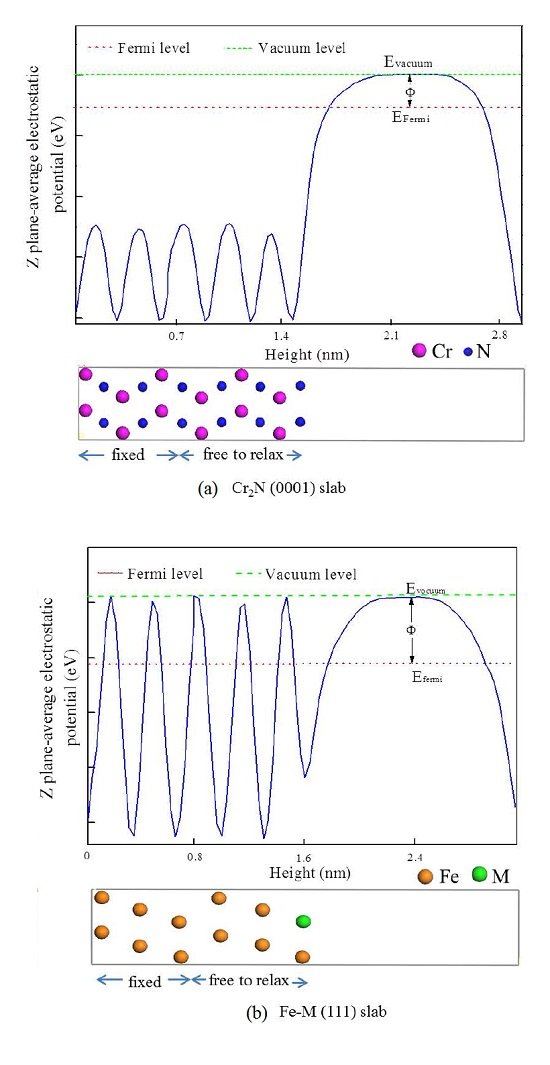
| Terminated Plane | Work Function (eV) | VPD Relative to the Cr2N (V) |
|---|---|---|
| Clean Fe | 5.046 | −0.218 |
| Fe-Mn | 5.063 | −0.201 |
| Fe-V | 4.710 | −0.554 |
| Fe-Ti | 4.931 | −0.333 |
| Fe-Mo | 4.916 | −0.348 |
| Fe-Ni | 4.811 | −0.453 |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, H.; Zhang, C.; Liu, J.; Li, Y.; Fang, X.; Li, J.; Han, P. First-Principles Study on the Structural Stability and Segregation Behavior of γ-Fe/Cr2N Interface with Alloying Additives M (M = Mn, V, Ti, Mo, and Ni). Metals 2016, 6, 156. https://doi.org/10.3390/met6070156
Huang H, Zhang C, Liu J, Li Y, Fang X, Li J, Han P. First-Principles Study on the Structural Stability and Segregation Behavior of γ-Fe/Cr2N Interface with Alloying Additives M (M = Mn, V, Ti, Mo, and Ni). Metals. 2016; 6(7):156. https://doi.org/10.3390/met6070156
Chicago/Turabian StyleHuang, Hui, Caili Zhang, Jie Liu, Yue Li, Xudong Fang, Jianchun Li, and Peide Han. 2016. "First-Principles Study on the Structural Stability and Segregation Behavior of γ-Fe/Cr2N Interface with Alloying Additives M (M = Mn, V, Ti, Mo, and Ni)" Metals 6, no. 7: 156. https://doi.org/10.3390/met6070156
APA StyleHuang, H., Zhang, C., Liu, J., Li, Y., Fang, X., Li, J., & Han, P. (2016). First-Principles Study on the Structural Stability and Segregation Behavior of γ-Fe/Cr2N Interface with Alloying Additives M (M = Mn, V, Ti, Mo, and Ni). Metals, 6(7), 156. https://doi.org/10.3390/met6070156