
Copper Recovery from Polluted Soils Using Acidic Washing and Bioelectrochemical Systems


Abstract
:1. Introduction
2. Experimental Section
2.1. Soil Samples
Pre-Treatment of the Soil Samples
2.2. Leaching Experiments
2.2.1. Optimizing Leaching Experiments
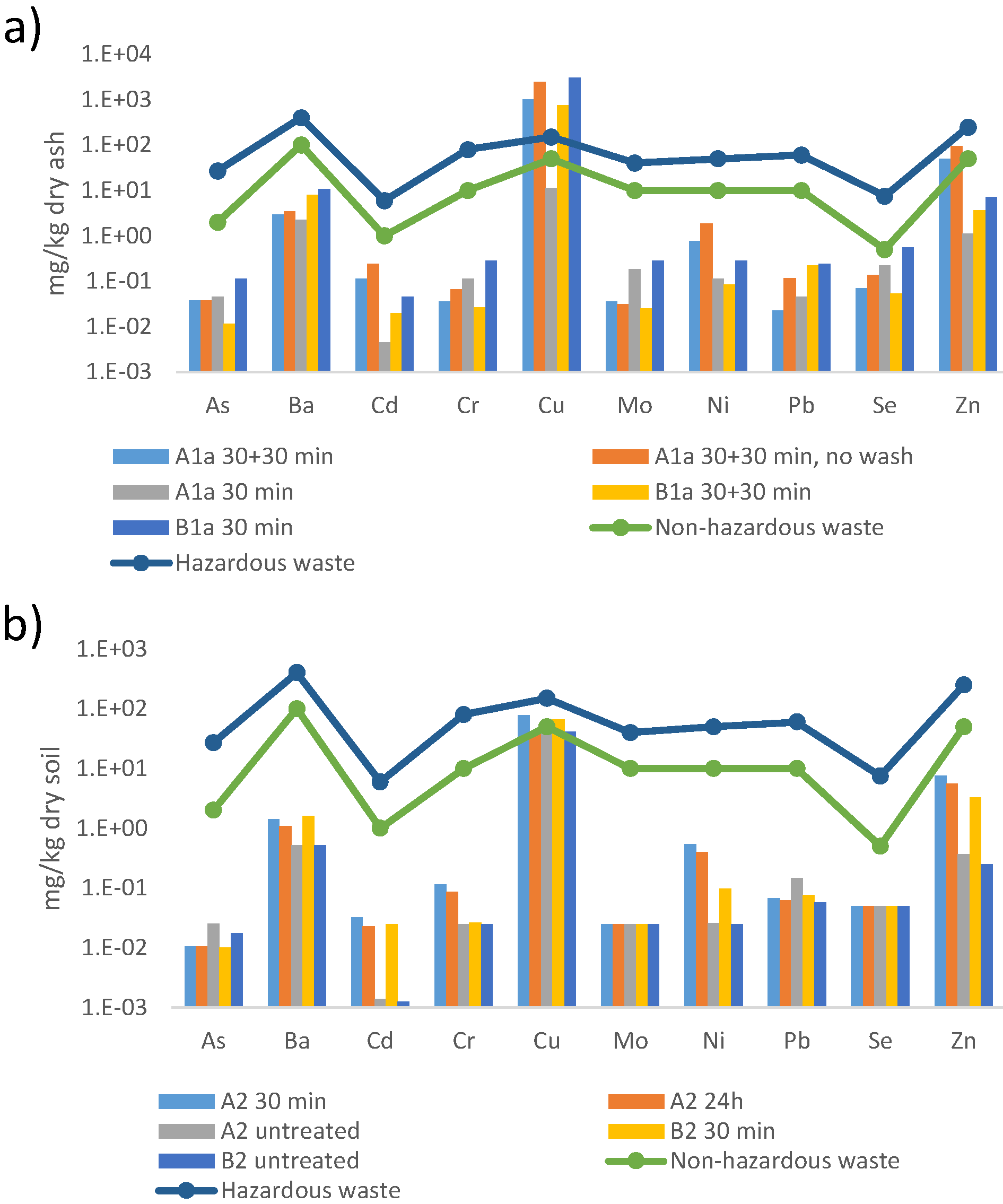
2.2.2. SS-EN-12457-3 Leaching Tests
2.3. Bioelectrochemical Experiments
2.4. Leaching and Degradation of PAHs
2.5. Analytical Methods
3. Results and Discussion
3.1. Characterization of Original Samples
Total Amounts

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Element | A1 (bark) | A1aoven (Bark Ash) | A1aincin (Bark Ash) | A2 (Soil) | B1 (Bark) | B1a (Bark Ash) | B2 (Soil) | Sensitive Land Use a (KM) | Less Sensitive Land Use a (MKM) |
|---|---|---|---|---|---|---|---|---|---|
| pH(H2O) | 4.9 b | 12.1 b | – c | 5.5 | – | – | 5.2 | ||
| wt. % DS | |||||||||
| SiO2 | 1.9 | 54 | 8.4 | 68 | 26 | 57 | 76 | * d | * |
| Al2O3 | 0.7 | 10 | 4.6 | 12 | 5.3 | 11 | 13 | * | * |
| CaO | 0.8 | 4.5 | 26 | 1.8 | 0.5 | 1.3 | 0.8 | * | * |
| Fe2O3 | 0.5 | 4.0 | 1.8 | 2.5 | 2.0 | 4.4 | 1.1 | * | * |
| K2O | 0.1 | 2.7 | 6.9 | 2.4 | 1.6 | 3.5 | 4.2 | * | * |
| MgO | 0.06 | 0.7 | 1.7 | 0.6 | 0.1 | 0.2 | 0.1 | * | * |
| MnO | 0.01 | 0.07 | 0.01 | 0.04 | 0.01 | 0.03 | 0.02 | * | * |
| Na2O | 0.08 | 2.0 | 7.3 | 2.6 | 1.0 | 2.5 | 2.7 | * | * |
| P2O5 | 0.1 | 0.6 | 29 | 0.04 | 0.2 | 0.4 | 0.01 | * | * |
| TiO2 | 0.02 | 0.4 | 0.9 | 0.5 | 0.2 | 0.2 | 0.2 | * | * |
| mg/kg DS | |||||||||
| As | 2.0 | 11 | <3 | 0.6 | 7.8 | 22 | 0.3 | 10 | 25 |
| Ba | 110 | 930 e | 430 | 500 | 620 | 230 | 860 | 200 | 300 |
| Be | 0.1 | 1.5 | <0.5 | 1.4 | 0.7 | 1.5 | 1.9 | * | * |
| Cd | 0.3 | 1.3 | 0.1 | 0.01 | 0.1 | 0.3 | 0.03 | 0.5 | 15 |
| Co | 1.2 | 7.5 | 2.3 | 2.2 | 0.9 | 2.2 | 0.3 | 15 | 35 |
| Cr | 3.9 | 50 | 71 | 42 | 9.0 | 24 | 25 | 80 | 150 |
| Cu | 11,000 | 130,000 | 19,000 | 1100 | 15,000 | 110,000 | 720 | 80 | 200 |
| Hg | 0.1 | <0.01 | <0.01 | <0.04 | 0.4 | <0.01 | 0.1 | 0.25 | 2.5 |
| Mo | 0.2 | 2.4 | 3.1 | 0.3 | 0.3 | 1.4 | 0.2 | 40 | 100 |
| Nb | 0.4 | 8.8 | 5.4 | 9.0 | 7.0 | 6.4 | 5.9 | * | * |
| Ni | 3.5 | 27 | 21 | 3.1 | 2.6 | 7.9 | 0.3 | 40 | 120 |
| Pb | 31 | 360 | 61 | 7.9 | 39 | 150 | 3.2 | 50 | 400 |
| S | 570 | 4900 | 2700 | 76 | 510 | 2400 | <50 | * | * |
| Sb | – | 6.7 | 8.9 | 0.2 | – | 16 | 0.1 | 12 | 30 |
| Sc | 0.6 | 5.9 | <1 | 7.7 | 1.2 | 2.9 | 1.9 | * | * |
| Sn | – | 19 | 33 | – | – | 19 | – | * | * |
| Sr | 30 | 230 | 130 | 190 | 95 | 260 | 200 | * | * |
| V | 6.7 | 38 | 12 | 44 | 7.0 | 15 | 9.7 | 100 | 200 |
| W | 0.6 | <50 | <50 | 1.2 | 0.6 | <50 | 0.7 | * | * |
| Y | 2.3 | 19 | 2.9 | 17 | 6.9 | 8.0 | 5.4 | * | * |
| Zn | 44 | 260 | 1500 | 14 | 36 | 160 | 3.5 | 250 | 500 |
| Zr | 5.7 | 150 | 13 | 230 | 42 | 95 | 113 | * | * |

3.2. Optimization of Leaching Parameters
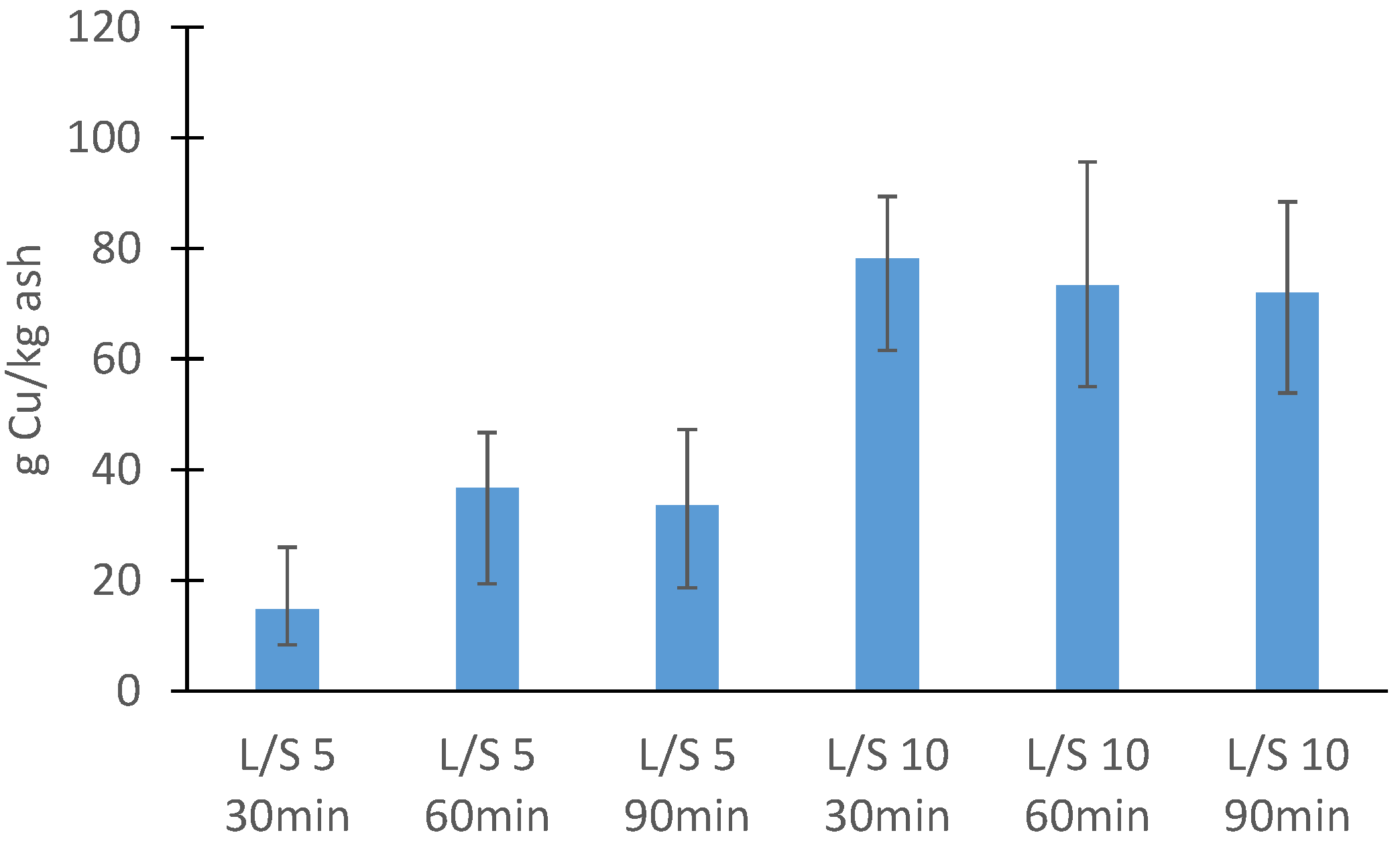
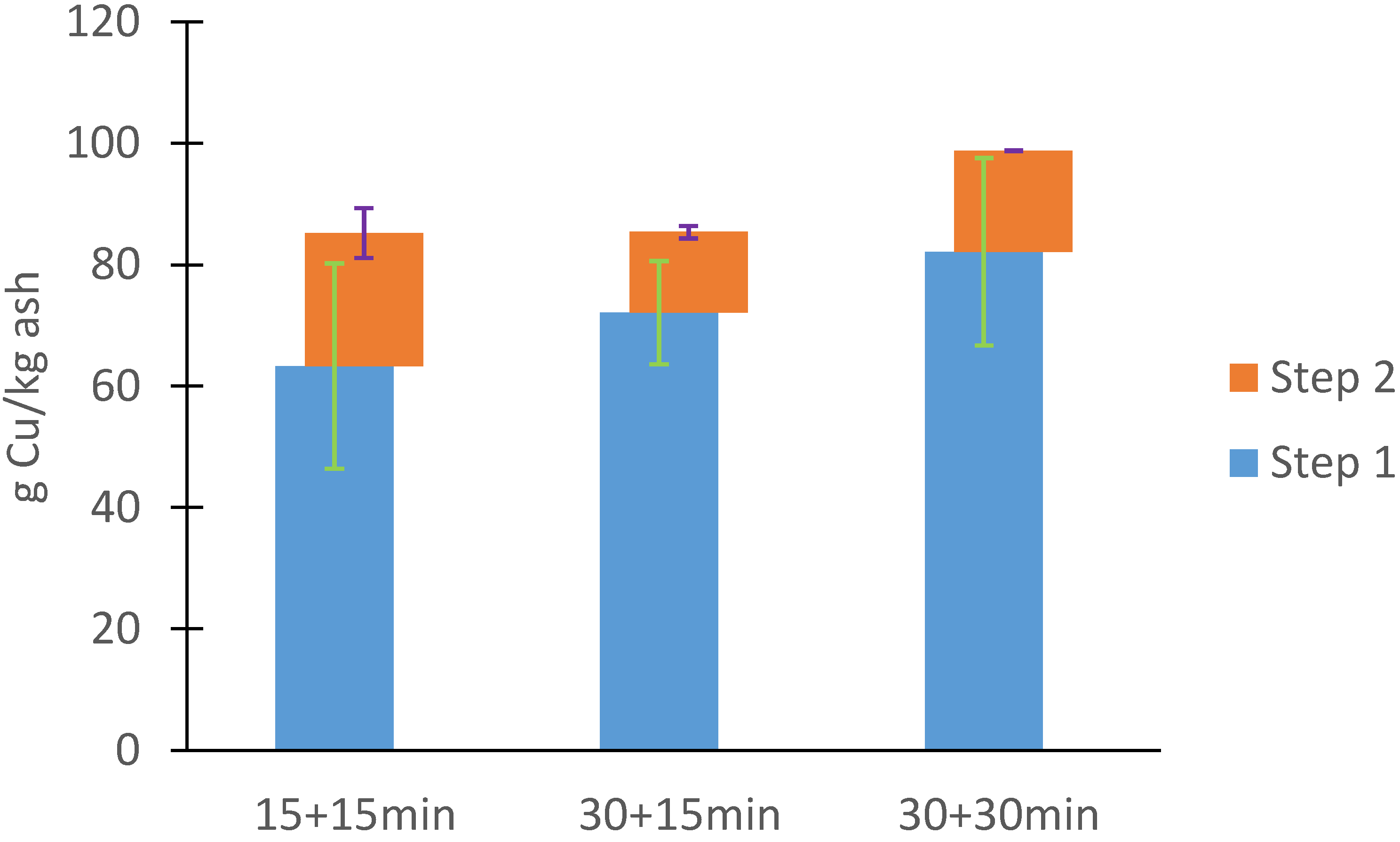
3.2.1. Ash

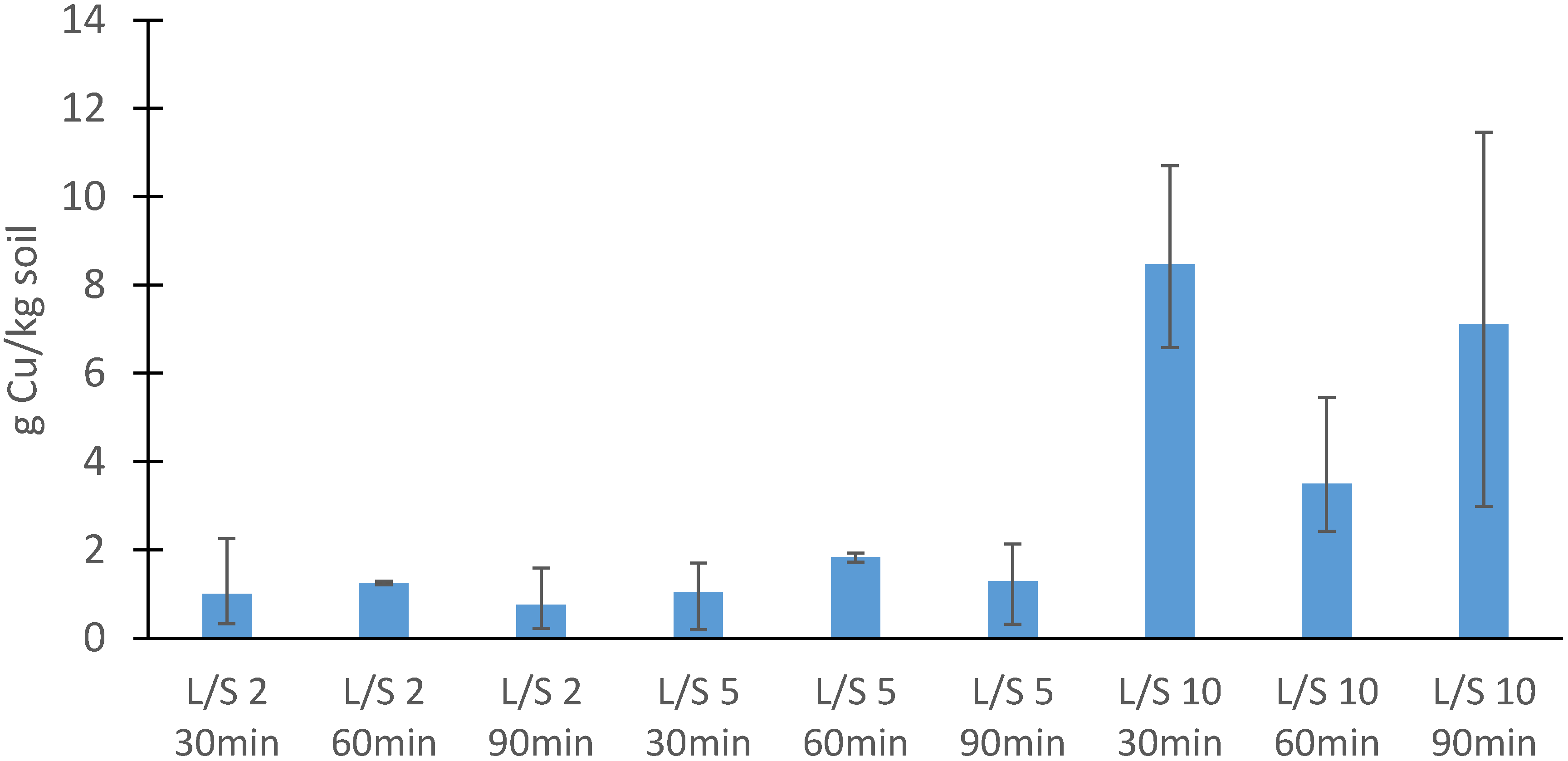
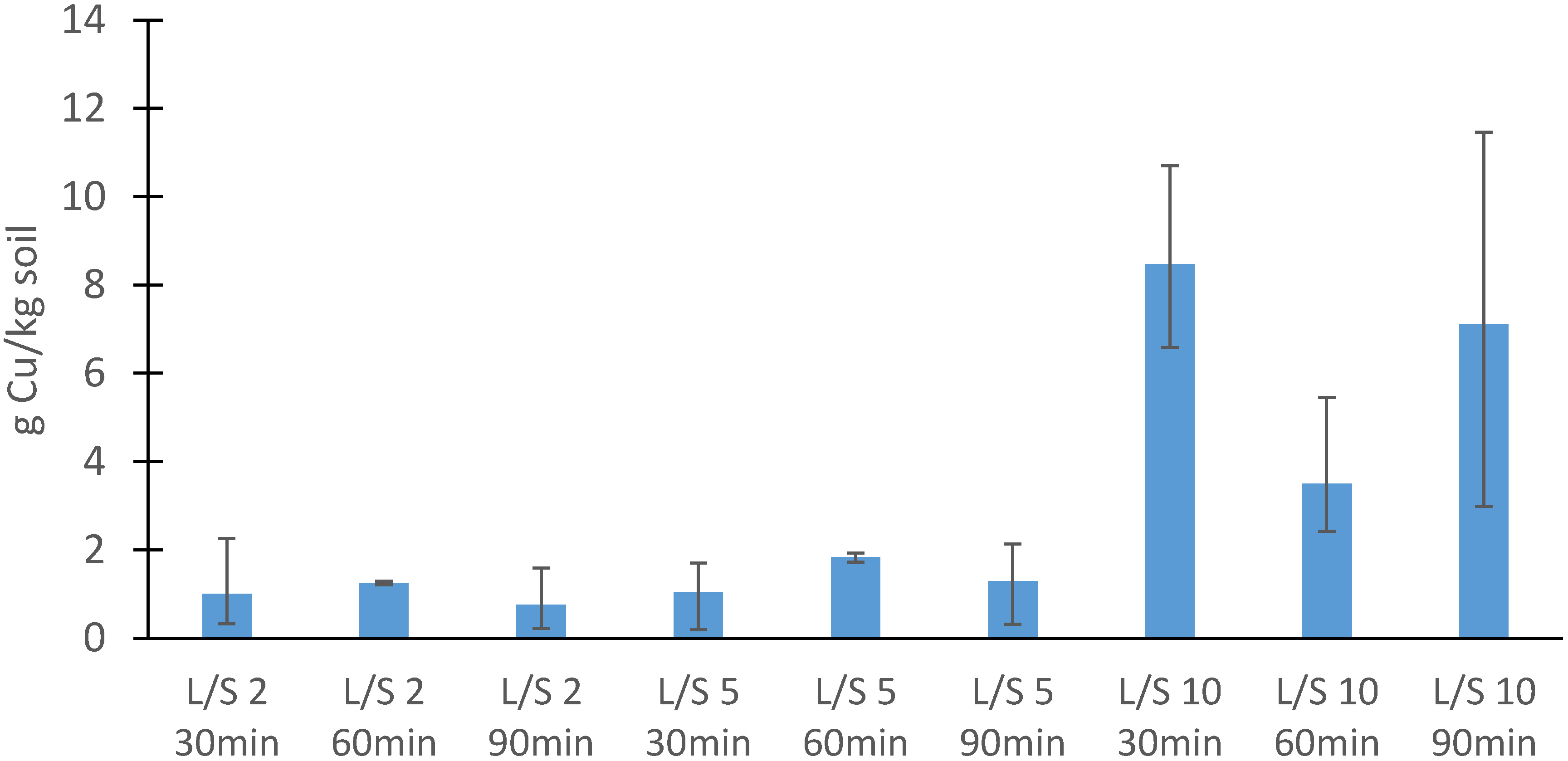
3.2.2. Soil
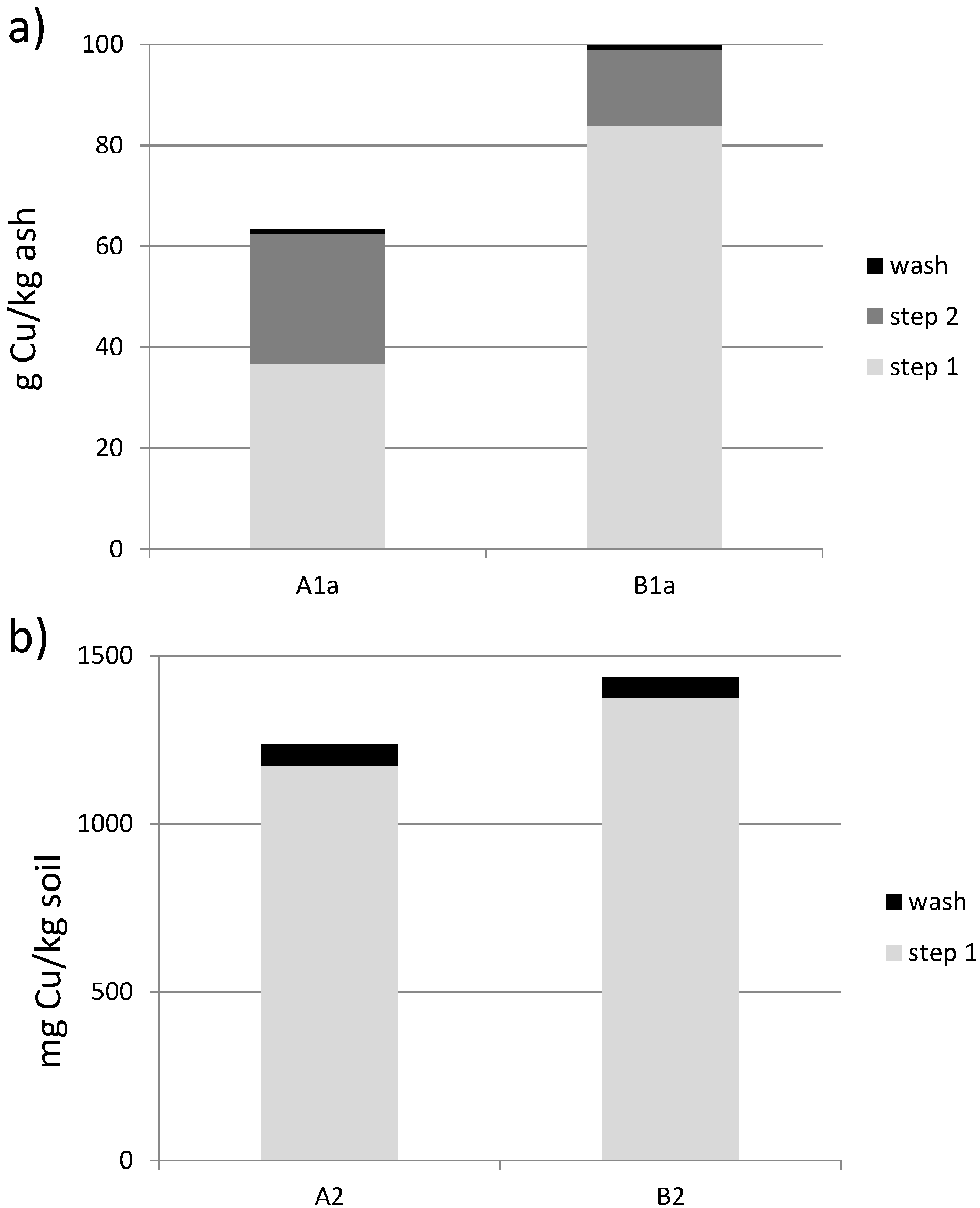
3.3. Batch Leaching
| Element | A1a (Bark Ash) a | B1a (Bark Ash) a | A2 (Soil) a | B2 (Soil) a | ||
|---|---|---|---|---|---|---|
| Step 1 b | Step 2 c | Step 1 b | Step 2 c | Step 1 b | Step 1 b | |
| mg/L | ||||||
| Al | 820 | 340 | 96 | 52 | 150 | 560 |
| As | 0.7 | 0.3 | 2.5 | 0.4 | 0.2 | 0.2 |
| Ba | 5.2 | 2.1 | 1.3 | 1.5 | 2.0 | 1.7 |
| Pb | 21 | 12 | 11 | 11 | 11 | 9.3 |
| Cd | 0.3 | 0.3 | 0.3 | 0.3 | 0.3 | 0.3 |
| Co | 0.1 | <0.1 | <0.1 | <0.1 | <0.1 | <0.1 |
| Cu | 3400 | 2400 | 8400 | 1500 | 120 | 140 |
| Cr | 0.8 | 0.2 | 0.1 | 0.1 | 0.1 | 0.1 |
| Li | 0.6 | 0.08 | 0.06 | 0.04 | 0.06 | 0.0 |
| Mn | 20 | 5.0 | 6.9 | 4.5 | 4.9 | 5.4 |
| Mo | 0.2 | <0.1 | <0.1 | <0.1 | <0.1 | <0.1 |
| Ni | 6.6 | 2.5 | 9.4 | 0.7 | 0.1 | 0.1 |
| Se | 0.1 | <0.1 | <0.1 | <0.1 | 0.1 | <0.1 |
| Sr | 13 | 2.2 | 4.3 | 1.1 | 0.7 | 0.6 |
| V | 0.7 | 0.1 | 0.5 | <0.1 | 0.2 | 0.1 |
| Zn | 430 | 90 | 70 | 60 | 60 | 50 |
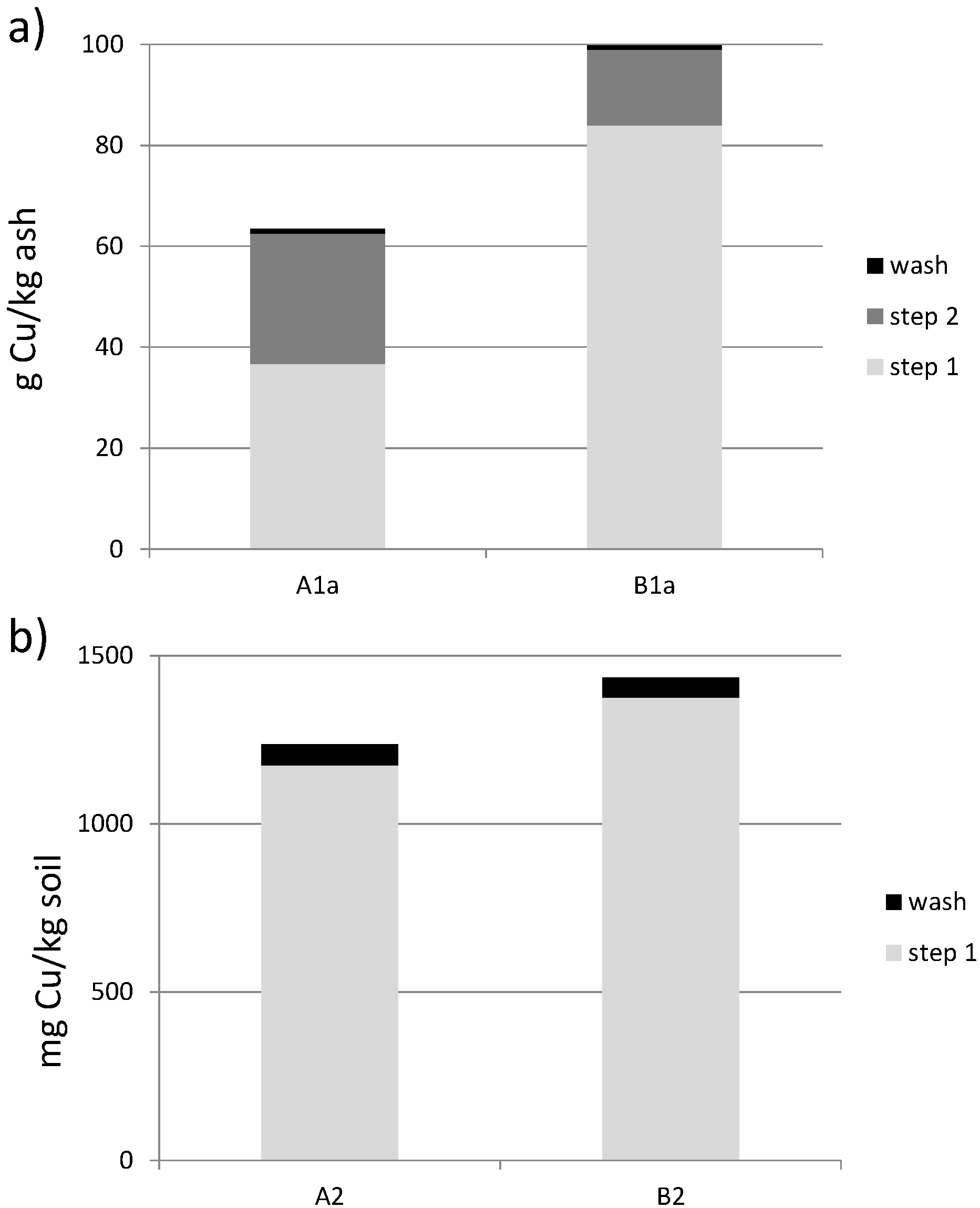
3.4. Cu Recovery Using BES
| Run | Duration (h) | Resistor (Ω) | Final Cu Conc. a (mg/L) | Theoretical Cu Recovery b (mg) | Cu removal from Catholyte (mg) | Measured Cu Recovery (mg) | Measured Cu Recovery c (%) |
|---|---|---|---|---|---|---|---|
| 1 | 19 | 1000 | 1500 | 6.4 | 7.4 | 4.9 | 23 |
| 2 | 29 | 100 | 420 | 21 | 17 | 13 | 64 |
| 3 | 22 | 1000 | 1500 | 8.1 | 7.2 | 4.2 | 20 |
| 4 | 72 | 100 | 160 | 27 | 20 | 17 | 79 |
3.5. Characterization of Leached Soil Samples
3.5.1. Metal Content
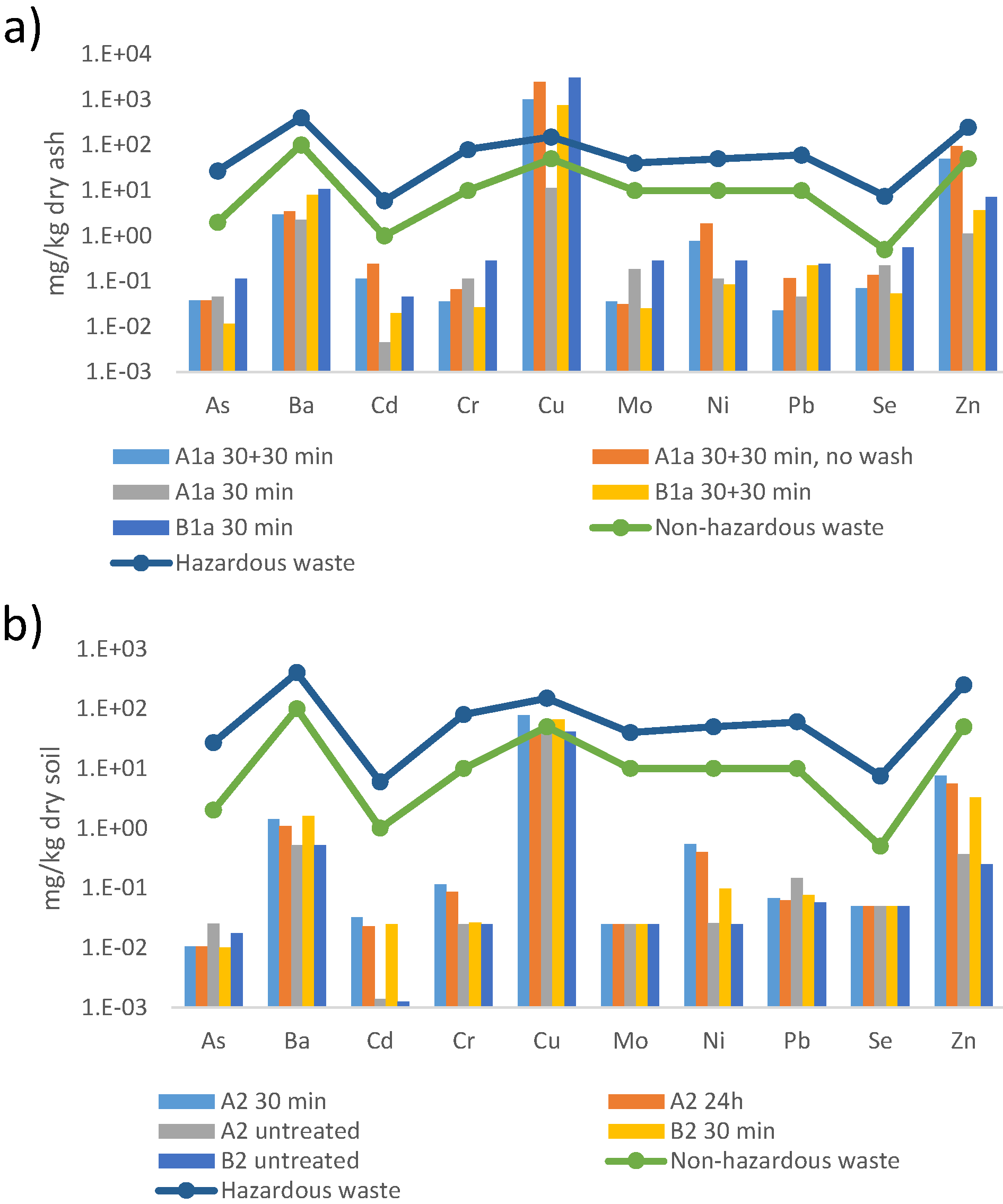
| Element | A1a Bark Ash Residue | A2 Soil Residue |
|---|---|---|
| wt. % DS | ||
| SiO2 | 54 | 71 |
| Al2O3 | 12 | 11 |
| CaO | 1.0 | 1.9 |
| Fe2O3 | 5.6 | 2.2 |
| K2O | 4.6 | 2.4 |
| MgO | 0.8 | 0.5 |
| MnO | <0.1 | <0.1 |
| Na2O | 1.8 | 2.6 |
| P2O5 | 1.1 | <0.1 |
| TiO2 | 8.1 | 0.6 |
| mg/kg DS | ||
| As | <6 | 0.8 |
| Ba | 2200 | 450 |
| Be | 1.9 | 1.3 |
| Cd | 0.7 | 0.1 |
| Co | 7.2 | 1.7 |
| Cr | 220 | 20 |
| Cu | 10,000 | 210 |
| Hg | 68 | 3.6 |
| Mo | 4.2 | 0.4 |
| Nb | 34 | 8.5 |
| Ni | 66 | 3.9 |
| Pb | 370 | 8.4 |
| S | 900 | 220 |
| Sb | 64 | 2.7 |
| Sc | 7.2 | 7.5 |
| Sn | 180 | 2.5 |
| Sr | 82 | 180 |
| V | 37 | 41 |
| W | <50 | 1.1 |
| Y | 10 | 14 |
| Zn | 1000 | 28 |
| Zr | 140 | 220 |
3.5.2. Metal Leaching
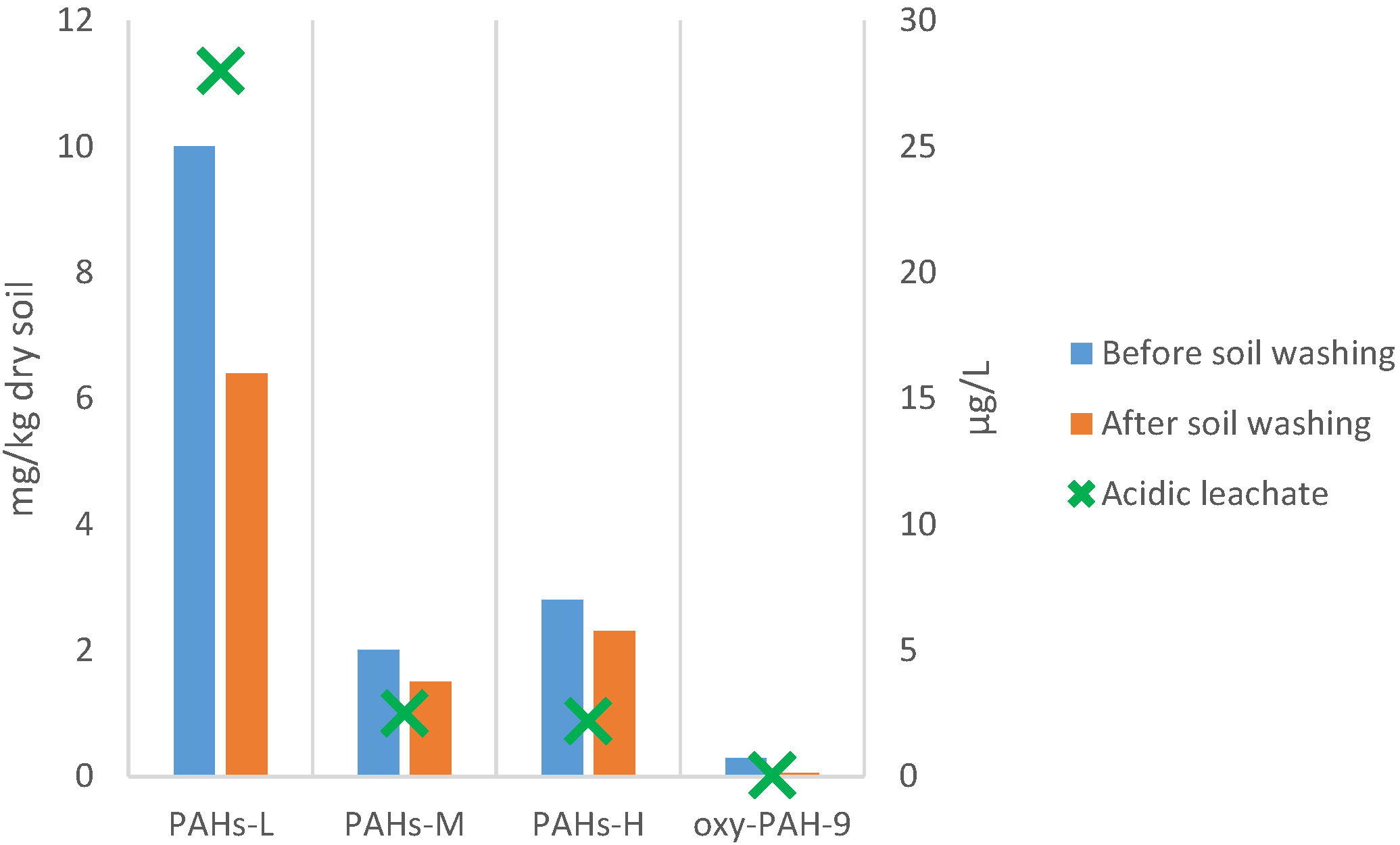
3.5.3. PAHs and Oxy-PAHs Content
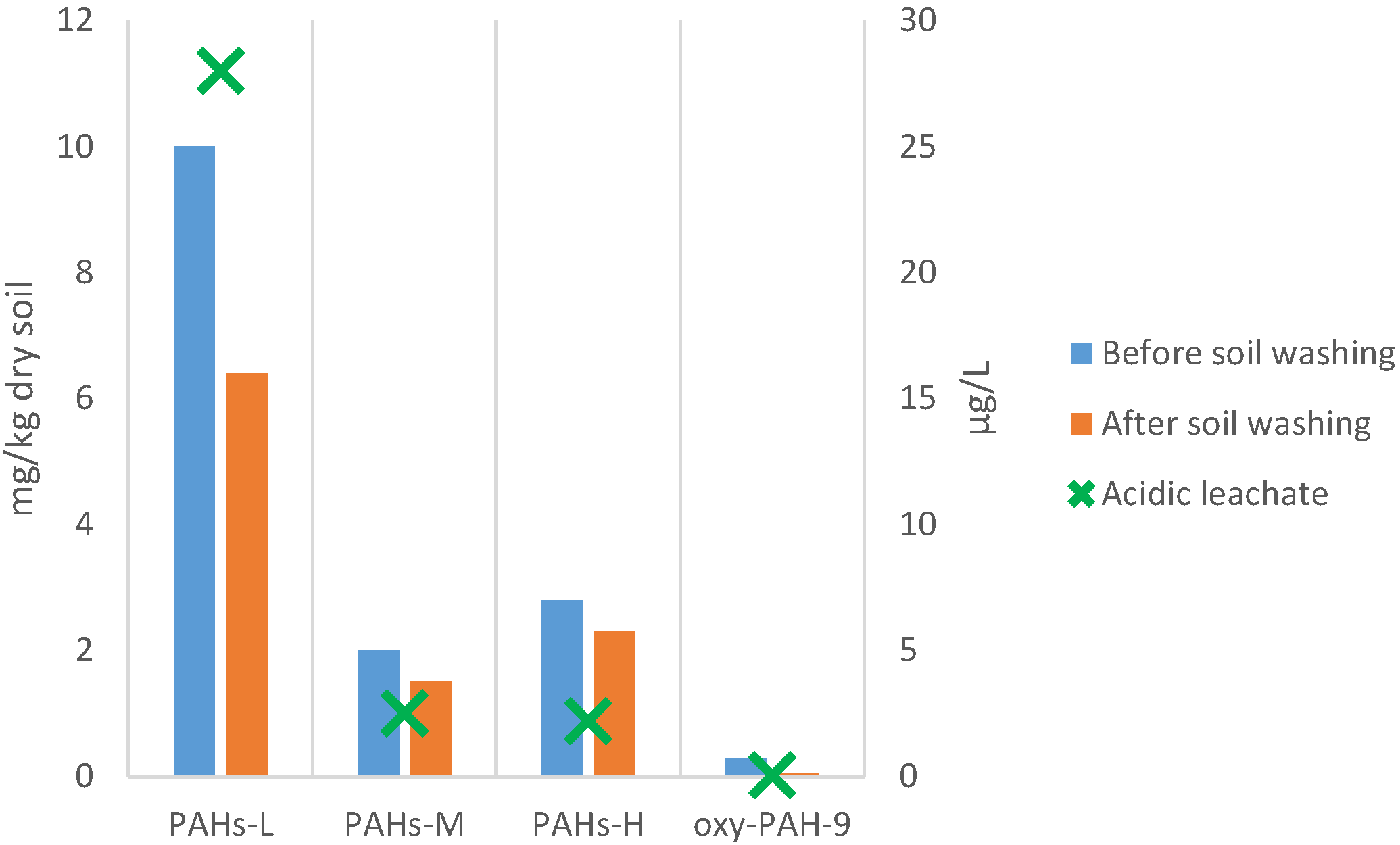
4. Conclusions
Supplementary Files
Supplementary File 1Acknowledgments
Author Contributions
Conflicts of Interest
References
- Sierra, C.; Menendez-Aguadoa, J.M.; Afifb, E.; Carreroc, M.; Gallegoa, J.R. Feasibility study on the use of soil washing to remediate the As-Hg contamination at an ancient mining and metallurgy area. J. Hazard. Mater. 2011, 196, 93–100. [Google Scholar] [CrossRef] [PubMed]
- Bisone, S.; Blais, J.; Drogui, P.; Mercier, G. Toxic metal removal from polluted soil by acid extraction. Water Air Soil Pollut. 2012, 223, 3739–3755. [Google Scholar] [CrossRef]
- Moon, D.H.; Lee, J.R.; Wazne, M.; Park, J.H. Assessment of soil washing for Zn in contaminated soils using various washing solutions. J. Ind. Eng. Chem. 2012, 18, 822–825. [Google Scholar] [CrossRef]
- Sierra, C.; Martínez-Blanco, D.; Blanco, J.A.; Gallego, J.R. Optimization of the concentration operation via magnetic separation: A case study for soil washing. Chemosphere 2014, 107, 290–296. [Google Scholar] [CrossRef] [PubMed]
- Sierra, C.; Martínez, J.; Menéndez-Aguado, J.M.; Afif, E.; Gallego, J.R. High intensity magnetic separation for the clean-up of a site polluted by lead metallurgy. J. Hazard. Mater. 2013, 248–249, 194–201. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, M.; Singh, S.P. A review on phytoremediation of heavy metals and utilization of its byproducts. Ecol. Environ. Res. 2005, 3, 1–18. [Google Scholar] [CrossRef]
- Camenzuli, D.; Gore, D.B.; Stark, S.C. Immobilisation of metals in contaminated landfill material using orthophosphate and silica amendments: A pilot study. Int. J. Environ. Pollut. Remediat. 2015, 3, 27–32. [Google Scholar] [CrossRef]
- Chigbo, C.; Batty, L. Chelate-assisted phytoremediation of Cu-pyrene-contaminated soil using Z. Mays. Water Air Soil Pollut. 2015, 226. [Google Scholar] [CrossRef]
- Travar, I.; Kihl, A.; Kumpiene, J. The release of As, Cr and Cu from contaminated soil stabilized with apc residues under landfill conditions. J. Environ. Manag. 2015, 151, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Voglar, D.; Lestan, D. Chelant soil-washing technology for metal-contaminated soil. Environ. Technol. 2014, 35, 1389–1400. [Google Scholar] [CrossRef] [PubMed]
- Pociecha, M.; Lestan, D. Washing of metal contaminated soil with edta and process water recyling. J. Hazard. Mater. 2012, 235–236, 384–387. [Google Scholar] [CrossRef] [PubMed]
- Karlfeldt Fedje, K.; Yillin, L.; Strömvall, A.M. Remediation of metal polluted hotspot areas through enhanced soil washing—Evaluation of leaching methods. J. Environ. Manag. 2013, 128, 489–496. [Google Scholar]
- Achugasim, O.; Ojinnaka, C.; Osuji, L. Assessment of ability of three chemcial oxidants to remove hydrocarbons from soils polluted by bonny light crude oil. Eur. Chem. Bull. 2013, 2, 226–230. [Google Scholar]
- Xu, C.B.; Dong, D.B.; Meng, X.L.; Su, X.; Zheng, X.; Li, Y.Y. Photolysis of polycyclic aromatic hydrocarbons on soil surfaces under uv irradiation. J. Environ. Sci. China 2013, 25, 569–575. [Google Scholar] [CrossRef]
- Baboshin, M.A.; Golovleva, L.A. Aerobic bacterial degradation of polycyclic aromatic hydrocarbons (PAHs) and its kinetic aspects. Microbiology 2012, 81, 639–650. [Google Scholar] [CrossRef]
- Lundstedt, S.; White, P.A.; Lemieux, C.L.; Lynes, K.D.; Lambert, L.B.; Oberg, L.; Haglund, P.; Tysklind, M. Sources, fate, and toxic hazards of oxygenated polycyclic aromatic hydrocarbons (PAHs) at PAH-contaminated sites. Ambio 2007, 36, 475–485. [Google Scholar] [CrossRef]
- Jenkins, J.; Dodge, P.; Morenci, A. Solvent Extraction Transfer of Cu from Leach Solution to Electrolyte; Pergamon: New York, NY, USA, 2002; pp. 307–325. [Google Scholar]
- Nancharaiah, Y.V.; Venkata Mohan, S.; Lens, P.N. Metals removal and recovery in bioelectrochemical systems: A review. Bioresour. Technol. 2015. [Google Scholar] [CrossRef] [PubMed]
- Modin, O.; Fedje, K.K. Combined Wastewater Treatment and Recovery of Copper from Ash Leachate. In Proceedings of the IWA World Water Congress & Exhibition, Busan, Korea, 16–31 September 2012.
- Tao, H.C.; Liang, M.; Li, W.; Zhang, L.J.; Ni, J.R.; Wu, W.M. Removal of copper from aqueous solution by electrodeposition in cathode chamber of microbial fuel cell. J. Hazard. Mater. 2011, 189, 186–192. [Google Scholar] [CrossRef] [PubMed]
- Ter Heijne, A.; Liu, F.; Weijden, R.V.D.; Weijma, J.; Buisman, C.J.N.; Hamelers, H.V.M. Copper recovery combined with electricity production in a microbial fuel cell. Environ. Sci. Technol. 2010, 44, 4376–4381. [Google Scholar] [CrossRef] [PubMed]
- Modin, O.; Wang, X.; Wu, X.; Rauch, S.; Fedje, K.K. Bioelectrochemical recovery of Cu, Pb, Cd, and Zn from dilute solutions. J. Hazard. Mater. 2012, 235–236, 291–297. [Google Scholar] [CrossRef] [PubMed]
- Swedish Standard Institute. SS-EN-12457-3 characterization of waste—Leaching—Compliance test for leaching of granular waste materials and sludges. In Part 3: Two Stage Batch Test at a Liquid to Solid Ratio of 2 L/kg and 8 L/kg for Materials with High Soild Content and with Particle Size Below 4 mm (witout or with Size Reduction); Swedish Standard Institute: Stockholm, Sweden, 2003. [Google Scholar]
- Mongomery, D.C. Design and Analysis of Experiments, 7th ed.; John Wiley & Sons Pte Led (Asia): Hongkong, China, 2009. [Google Scholar]
- SEPA. Riktvärden för Förorenad Mark Modellbeskrivning och Vägledning Rapport 5976 (Guidelines for Contaminated Soil Model Description and Guidance Report 5976); SEPA: Stockholm, Sweden, 2009. [Google Scholar]
- Springfors, A. Kartläggning av Föroreningssituationen i Mark, Grundvatten och Sediment, Förenklad Riskbedömning, Översiktlig Åtgärdsutredning Samt Riskbedömning för Föreslagna Efterbehandlingsåtgärder för Långön, Köpmannebo; ÅF-IPK AB: Göteborg, Sweden, 2001. [Google Scholar]
- Florell, S.E.; Stubdrup, O.P.; Arnér, M. Rapport, Televerket Björkhult, Huvudstudie (Report, Televerket Björkhult); WSP: Linköping, Sweden, 2008. [Google Scholar]
- Naturvårdsverkets föreskrifter om deponering, kriterier och förfaranden för mottagning av avfall vid anläggningar för deponering av avfall. Available online: http://www.naturvardsverket.se/Stod-i-miljoarbetet/Rattsinformation/Foreskrifter-allmanna-rad/NFS/2004/NFS-200410---Mottagning-av-avfall-for-deponering/ (accessed on 17 July 2015).
- Yip, T.C.M.; Tsang, D.C.W.; Ng, K.T.W.; Lo, I.M.C. Empirical modeling of heavy metal extraction by edds from singel-metal and mulit-metal contaminated soils. Chemosphere 2008, 74, 301–307. [Google Scholar] [CrossRef] [PubMed]
- Udovic, M.; Lestan, D. Edta and hcl leaching of calcareous and acidic soils polluted with potentially toxic metals: Remediation efficiency and soil impact. Chemosphere 2012, 88, 718–724. [Google Scholar] [CrossRef] [PubMed]
- Karlfeldt, K.; Steenari, B.M. Assessment of metal mobility in msw incineration ashes using water as the reagent. Fuel 2007, 86, 1983–1993. [Google Scholar] [CrossRef]
- Kalmykova, Y.; Strömvall, A.M.; Steenari, B.M. Alternative materials for removal of heavy metals and petroleum hydrocarbons from contaminated leachates. Environ. Technol. 2008, 29, 111–122. [Google Scholar] [CrossRef] [PubMed]
- Swedish Environmental Protection Agency. Generic Guideline Values for Contaminated Soils. Availble online: http://www.swedishepa.se/en/In-English/Start/Operations-with-impact-on-the-environment/Remediation-of-contaminated-areas/Classification-and-risk-assessment/Generic-guideline-values-for-contaminated-soils/ (accessed on 29 May 2012).
- Bandowe, B.A.M.; Wilcke, W. Analysis of polycyclic aromatic hydrocarbons and their oxygen-containing derivatives and metabolites in soils. J. Environ. Qual. 2010, 39, 1349–1358. [Google Scholar] [CrossRef] [PubMed]
- Jonsson, S.; Persson, Y.; Frankki, S.; van Bavel, B.; Lundstedt, S.; Haglund, P.; Tysklind, M. Degradation of polycyclic aromatic hydrocarbons (PAHs) in contaminated soils by Fenton’s reagent: A multivariate evaluation of the importance of soil characteristics and pah properties. J. Hazard. Mater. 2007, 149, 86–96. [Google Scholar] [CrossRef] [PubMed]
- Mao, D.; Lookman, R.; Van de Weghe, H.; Weltens, R.; Vanermen, G.; De Brucker, N.; Diels, L. Combining HPLC-GCXGC, GCXGC/TOF-MS, and selected ecotoxicity assays for detailed monitoring of petroleum hydrocarbon degradation in soil and leaching water. Environ. Sci. Technol. 2009, 43, 7651–7657. [Google Scholar] [CrossRef] [PubMed]
- Kalmykova, Y.; Bjoerklund, K.; Stroemvall, A.-M.; Blom, L. Partitioning of polycyclic aromatic hydrocarbons, alkylphenols, bisphenol A and phthalates in landfill leachates and stormwater. Water Res. 2013, 47, 1317–1328. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, K.; Kalmykova, Y.; Strömvall, A.M.; Baun, A.; Eriksson, E. Particle phase distribution of polycyclic aromatic hydrocarbons in stormwater—Using humic acid and iron nano-sized colloids as test particles. Sci. Total Environ. 2015, 532, 103–111. [Google Scholar] [CrossRef] [PubMed]
- Tunega, D.; Gerzabek, M.H.; Haberhauer, G.; Totsche, K.U.; Lischka, H. Model study on sorption of polycyclic aromatic hydrocarbons to goethite. J. Colloid Interface Sci. 2009, 330, 244–249. [Google Scholar] [CrossRef] [PubMed]
- Oren, A.; Chefetz, B. Successive sorption-desorption cycles of dissolved organic matter in mineral soil matrices. Geoderma 2012, 189–190, 108–115. [Google Scholar] [CrossRef]
- Yap, L.C.; Gan, S.; Ng, H.K. Fenton based remediation of polycyclic aromatic hydrocarbons-contaminated soils. Chemosphere 2011, 83, 1414–1430. [Google Scholar] [CrossRef] [PubMed]
- Tran, L.H.; Drogui, P.; Mercier, G.; Blais, J.F. Electrochemcial degradation of polycyclic aromatic hydrocarbons in creostoe solution using ruthenium oxid on titanium expanded mesh anod. J. Hazard. Mater. 2009, 164, 1118–1129. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.J.; Blais, J.F.; Mercier, G.; Bergeron, M.; Drogui, P. PAH removal from spiked municipal wastewater sewage sludge using biological, chemical and electrochemical treatments. Chemosphere 2007, 68, 1143–1152. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fedje, K.K.; Modin, O.; Strömvall, A.-M. Copper Recovery from Polluted Soils Using Acidic Washing and Bioelectrochemical Systems. Metals 2015, 5, 1328-1348. https://doi.org/10.3390/met5031328
Fedje KK, Modin O, Strömvall A-M. Copper Recovery from Polluted Soils Using Acidic Washing and Bioelectrochemical Systems. Metals. 2015; 5(3):1328-1348. https://doi.org/10.3390/met5031328
Chicago/Turabian StyleFedje, Karin Karlfeldt, Oskar Modin, and Ann-Margret Strömvall. 2015. "Copper Recovery from Polluted Soils Using Acidic Washing and Bioelectrochemical Systems" Metals 5, no. 3: 1328-1348. https://doi.org/10.3390/met5031328
APA StyleFedje, K. K., Modin, O., & Strömvall, A.-M. (2015). Copper Recovery from Polluted Soils Using Acidic Washing and Bioelectrochemical Systems. Metals, 5(3), 1328-1348. https://doi.org/10.3390/met5031328