1. Introduction
Many advanced engineering applications need alloys with a combination of excellent surface hardness, corrosion resistance, wear resistance, and high-cycle fatigue life. Alloy properties are most critical where environmental impact occurs: at the alloy surface. An approach to make alloy parts with superior performance and lifetime consists of “surface engineering” alloy parts after they first have been shaped and undergone surface machining. A particularly potent method of such alloy surface engineering is the infusion of concentrated interstitial solute, e.g., carbon or nitrogen from a gas phase, performed at low temperature. “Low” temperature, in this context, means a temperature that is high enough to enable rapid diffusion of interstitial solute, but, at the same time, low enough to suppress precipitation of undesired phases within the processing time. Under these conditions, the concentration of interstitial solute can become orders of magnitude higher than it could be in thermodynamic equilibrium [
1,
2,
3,
4,
5,
6,
7,
8,
9,
10]. Useful methods of surface engineering by gas-phase-based infusion of concentrated interstitial solute include low-temperature carburization, -nitridation, or -nitrocarburization [
7,
8,
10,
11,
12,
13,
14]. An industrial process has been established for gas-phase-based low-temperature carburization, optimized particularly for ferrules (
Figure 1) made from AISI-316L (austenitic Fe–Cr–Ni stainless steel containing Mo) [
6,
15,
16,
17,
18]. Within a carbon infusion processing time ≈80 ks (1 day), this process can generate a highly conformal carbon-rich sub-surface zone (“case”) with carbon fractions up to
and a carbon diffusion profile that, absent a sharp interface to the underlying alloy core, has an apparent “depth” of ≈25 μm in metallographic images. Similar methods have been developed for low-temperature nitriding and -nitro-carburization [
9,
19,
20,
21,
22,
23,
24,
25,
26,
27]. Even low depths of concentrated interstitial carbon and/or nitrogen impart significantly improved corrosion resistance in chlorine-rich environments (saltwater). Infusion of interstitial solute to greater depths also significantly increases surface hardness, wear resistance, and fatigue life, leading to numerous technical applications that may benefit from such surface engineering [
1,
3,
5,
14,
28,
29,
30,
31].
The usefulness of many important “corrosion resistant” austenitic alloys originates from their ability to spontaneously form a surface film of a passivating oxide. Representative alloys are AISI-316L, Al-6XN
®, Inconel
® 625, Incoloy
® 825, and Hastelloy
® C-22. The oxide protects the alloy against further oxidation and against corrosive attack from the environment. The oxide is typically a Cr-rich mixed oxide with a thickness that can be below 1 nm [
32].
While the passivating oxide is beneficial in applications, it constitutes a diffusion barrier to carbon and nitrogen at the temperatures required by low-temperature infusion of interstitial solute. To enable the infusion of interstitial solute, an extra step is required to remove the passivating surface oxide film, or make it permeable to carbon or nitrogen. This critical step is known as “surface activation”. An activated surface is one that was made permeable to carbon and nitrogen, so carbon and nitrogen adsorbed on the alloy surface can dissolve into the underlying alloy. Several methods have been designed for surface activation over the development of low-temperature carburization, nitrocarburization, and nitridation, including the following:
Immersion in acidic solutions [
33,
34,
35,
36].
Plasma-based processes [
3,
7,
19,
29].
More recent developments of solid-reagent pyrolysis processes [
24,
36,
40,
41,
42,
43,
44,
45].
Each of these methods have benefits and detriments in terms of effectiveness, safety, overall process complexity, and applicability to different alloys. Surface activation is also not typically a process with binary results. Through certain mixtures of activation and infusion processes, one can produce, e.g., deep and even infusion or areas of varying infusion depth and interstitial level. The general understanding of how these methods activate the surface has been the same as for high-temperature carburization or nitriding: by removing the passivating oxide film, so that carbon or nitrogen can be infused through an oxide-free metal surface. However, our present work demonstrates that the earlier-derived understanding is incomplete.
In this work, we demonstrate that surface activation for low-temperature carburization can be more complex than expected from previous work. To this end, we study and compare the composition and structure of alloy surfaces activated by two different processes. The fundamental difference between them is that the established “double HCl” activation sequentially applies steps of surface etching and initial infusion of interstitial solute, whereas our newly developed method, i.e., “solid-reagent pyrolysis” activation, applies initial infusion of solute in tandem with surface etching. In the following, we abbreviate these two categories as AS (in-sequence etching and infusion) and AT (in-tandem etching and infusion). For both activation schemes, we study the resulting alloy surface immediately after activation as well as after following up with IC (continuous infusion of interstitial solute without further surface activation). The specific processes we studied for each category are these:
- AS
Activation by etching and initial solute infusion performed in 3 sequential steps, realized by directly applying specific gases from a gas reservoir: (i) etching with HCl gas, (ii) initial low-temperature carburization with CO gas, and (iii) repeating the first step. This is an established process, currently practiced by industry and known as “double HCl” activation.
- AT
Activation by gas generated by
pyrolysis of a solid reagent, in this case NH
4Cl. Etching and initial solute infusion performed
in tandem in a single step. This is a new kind of activation process, first practiced by Christiansen, Somers et al. [
43,
46] as an “open-vessel” process with urea as solid reagent and recently developed further, including as “closed-vessel” process, with optimized solid reagents by our group [
24,
40,
47].
- IC
Continuous infusion of interstitial solute, carried out as low-temperature carburization, with no further activation.
We present results revealing the respective microstructure and spatial distribution of atom species obtained by high-resolution imaging (FIB (focused ion beam) micromachining, SEM (scanning electron microscopy)), spatially-resolved spectrometry (XEDS (X-ray energy-dispersive spectrometry), ToF-SIMS (time-of-flight secondary-ion mass spectrometry)), and diffractometry (XRD (X-ray diffractometry)). Further, we compare surface hardenability by low-temperature carburization enabled by each activation method, measured by microhardness and effective lattice parameter expansion. Based on these data, we propose models for the microscopic mechanisms of surface activation that will enable future systematic development toward more effective process schemes.
2. Experimental Methods and Procedures
Table 1 shows 6 different kinds of specimens that were prepared and studied in this work. Processing consisted of first exposing—or not exposing—the specimen to one of two different methods of surface activation,
AS and
AT. Each one of these three options was followed—or not followed—by
IC, carried out as low-temperature carburization, with no further surface activation.
Table 1 introduces two-letter codes we use for distinguishing these six different treatments.
AS is currently industrially used as “double-HCl” process [
6,
15,
16,
17,
18,
48]. It involves
direct control of the gas composition by gas supply from a reservoir. The first step, HCl(1), consists of flowing a HCl/N
2 gas mixture around the samples at 523 K (250 °C) for 11 ks (3 h). It is followed by a short low-temperature carburization step, Carb(1), applying a CO/H
2/N
2 mixture at around 723 K (450 °C) for 11 ks (3 h). Then, HCl(1) is repeated as HCl(2). After this activation process, the furnace was purged with N
2 and cooled.
AT constitutes a new approach to surface activation, using gas evolving from
pyrolysis of a solid reagent. The composition of the gas is controlled indirectly via the composition of the solid reagent, the amount of reagent relative to chamber volume and specimen surface area, and the temperature (potentially different for reagent and specimen).
AT can be carried out in an “open vessel” setup, as seen in
Figure 2 and described extensively in [
36]. An analogous “closed-vessel”setup can also be used and produces similar outcomes [
24,
40]. In our study, the solid reagent is NH
4Cl. All experiments were carried out with the same mass of NH
4Cl per alloy specimen surface area: 0.061 mg/mm
2. The vessel containing the specimen and the NH
4Cl was heated to 563 K within 0.1 ks and kept at this temperature for 2.7 ks (45 min). Before exposing the specimen to air, it was allowed to cool for 1 ks.
For
IC, continuous low-temperature carburization after surface activation, we used the same furnace as for
AS. After activation, before sealing and purging the furnace for performing carburization, we extracted “
SX” samples for external testing and/or added “
T” samples, i.e., samples activated via
AT, which had intermittently been exposed to air for storage and transport. Then, the activated samples underwent a longer (roughly 0.11 Ms (30 h) low-temperature carburization step
C. Further, in-depth description of this processing step may be found in [
48].
We studied AISI-316L specimens of two different shapes: “ferrules” and “coupons”. An example of ferrule is shown in
Figure 1. Ferrules were chosen as specimens because they are produced under well-controlled and reproducible conditions. This means that property changes observed after carburization under different conditions can uniquely be attributed to the variation in processing conditions, rather than e.g., be caused by unknown changes in the microstructure between different samples from a less-defined material. For this work, we used as-machined Swagelok
® ferrules with an inner diameter of 6.4 mm (0.25 inch). The AISI-316L from which they are made has a somewhat (1.05 times) higher Cr atom and a significantly (1.2 times) higher Ni-fraction than standard AISI-316L—both within the allowed compositional range of AISI-316L. More precisely, the specification requires mass fractions
and
for Cr and Ni, respectively, while typical values for standard AISI-316L are
and
.
Table 2 shows the composition of the Ni- and Cr-rich AISI-316L by
atom fractions. The AISI-316L raw material was in a strain-hardened state (1/8 hard, as defined in [
49]) prior to machining.
The second kind of specimens where AISI-316L foil coupons with a thickness of 100 μm and lateral dimensions of 14 mm by 59 mm. These samples were used in experiments on low-temperature nitriding via gas from solid-reagent (NH4Cl) pyrolysis.
At the beginning, all samples were cleaned in a series of ultrasonic baths with deionized water, isopropanol, and acetone before drying in an oven at 378 K (105 °C).
Then, the samples were—or were not—surface-activated by either
S or
T, followed—or not followed—by low-temperature carburization
C to produce all the sample variants shown in
Table 2.
Figure 2 depicts the entire process for the example of “Hybrid Processing”,
TC in
Table 1. This includes surface activation
T by gas evolved from pyrolyzing NH
4Cl and subsequent low-temperature carburization, carried out in
separate vessels. Processing surface activation and carburization in different vessels makes the surface activation “ex situ”. For surface activation, the sample and a solid reagent (NH
4Cl or
15N
2H
4Cl) were held within a clean glass test tube, attached to a manifold for inert gas purging and exhausting of reagent pyrolysis products during and after the process. The test tubes and samples were immersed into a bath of molten Sn, thermostatically heated to the desired activation temperature (e.g., 563 K) for the prescribed activation time (e.g., 2.7 ks (45 min)), then removed and cooled. Subsequently, during transit to various analyses of the activated surface or to a gas carburization furnace, the samples were exposed to the atmosphere.
Some alloy samples were activated with reagents enriched with long-term stable isotopes, (e.g., 15N2H4Cl) so the presence of hydrogen (as deuterium) and larger ion fragments could be mapped with ToF-SIMS. After surface activation, samples were prepared as follows. First, we performed FIB milling to reveal the activated sub-surface zone. For this step, we used an FEI Helios dual-beam (scanning electron beam and scanning focused ion beam) system (Thermo Fisher Scientific, Waltham, MA, USA). Prior to milling, layers of Pt were deposited by ion-beam-induced decomposition of organo-metallic precursor molecules on part of the sample to preserve the original surface structure. The sample was fixtured and rotated relative to the incident gallium ion beam to cut into the surface at an angle of against the original surface. In addition to providing excellent accessibility for imaging, such angled cutting doubles the magnification by which the microstructure appears in plan-view images along the direction into the alloy. After FIB milling, samples were imaged in both SE (secondary electron) and BSE (backscattered electrons) mode as well as evaluated with XEDS for mapping and line scans through the activated sub-surface zones.
After preparation, the samples were extracted and transferred for ToF-SIMS analysis in a PHI TRIFT V nano-ToF (PHI, Chanhassen, MN, USA). The instrument operates with a pulsed ion beam, which liberates secondary ion- and neutral fragments (i.e., neutral and charged atoms and atom clusters) from the sample surface. Applying electric charge to the specimen will cause secondary ions of the same polarity to be repelled toward the detector and enable determination of their mass-to-charge ratio from the time of flight. After standard mass and spatial resolution calibration, the samples were evaluated at various magnifications in positive and negative polarity modes. A Ga+ primary ion source in unbunched mode was used. Before the actual analysis, we sputtered the samples remove potential surface contamination from transfer between instruments. Because 1H is an unavoidable contaminant even in ultra-high vacuum, a 2H- (deuterium-) enriched reagent was used for the TX (activation T, no C) samples. Additionally, 15N-enrichment in the reagent was chosen to trace nitrogen-bearing species in the sample.
To acquire hardness–depth profiles resulting from infusion of interstitial solute, we mounted the samples and polished them to a 3 μm diamond suspension finish for microhardness indentation and optical microscopy. Microhardness–depth profiles were acquired by performing series of hardness indents at depths from the surface with constant depth increment Δz. For these measurements, we used a Clemex automated microhardness indentation system (Clemex, Stanford, CA, USA) with a Vickers indenter operating with a load of 0.25 N (25 gf) and a dwell time of th = 10 s. To address scatter, each carburized sample set had three pieces evaluated with two sets of 6 indents placed at each .
XRD was conducted to observe induced changes in atomistic structure, especially lattice parameter expansion from carburization, and detect additional phases potentially forming during activation processes. XRD was performed with a Discover D8 (Bruker, Madison, WI, USA) with a 2D VANTEC-500 solid state detector and a Co Kα source. Measurements were performed between
5° and
95° in 4 frames with 10° overlap between frames and 0.5 ks per frame. In order to display diffractograms independent of the wavelength of the X-ray source, these data were converted to spatial frequencies
, where
denotes the spacing of the {
hkl} planes. Relative shifts between peaks, e.g.,
and
, were evaluated by the cross-correlation function
discretized corresponding to the discretization of the XRD data. Especially, this yielded the shifts of peaks positions after carburization relative to the peak positions of non-treated AISI-316L. Evaluating the positions of the {111}, {200}, and {220} peaks after carburization, we also determined a best estimate for the lattice parameter of the carburized material. This evaluation is subject to two problems. One problem is that the X-rays with the wavelength we employed penetrate the specimen to a depth of typically 5 μm. Therefore, the resulting peak shapes and positions must be understood as a weighted average over depths of different fractions of interstitial solute. Second, small errors in the specimen height are known to cause peak shifts, such that the lattice parameters that can be calculated from each individual peak position appear to be different. This artifact can be eliminated by the “Nelson–Riley” correction, which we have performed by rescaling of the spatial frequency coordinate with the Nelson–Riley function and linear regression analysis [
50]. To identify product phases from the
P activation method, we performed peak matching with database powder diffractograms from the ICDD’s Powder Diffraction File™ (PDF
®).
Lastly, we performed a simple experiment to determine if NH4Cl can successfully infuse interstitial nitrogen without an additional treatment gas supply. The equipment used for this experiment was a Carbolite Gero HZS 12/900 horizontal tube furnace (Carbolite Gero USA Verder Scientific, Newtown, PA, USA) with a 150 mm long fused silica sample vessel inside the work tube. In this experiment, we heated foil samples in an N2-purged tube furnace with 0.25 mg/mm2 NH4Cl to 773 K (500 °C) for 1.8 ks (30 min). After cooling, samples were polished, etched, and imaged to determine whether infusion had occurred. One sample was also analyzed by XRD to determine whether the treatment had produced expanded austenite and/or caused precipitation of nitrides.
3. Results
3.2. Microstructure–Imaging
Toward the goal of understanding what constitutes an “activated” surface at the microscopic level, we performed several complementary methods of microcharacterization: microscopy, elemental mapping, and diffractometry. We analyzed one sample from each group of
SX (activation
S, no
C) and
TX, i.e., samples that were exposed to one of the activation treatments, but no
subsequent carburization (
SX, of course, does involve a brief intermediate step of carburization, Carb(1) in
Table 1).
As a new method in this area of research, we used FIB-cutting into activated sample surfaces to reveal the microstructure directly below the surface with a minimum of disruptions and artefacts that plague more traditional sample preparation methods.
Figure 4A shows a FIB-cut 30° inclined cross-section of the sub-surface zone in a
SX sample. (Recall that owing to the FIB-cut angle
, the depth
z into the specimen is projected to the
y-coordinate with a scaling factor of Sin
. The vertical lines in the region of the alloy core are an artefact of FIB preparation, known as “curtaining”. The specimen surface surrounding the FIB-cut region exhibits soot particles with diameters on the order of 10 μm (
Figure 4A.0) as well as a layer of loosely packed soot particles with diameters up to 3 μm (
Figure 4A.2). The soot layer is covered by the Pt layer deposited for FIB preparation. The Pt layer appears with highest brightness. Between the Pt-covered soot layer and the alloy below (light-grey), the image features a dark-grey zone with a thickness of about 0.5 μm (
Figure 4A.2).
In the following, we denote this zone as RL-S (reaction layer induced by treatment S).
It appears that the RL-S consists of a single phase. The reduced brightness in the BSE image A.1 (compared to the brightness of the alloy core below) suggests that this zone has a lower mass density than the alloy core. Additionally, the interface between the dark-grey zone layer and alloy core exhibits high fractality.
The images show that (even) the double-HCl activation process does not simply “remove” the surface oxide film, but actually produces a continuous reaction layer with a thickness of ≈0.5 μm over the alloy surface.
As to surface activation via gas from solid reagent pyrolysis,
Figure 4B shows SE-(top) and BSE images (bottom) for sample
TX, activated with
15N
2H
4Cl at 563 K for 2.7 ks. Stable imaging of this and similarly activated samples with high resolution required a low accelerating voltage and a low beam current. Initial imaging in a separate region with higher voltage and current caused gross damage to the surface material that appeared as material evaporating and topographic disruption within a few seconds. The low voltage (e.g., 5 kV) and current helped reduce damage, but there was still some (minor) disruption in the region that underwent XEDS mapping owing to long analysis times. The damage shows as [H]
+ depletion in ToF-SIMS maps, see
Section 3.3.2. This interesting effect, paired with some charging observed during SEM imaging, not only guided our analytical technique, but also revealed that the surface layer material is less electrically and thermally conductive than the alloy core. This suggests that the dark-grey zone in B.1 is a distinct
reaction layer not just the topmost sub-surface zone of the alloy with infused interstitials (see below). Compared to the alloy core, the reaction layer has low mass density.
In the following, we denote this layer as RL-T (reaction layer induced by treatment T).
The BSE image
Figure 4B.2 and
Figure 5C, owing to its high magnification, reveals much detail. The image reveals 5 distinguishable grey shades. According to the characteristics of BSE imaging, higher intensity corresponds to higher mass density. The highest mass density is observed on the top surface of the specimen. It originates from the Pt layer that was deposited on the specimen for FIB preparation. In the angled cross-section view, the Pt layer has a thickness of 0.1 μm. The second highest mass density is observed in the alloy core—in the bottom half of the image. This zone of alloy core, denoted as Zone 0, shows a uniform grey level, as expected for a homogeneous solid solution.
Between the alloy core and the Pt layer, image B.1 reveals a complex multi-layer structure in the RL-T. The total thickness of this structured reaction layer is about ≈5 μm, accounting for the cut angle . The inner architecture of the RL-T consists of three distinguishable zones:
Zone 1, the lowermost zone, right above the alloy core, exhibits two phases, distinguishable by the grey level they exhibit in the image: A low-density (dark-grey) phase and a mid-density (mid-grey) phase, denser than the former one, but less dense than the alloy core. Both phases exhibit a highly fractal morphology. The volume fraction of the mid-density phase is somewhat larger than that of the low-density phase. From the interface to the alloy core to the top of Zone 1, the volume fractions do not change significantly. The mid-density phase appears more globular, while the low-density phase occupies the space between the globular particles of the mid-density phase. The partitioning of the volume into regions of mid-density and low-density is fine-scaled immediate to the layer–alloy interface, but becomes coarser with increasing height above the interface. The layer–alloy interface appears to be highly fractal and rough, indicating etch attack of the alloy. Along the interface, both the mid-density phase and the low-density phase are in contact with the alloy.
In Zone 2, on top of the two-phase Zone 1, the SEM images show a uniform layer of the mid-density (mid grey) phase. Over a longer section of the (virtual) interface between Zone 2 and Zone 3 exhibits a crack, suggesting the relaxation of internal stress, possibly during cooling. Accounting for the cut angle , the thickness of Zone 2 is ≈1.5 μm.
Zone 3 consists of the same mid-density phase as Zone 2, but here it only fills the volume to about one half. The other half of the volume consists of voids with a typical lateral extension of ≈1 μm. The voids tend to be elongated parallel to the surface, even more so than it appears because of the magnification anisotropy introduced by the cut-angle .
In all three zones, the low-density phase and the mid-density phase appear with constant grey level in the BSE image. This suggests that their composition does not significantly vary as a function of distance z from the surface.
To evaluate the temporal evolution of the RL-T at the activation temperature, additional samples similar to
TX were prepared at 563 K for 0.3 and 0.9 ks. All samples seen in
Figure 5 show the same multi-zone structure described above. The total layer thickness increases
linearly with time: 2.7 ks: ≈ 5.5 μm, 0.9 ks: ≈ 2 μm, and 0.3 ks: ≈ 1 μm. In the 0.3 ks sample, Zone 3 is significantly less porous than the same region of the samples activated for longer times. Additionally, the porous zone is bordered by the mid-density phase and a network of smaller pores within the low-density (dark-grey) phase.
3.4. Microstructure—Diffractometry
Aiming to understand the phases formed during reagent-pyrolysis- and double-HCl activation processes, we performed XRD.
Figure 12 shows the diffractogram of sample
TX. The main peaks originate from the alloy core, ammonium–iron chlorides, chromium ammine chloride, chromium chloride, residual NH
4Cl, and SiO
2. According to peak matching with library data, the latter likely originates from fiber insulation used while processing this sample. The NH
4Cl is present because the NH
4Cl was still undergoing pyrolysis at the end of the 2.7 ks isotherm. Apparently, the remaining NH
3 (ammonia) and HCl gases condensed back onto the sample surface during furnace cooling, decorating it with small crystallites. The peaks from ammonium iron chlorides, chromium ammine chloride, and chromium chloride species corroborate the results from SEM, ToF-SIMS, and XEDS. FeCl
3 is
not among the apparent species. Thermodynamically, it could form from Fe and HCl, but it appears that the ammonium iron chlorides are the favored product, owing to the presence of NH
3 and HCl.
Figure 13 shows the diffractograms for samples
XX (no activation, nor carburization),
SX (activation
S, no
C),
SC (activation
S, then carburization
C), and
TC (activaton
T, then carburization
C). Compared to
XX, sample
SX shows the austenite peaks shifted toward lower spatial frequencies. This indicates a lattice parameter expansion caused by carbon infusion during the short carburization Carb(1) between the HCl gas steps HCl(1) and HCl(2). Furthermore, the expanded-austenite peaks appear distinct from the still-present original austenite peaks in the diffractogram, rather than a single asymmetric shifted peak that is often observed after full low-temperature carburization. This indicates that the XRD beam substantially probed into the deeper, non-expanded austenite of the alloy core. Additionally, non-austenite peaks are observed at low spatial frequencies. They originate from oxides and hydrated/oxidized chlorides of the matrix metals. During the experiments, we observed carbon soot on the alloy surface, which may explain some of the peaks observed at small spatial frequencies.
To explore how these samples with different surface activation respond to subsequent low-temperature carburization, compare the diffractograms of samples SC and TC. The diffractogram of sample TC exhibits significantly shifted austenite peaks, indicative of considerably high carbon level and a mean depth of carburization comparable to the penetration depth of the Co-Kα X-rays (≈5 μm). The diffractogram of sample SC also exhibits these peak shifts, but to a lesser extent. The diffractograms of both samples feature the usual asymmetric and shifted austenite peaks without distinct peaks for the original austenite like those seen in the diffractogram of sample SX.
As mentioned, the peak shifts indicate an expansion of the spacings between metal atoms. The
effective lattice parameter observed in X-ray diffractograms positively correlates with the carbon penetration [
40]
However, the exact relationship between peak shift, peak shape, and the carbon-fraction–depth profile is complicated. Part of the complication is that absorption of the X-ray beam, exponentially dependent on path length, but also on density, thus carbon fraction XC, leads to reduced X-ray beam intensity in greater depths. Moreover, the intensity as a function of depth depends on the diffraction angle , as this determines the relationship between depth and path length. What appears as a peak in X-ray diffractograms can be understood as weighed superposition (integral) of peaks originating from different depths z with correspondingly different carbon fractions and weighing factors that correspond on the diffraction angle .
Table 3 shows XRD data for the tested samples. Using the data from specimen
XX (non-treated) as a baseline, the peak shifts for the treated samples were determined from the location
of the maximum of the cross-correlation function (1). Effective lattice parameters were calculated for each peak
and an overall effective lattice parameter
determined by the Nelson–Riley correction, i.e., extrapolating the linear regression line of the lattice parameters
versus the Nelson–Riley function
to obtain
. In each sample, the effective lattice parameter
is found increased versus that of the alloy core, owing to carbon infusion. Sample
SX (activation
S, no
C), as it had only a brief carburization, exhibits only a 1.1% expansion of the effective lattice parameter. The diffractograms of sample
SC (activation
S, then carburization
C) exhibits a larger effective lattice parameter expansion of 2.6%. Sample
TC (activaton
T, then carburization
C) shows the largest peak shift of all, indicating a 3.1% expansion of the effective lattice parameter. Although sample
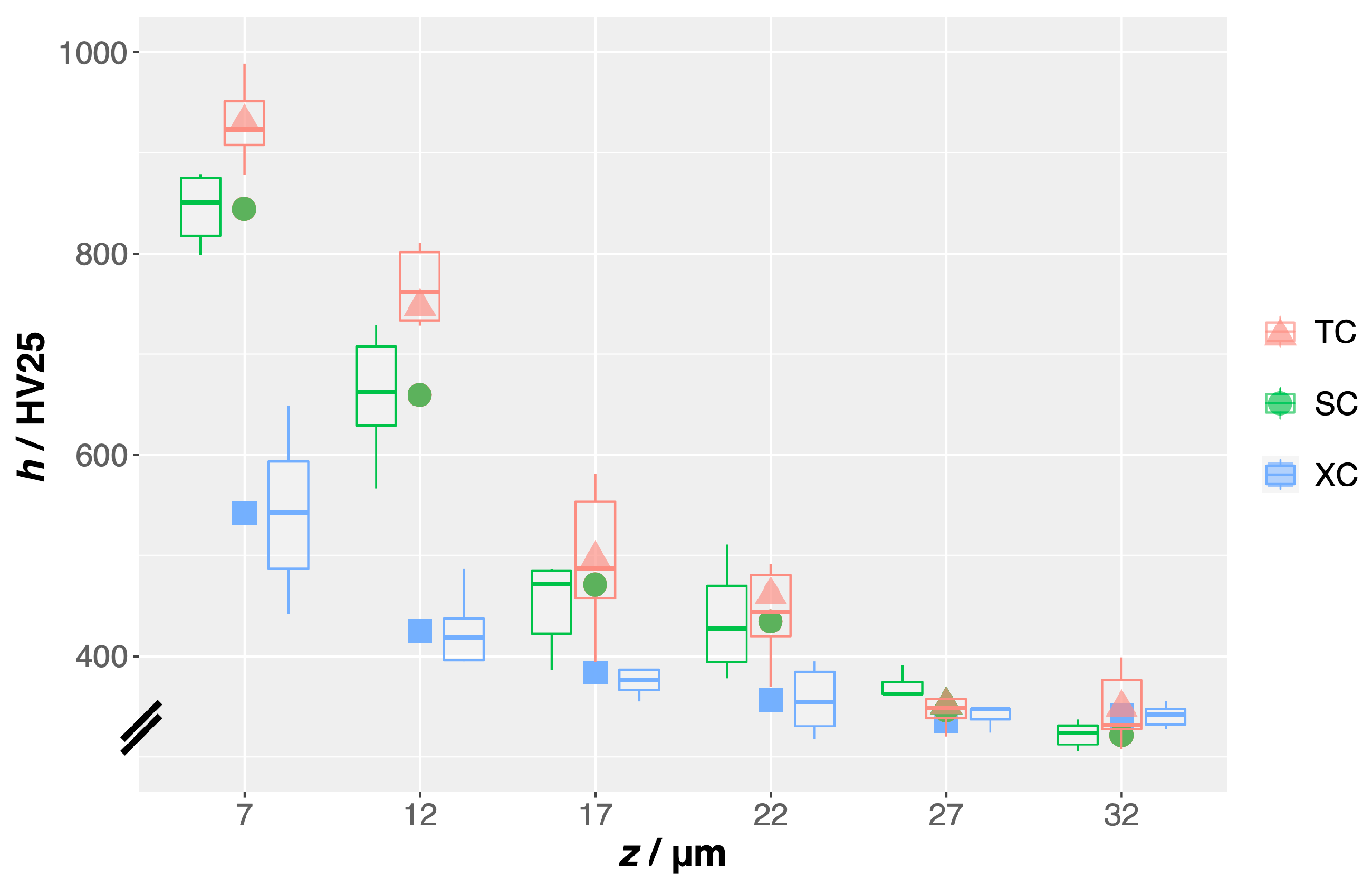
TC only had the second-long-carburization step, it appears to have absorbed more carbon than the double-HCl-activated-and-carburized sample with its longer total exposure to low-temperature carburization.
The expansion of the effective lattice parameter measured by XRD correlates with the observed increase in microhardness. Of all tested samples, the NH4Cl-activated-and-carburized sample TC exhibits the most pronounced peak shifts and effective lattice parameter expansion as well as the highest surface hardness.
4. Discussion
Our data show that the native oxide on alloys such as AISI-316L constitutes a diffusion barrier to carbon and nitrogen. This is evident from the inconsistency and the overall low level of hardening exhibited by samples XC, carburized without prior surface activation. Therefore, surface engineering of such alloys by infusion of interstitial solute (carbon or nitrogen) requires surface activation, meaning removal of the oxide film or making it permeable to carbon and nitrogen.
For the first time, we have observed that surface activation can involve considerable microscopic complexity in addition to removing the oxide film (or perforating it or making it permeable). Our results of heat treatments and surface analysis reveal that an alloy surface that is “activated” for infusion of carbon (or nitrogen) can differ much from a “bare” alloy surface. Instead of the latter, we find that an activated alloy surface can be covered by a non-metallic substance with a complex microstructure and phase composition. We could neither directly observe the initial oxide film on the as-received alloy, nor its removal or modification toward increased permeability. However, since the activation methods we applied enable effective infusion of interstitial solute, carbon in this work, we conclude that the passivating oxide film that initially covers the alloy surface is removed, perforated, or, at least, made permeable to carbon (and nitrogen) as non-metallic reaction layer (RL-S or RL-T) forms on the surface.
4.2. AT–Activation by Gas from Solid-Reagent Pyrolysis—Model
For activation by gas from pyrolysis of NH
4Cl, our results suggest a different microscopic mechanism, illustrated in
Figure 15: The gas that results from NH
4Cl reacts with the alloy surface. The reaction products build up the RL-T, a multi-phase product layer with a complex architecture and microstructure, predominantly consisting of the observed metal chlorides and ammonium (
Figure 4,
Figure 5,
Figure 6,
Figure 7,
Figure 8,
Figure 9,
Figure 10 and
Figure 11). NH
4Cl (and
15N
2H
4Cl), in particular, pyrolyze to give off NH
3 and HCl [
36]. Both are known to attack Cr-rich oxide. We conclude that these molecules perforate/remove the passivating oxide film that initially covers the alloy surface.
The hardness–depth profiles
in
Figure 3 and XRD results in
Table 3 obtained after low-temperature carburization
quantitatively show that surface activation via gas from solid-reagent pyrolysis is somewhat more effective than what can currently be accomplished by “double-HCl” activation. Moreover, the RL-T generated by activation via gas from solid-reagent pyrolysis can even
keep the surface activated after air exposure to the extent that specimens can be transferred to another furnace through air and be carburized or nitrided at low temperature without further surface activation. This is similar to the “provisional” passivation we observed earlier in the context of liquid-HCl activation [
34].
The temporal evolution of the RL-T can be understood by considering the microstructure of samples activated for different times. If the layer growth were limited by diffusion, the layer thickness
w should be of the form
The data on the temporal evolution of the RL-T presented in Results
Section 3.2, in contrast, more suggest a linear form,
where
κ = (2.5 ± 0.8) nm/s. Generally, a linear growth law suggests that the growth rate is not limited by diffusion, but, instead, by the reaction rate at an interface. The interface in question is the fractal interface between Zone 0 (alloy core) and Zone-1 (RL-T). This means that the RL-T with a thickness in the range we observe here does
not passivate the alloy surface. The linear growth law suggests that the transport rate of nitrogen (ammine) and chlorine species to the RL-T–alloy interface is faster than the layer can form at this interface.
The different degrees of surface voids and porosity at different stages provide further information about the temporal evolution of the layer. For example, the
TX sample activated for only 0.3 ks shows minor porosity at the top surface (Zone 3), while the two samples activated for longer times show more distinct and coarse voids (
Figure 5). This difference can be understood as progressing void formation in situ, in a pre-existing layer. The voids observed after longer activation, e.g., in
Figure 4B.2, may have grown as Cr in the dark-grey-looking regions reacted with HCl (g) and formed CrCl
3 (g). The interface between the voids and the two-phase region often end in a network of smaller pores within the dark-grey-looking phase. A reaction between these Cr-rich fields and HCl (g) would explain why the dark-grey-looking phase only volatilizes at sites reachable by gaseous species instead of volatilizing in all areas.
Alternatively, the voids can be understood as gaps between outward-growing “pillars” of layer material. This would require significant diffusion of metal atoms to supply the growths. Such diffusion would be unlikely in a bulk alloy, but BSE images show that the reaction layer has a considerably lower mass density than the alloy. Additionally, it is observed that the two-phase region has a coarser microstructure nearer to the surface and this coarsening (with segregation of Cr from Fe and Ni) suggests significant metal-atom diffusion. The low density of the RL-T may also explain why the reaction can continue and the reaction layer–alloy interface continues to propagate into the alloy: The low density of the layer allows for rapid diffusion of the relevant reaction species (e.g., NH3 and HCl) to migrate from the gas phase to the layer–alloy interface, where the reaction proceeds.
The findings of the present work on reactions between NH
4Cl and AISI-316L agree with the results of comparable studies in the literature. Meyer et al. discusses the use of NH
4Cl pyrolysis to produce more usable products from various rare-earth sesquioxides [
51]. They discuss that Y
2O
3 would not normally react with HCl gas in this temperature regime (575–675 K), but with NH
4Cl producing both HCl and NH
3, the oxide reacts to form a metal ammonium chloride with water as a byproduct. This “ammonium chloride route” requires both an acid–base reaction and the formation of a complex. Meyer et al. go on to suggest that H+, rather than HCl, acts as the acidic species. Other researchers have produced corroborating results of Fe- and Cr- alloy and oxide systems forming these metal-ammine- and metal-ammonium-chloride complexes in this temperature range [
52,
53,
54].




















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}