
Identification of Eastern United States Reticulitermes Termite Species via PCR-RFLP, Assessed Using Training and Test Data



Abstract
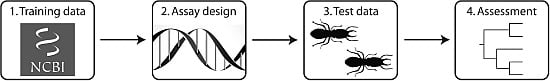
:
1. Introduction
2. Materials and Methods
2.1. Database Mining and PCR-RFLP Assay Design
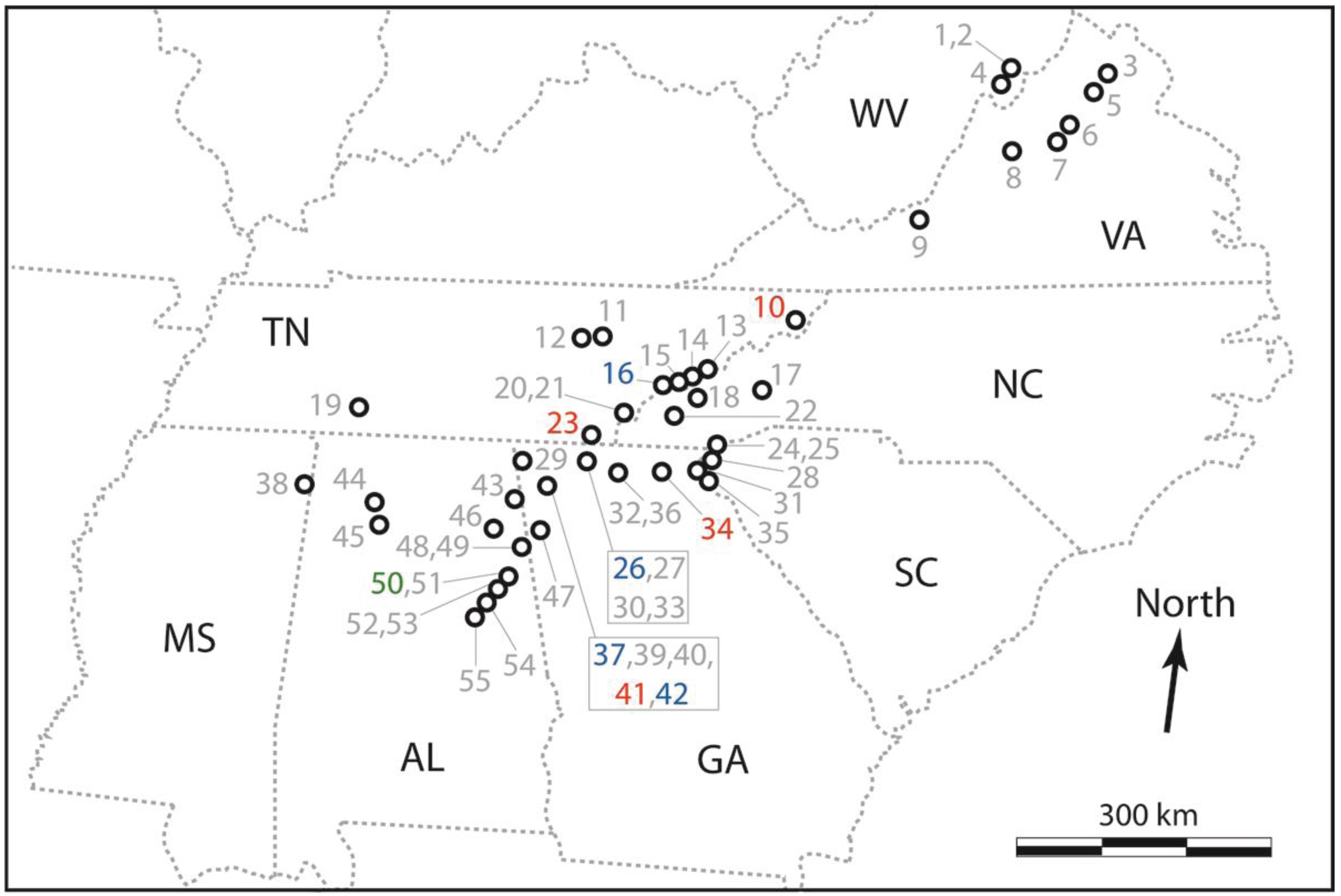
2.2. Empirical Application of the PCR-RFLP Assay to Novel Samples
2.3. Evaluation of Performance and Refinement of the Interpretive Framework
3. Results
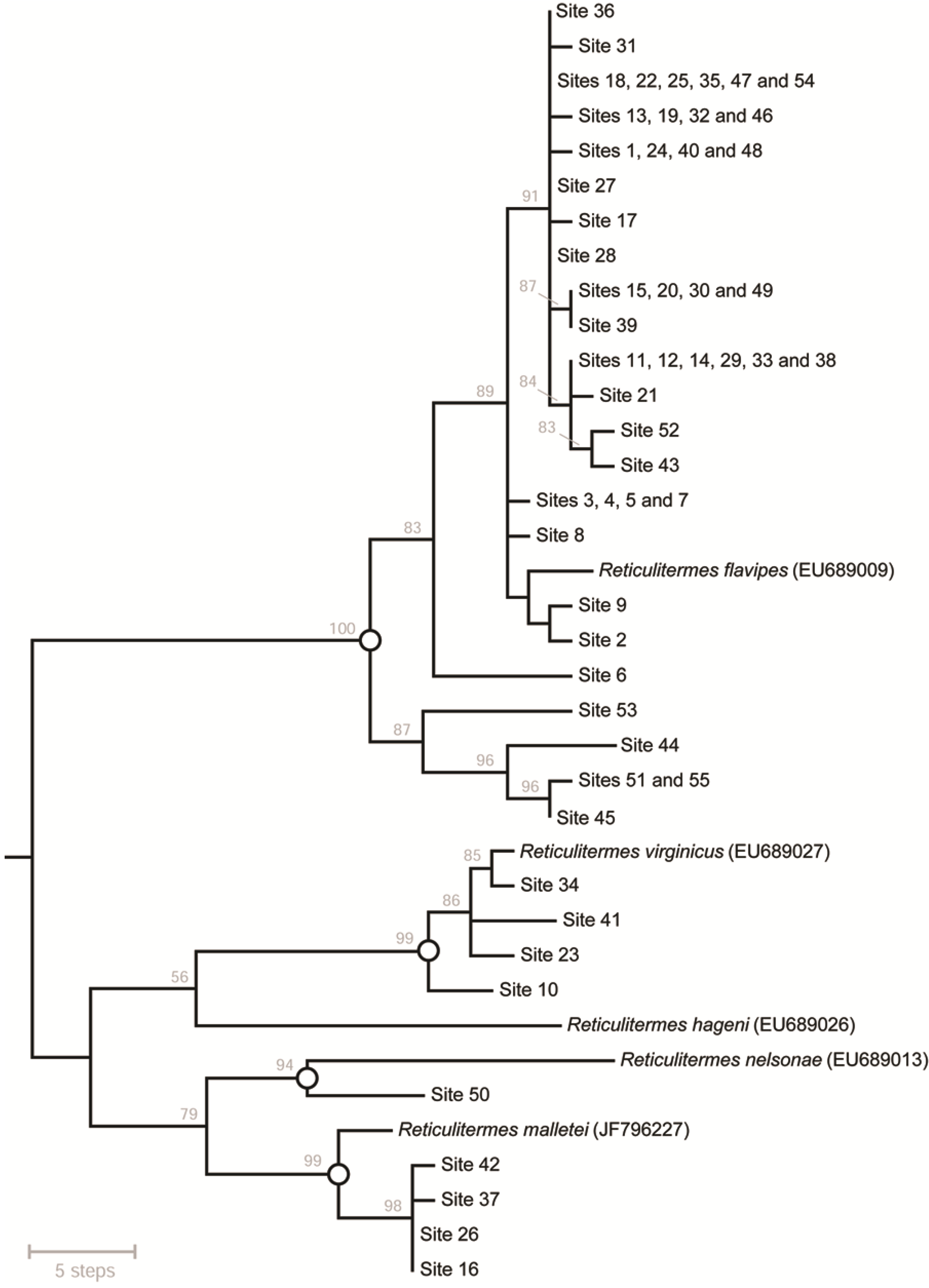
3.1. Database Mining and PCR-RFLP Assay Design
3.2. Empirical Application of the PCR-RFLP Assay to Novel Samples
3.3. Evaluation of Performance and Refinement of the Interpretive Framework

{kind=link}
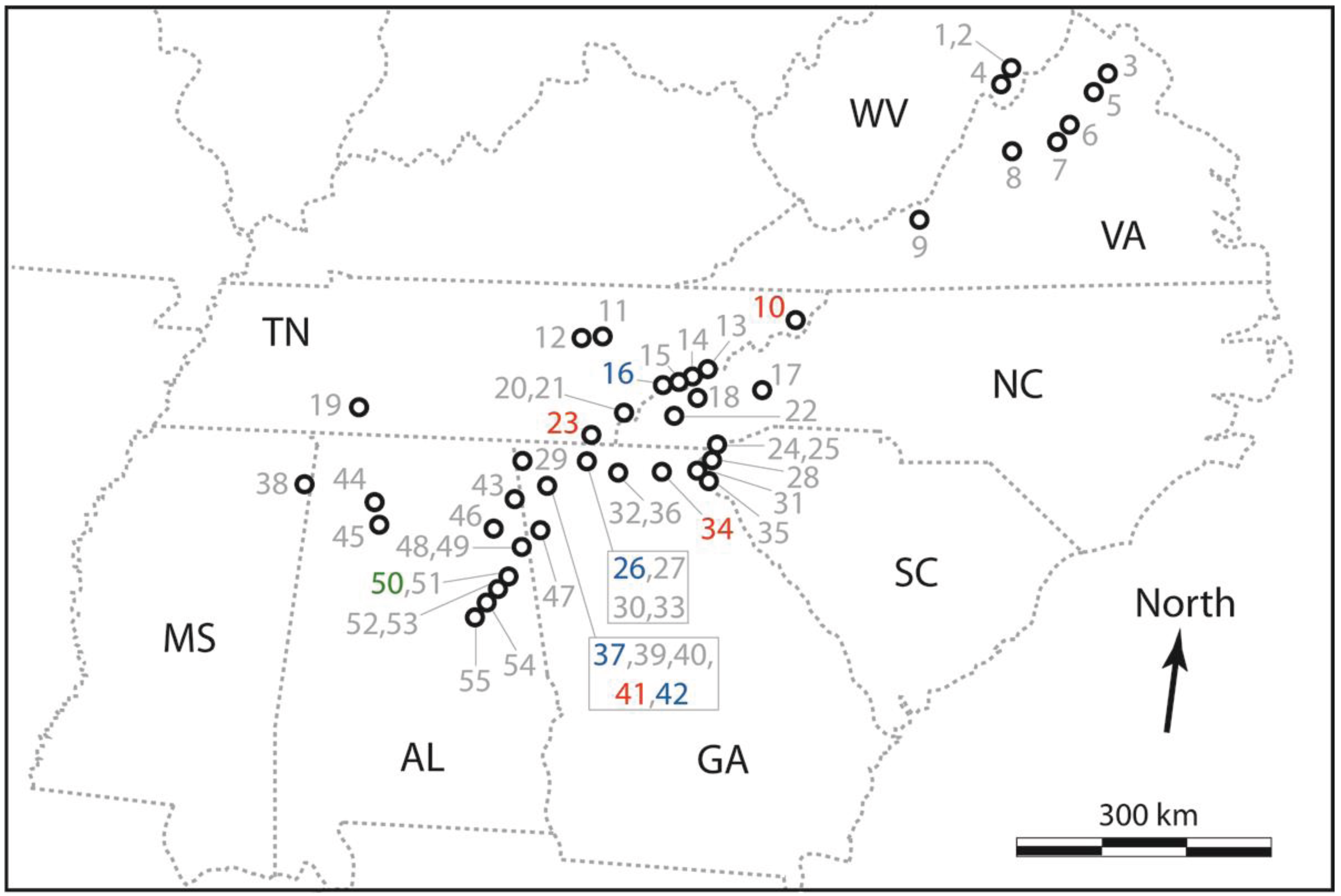{kind=link}
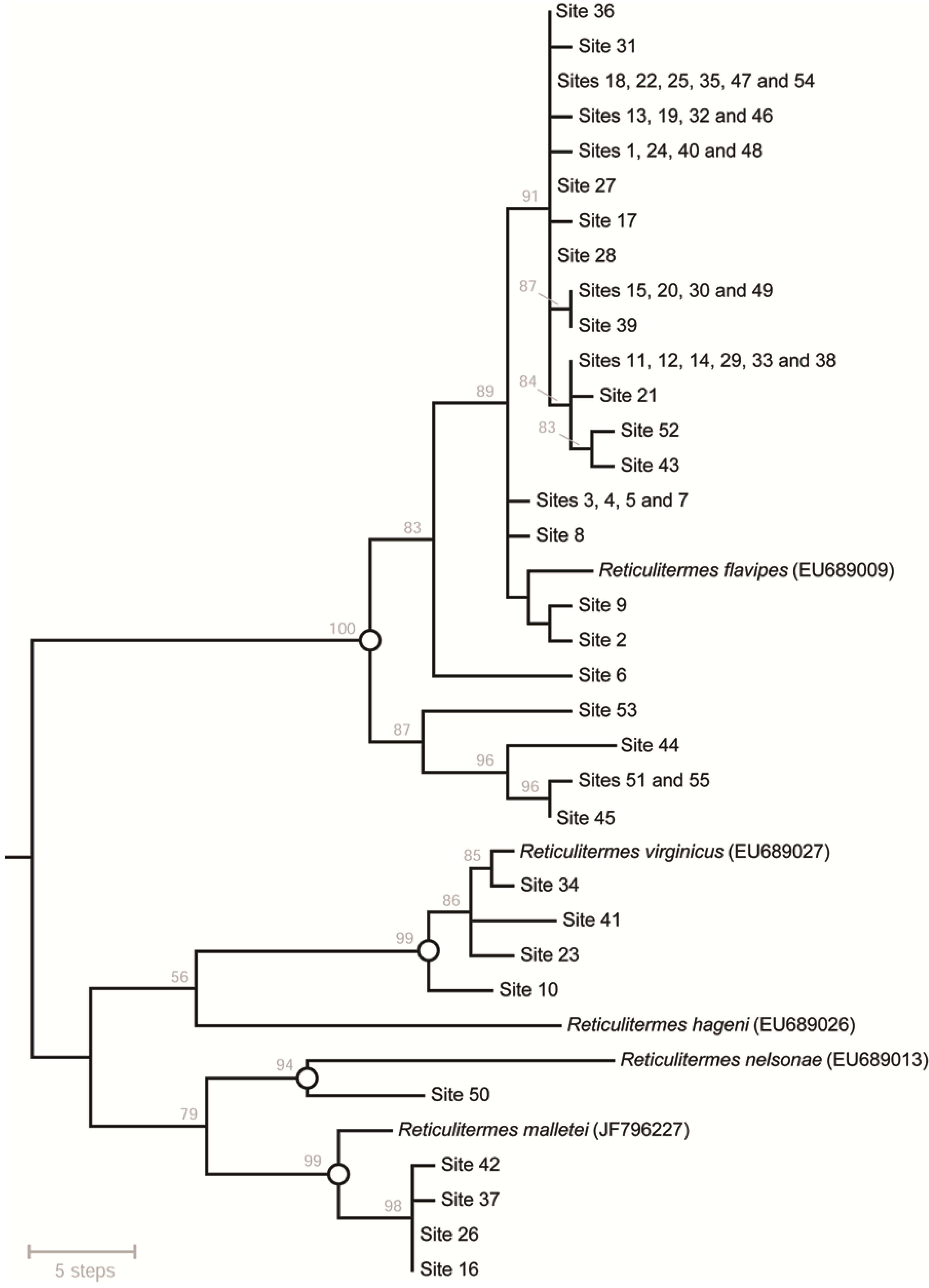{kind=link}
| Restriction Enzyme | Fragment | Species | ||||
|---|---|---|---|---|---|---|
| Sizes (bp) | R. flavipes | R. hageni | R. malletei | R. nelsonae | R. virginicus | |
| Rsa I | 175, 201 | ✓ | 〤 | 〤 | ✓ | ✓ |
| 48, 127, 201 | 〤 | 〤 | ✓ | ✓ | ✓ | |
| 86, 115, 175 | ✓ | ✓ | 〤 | 〤 | 〤 | |
| Taq I | 376 | 〤 | ✓ | ✓ | ✓ | ✓ |
| 153, 223 | 〤 | 〤 | 〤 | ✓ | 〤 | |
| 183, 193 | ✓ | 〤 | 〤 | 〤 | 〤 | |
| 67, 126, 183 | ✓ | 〤 | 〤 | 〤 | 〤 | |
| 30, 67, 126, 153 | ✓ | 〤 | 〤 | 〤 | 〤 | |
| Msp I | 376 | ✓ | 〤 | 〤 | ✓ | ✓ |
| 37, 339 * | ✓ | 〤 | ✓ | 〤 | 〤 | |
| 77, 299 | 〤 | ✓ | 〤 | ✓ | 〤 | |
| 37, 40, 299 | 〤 | 〤 | ✓ | 〤 | 〤 | |
4. Discussion
5. Conclusions
Supplementary Files
Supplementary File 1Acknowledgments
Author Contributions
Conflicts of Interest
References
- Ulyshen, M.D. Interacting effects of insects and flooding on wood decomposition. PLoS ONE 2014, 9, e101867. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.Y.; Forschler, B.T. Reticulitermes nelsonae, a new species of subterranean termite (Rhinotermitidae) from the southeastern United States. Insects 2012, 3, 62–90. [Google Scholar] [CrossRef]
- Szalanski, A.L.; Austin, J.W.; Owens, C.B. Identification of Reticulitermes spp. (Isoptera: Reticulitermatidae) from south central United States by PCR-RFLP. J. Econ. Entomol. 2003, 96, 1514–1519. [Google Scholar] [CrossRef] [PubMed]
- Vargo, E.L. Hierarchical analysis of colony and population genetic structure of the eastern subterranean termite, Reticulitermes flavipes, using two classes of molecular markers. Evolution 2003, 57, 2805–2818. [Google Scholar] [CrossRef] [PubMed]
- Vargo, E.L.; Husseneder, C. Biology of subterranean termites: Insights from molecular studies of Reticulitermes and Coptotermes. Annu. Rev. Entomol. 2009, 54, 379–403. [Google Scholar] [CrossRef] [PubMed]
- Hogg, I.D.; Hebert, P.D.N. Biological identification of springtails (Hexapoda: Collembola) from the Canadian Arctic, using mitochondrial DNA barcodes. Can. J. Zool. 2004, 82, 749–754. [Google Scholar] [CrossRef]
- Hajibabaei, M.; Janzen, D.H.; Burns, J.M.; Hallwachs, W.; Hebert, P.D.N. DNA barcodes distinguish species of tropical Lepidoptera. Proc. Natl. Acad. Sci. USA 2006, 103, 968–971. [Google Scholar] [CrossRef] [PubMed]
- Sheffield, C.S.; Hebert, P.D.; Kevan, P.G.; Packer, L. DNA barcoding a regional bee (Hymenoptera: Apoidea) fauna and its potential for ecological studies. Mol. Ecol. Resour. 2009, 9, 196–207. [Google Scholar] [CrossRef] [PubMed]
- Avise, J.C.; Arnold, J.; Ball, R.M.; Bermingham, E.; Lamb, T.; Neigel, J.E.; Reeb, C.A.; Saunders, N.C. Intraspecific phylogeography: The mitochondrial DNA bridge between population genetics and systematics. Annu. Rev. Ecol. Syst. 1987, 18, 489–522. [Google Scholar] [CrossRef]
- Folmer, O.; Black, M.; Hoeh, W.; Lutz, R.; Vrijenhoek, R. DNA primers for amplification of mitochondrial cytochrome c oxidase subunit I from diverse metazoan invertebrates. Mol. Mar. Biol. Biotechnol. 1994, 3, 294–299. [Google Scholar] [PubMed]
- Simon, C.; Frati, F.; Beckenbach, A.; Crespi, B.; Liu, H.; Flook, P. Evolution, weighting, and phylogenetic utility of mitochondrial gene sequences and a compilation of conserved polymerase chain reaction primers. Ann. Entomol. Soc. Am. 1994, 87, 651–701. [Google Scholar] [CrossRef]
- Baker, C.S.; Cipriano, F.; Palumbi, S.R. Molecular genetic identification of whale and dolphin products from commercial markets in Korea and Japan. Mol. Ecol. 1996, 5, 671–685. [Google Scholar] [CrossRef]
- Hebert, P.D.N.; Cywinska, A.; Ball, S.L.; deWaard, J.R. Biological identifications through DNA barcodes. Proc. R. Soc. Lond. B Biol. Sci. 2003, 270, 313–321. [Google Scholar] [CrossRef] [PubMed]
- Jarman, S.N.; Deagle, B.E.; Gales, N.J. Group-specific polymerase chain reaction for DNA-based analysis of species diversity and identity in dietary samples. Mol. Ecol. 2004, 13, 1313–1322. [Google Scholar] [CrossRef] [PubMed]
- Garrick, R.C.; Meadows, C.A.; Nicolas, A.N.; Nason, J.D.; Dyer, R.J. A set of polymorphic nuclear intron markers for conservation genetics and phylogeography of Euphorbia species (Pedilanthus clade). Conserv. Genet. 2008, 9, 1673–1676. [Google Scholar] [CrossRef]
- Garrick, R.C.; Rowell, D.M.; Simmons, C.S.; Hillis, D.M.; Sunnucks, P. Fine-scale phylogeographic congruence despite demographic incongruence in two low-mobility saproxylic springtails. Evolution 2008, 62, 1103–1118. [Google Scholar] [CrossRef] [PubMed]
- Garrick, R.C.; Meadows, C.A.; Nason, J.D.; Cognato, A.I.; Dyer, R.J. Variable nuclear markers for a Sonoran Desert bark beetle, Araptus attenuatus Wood (Curculionidae: Scolytinae), with applications to related genera. Conserv. Genet. 2009, 10, 1177–1179. [Google Scholar] [CrossRef]
- Garrick, R.C.; Nason, J.D.; Fernández-Manjarrés, J.F.; Dyer, R.J. Ecological coassociations influence species’ responses to past climatic change: An example from a Sonoran Desert bark beetle. Mol. Ecol. 2013, 22, 3345–3361. [Google Scholar] [CrossRef] [PubMed]
- Lancaster, M.L.; Gemmell, N.J.; Negro, S.; Goldsworthy, S.; Sunnucks, P. Ménage à trois on Macquarie Island: Hybridization among three species of fur seal (Arctocephalus spp.) following historical population extinction. Mol. Ecol. 2006, 15, 3681–3692. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Zhou, X.; Li, S.; Schwinghammer, M.; Scharf, M.E.; Buczkowski, G.; Bennett, G.W. Survey and identification of termites (Isoptera: Rhinotermitidae) in Indiana. Ann. Entomol. Soc. Am. 2009, 102, 1029–1036. [Google Scholar] [CrossRef]
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. MEGA6: Molecular Evolutionary Genetics Analysis version 6.0. Mol. Biol. Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef] [PubMed]
- Vincze, T.; Posfai, J.; Roberts, R.J. NEBcutter: A program to cleave DNA with restriction enzymes. Nucleic Acids Res. 2003, 31, 3688–3691. [Google Scholar] [CrossRef] [PubMed]
- Park, Y.C.; Maekawa, K.; Matsumoto, T.; Santoni, R.; Choe, J.C. Molecular phylogeny and biogeography of the Korean woodroaches Cryptocercus spp. Mol. Phylogenetics Evol. 2004, 30, 450–464. [Google Scholar] [CrossRef]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef] [PubMed]
- Sunnucks, P.; Hales, D.F. Numerous transposed sequences of mitochondrial cytochrome oxidase I–II in aphids of the genus Sitobion (Hemiptera: Aphididae). Mol. Biol. Evol. 1996, 13, 510–524. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Darriba, D.; Taboada, G.L.; Doallo, R.; Posada, D. jModelTest 2: More models, new heuristics and parallel computing. Nat. Methods 2012, 9, 772. [Google Scholar] [CrossRef] [PubMed]
- Hillis, D.M.; Bull, J.J. An empirical test of bootstrapping as a method for assessing confidence in phylogenetic analysis. Syst. Biol. 1993, 42, 182–192. [Google Scholar] [CrossRef]
- Germain, J.-F.; Chatot, C.; Meusnier, I.; Artige, E.; Rasplus, J.-Y.; Cruaud, A. Molecular identification of Epitrix potato flea beetles (Coleoptera: Chrysomelidae) in Europe and North America. Bull. Entomol. Res. 2013, 103, 354–362. [Google Scholar] [CrossRef] [PubMed]
- Ovalle, T.M.; Parsa, S.; Hernández, M.P.; Becerra Lopez-Lavalle, L.A. Reliable molecular identification of nine tropical whitefly species. Ecol. Evol. 2014, 4, 3778–3787. [Google Scholar] [CrossRef] [PubMed]
- Vesterlund, S.-R.; Sorvari, J.; Vasemägi, A. Molecular identification of cryptic bumblebee species from degraded samples using PCR-RFLP approach. Mol. Ecol. Resour. 2014, 14, 122–126. [Google Scholar] [CrossRef] [PubMed]
- Arimoto, M.; Iwaizumi, R. Identification of Japanese Lymantria species (Lepidoptera: Lymantriidae) based on PCR-RFLP analysis of mitochondrial DNA. Appl. Entomol. Zool. 2014, 49, 159–169. [Google Scholar] [CrossRef]
- Cameron, S.L.; Whiting, M.F. Mitochondrial genomic comparisons of the subterranean termites from the Genus Reticulitermes (Insecta: Isoptera: Rhinotermitidae). Genome 2007, 50, 188–202. [Google Scholar] [PubMed]
- Luchetti, A.; Mantovani, B.; Marini, M. Mitochondrial evolutionary rate and speciation in termites: Data on European Reticulitermes taxa (Isoptera: Rhinotermitidae). Insectes Soc. 2005, 52, 218–221. [Google Scholar] [CrossRef]
- Brower, A.V.Z. Rapid morphological radiation and convergence among races of the butterfly Heliconius erato inferred from patterns of mitochondrial DNA evolution. Proc. Natl. Acad. Sci. USA 1994, 91, 6491–6495. [Google Scholar] [CrossRef] [PubMed]
- Garrick, R.C.; Caccone, A.; Sunnucks, P. Inference of population history by coupling exploratory and model-driven phylogeographic analyses. Int. J. Mol. Sci. 2010, 11, 1190–1227. [Google Scholar] [CrossRef] [PubMed]
- Garrick, R.C.; Bonatelli, I.A.; Hyseni, C.; Morales, A.; Pelletier, T.A.; Perez, M.F.; Rice, E.; Satler, J.D.; Symula, R.E.; Thomé, M.T.; et al. The evolution of phylogeographic data sets. Mol. Ecol. 2015, 24, 1164–1171. [Google Scholar] [CrossRef] [PubMed]
- Sunnucks, P.; Blacket, M.J.; Taylor, J.M.; Sands, C.J.; Ciavaglia, S.A.; Garrick, R.C.; Tait, N.N.; Rowell, D.M.; Pavlova, A. A tale of two flatties: Different responses of two terrestrial flatworms to past environmental climatic fluctuations at Tallaganda in montane southeastern Australia. Mol. Ecol. 2006, 15, 4513–4531. [Google Scholar] [CrossRef] [PubMed]
- Garrick, R.C.; Rowell, D.M.; Sunnucks, P. Phylogeography of saproxylic and forest floor invertebrates from Tallaganda, south-eastern Australia. Insects 2012, 3, 270–294. [Google Scholar] [CrossRef]
- Bull, J.K.; Sands, C.J.; Garrick, R.C.; Gardner, M.G.; Tait, N.N.; Briscoe, D.A.; Rowell, D.M.; Sunnucks, P. Environmental complexity and biodiversity: The multi-layered evolutionary history of a log-dwelling velvet worm in montane temperate Australia. PLoS ONE 2013, 8, e84559. [Google Scholar] [CrossRef] [PubMed]
- Garrick, R.C. Montane refuges and topographic complexity generate and maintain invertebrate biodiversity: Recurring themes across space and time. J. Insect Conserv. 2011, 15, 469–478. [Google Scholar] [CrossRef]
- Szalanski, A.L.; Austin, J.W.; McKern, J.A.; Scheffrahn, R.H.; Owens, C.B.; Messenger, M.T. Molecular phylogeography of Reticulitermes (Isoptera: Rhinotermitidae) termites from Florida. Sociobiology 2008, 52, 619–632. [Google Scholar]
- Perdereau, E.; Bagnères, A.-G.; Bankhead-Dronnet, S.; Dupont, S.; Zimmermann, M.; Vargo, E.L.; Dedeine, F. Global genetic analysis reveals the putative native source of the invasive termite, Reticulitermes flavipes, in France. Mol. Ecol. 2013, 22, 1105–1119. [Google Scholar] [CrossRef] [PubMed]
- Elith, J.; Leathwick, J.R. Species distribution models: Ecological explanation and prediction across space and time. Annu. Rev. Ecol. Evol. Syst. 2009, 40, 677–697. [Google Scholar] [CrossRef]
- Edwards, D.L.; Garrick, R.C.; Tapia, W.; Caccone, A. Cryptic structure and niche divergence within threatened Galápagos giant tortoises from southern Isabela Island. Conserv. Genet. 2014, 15, 1357–1369. [Google Scholar] [CrossRef]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Garrick, R.C.; Collins, B.D.; Yi, R.N.; Dyer, R.J.; Hyseni, C. Identification of Eastern United States Reticulitermes Termite Species via PCR-RFLP, Assessed Using Training and Test Data. Insects 2015, 6, 524-537. https://doi.org/10.3390/insects6020524
Garrick RC, Collins BD, Yi RN, Dyer RJ, Hyseni C. Identification of Eastern United States Reticulitermes Termite Species via PCR-RFLP, Assessed Using Training and Test Data. Insects. 2015; 6(2):524-537. https://doi.org/10.3390/insects6020524
Chicago/Turabian StyleGarrick, Ryan C., Benjamin D. Collins, Rachel N. Yi, Rodney J. Dyer, and Chaz Hyseni. 2015. "Identification of Eastern United States Reticulitermes Termite Species via PCR-RFLP, Assessed Using Training and Test Data" Insects 6, no. 2: 524-537. https://doi.org/10.3390/insects6020524
APA StyleGarrick, R. C., Collins, B. D., Yi, R. N., Dyer, R. J., & Hyseni, C. (2015). Identification of Eastern United States Reticulitermes Termite Species via PCR-RFLP, Assessed Using Training and Test Data. Insects, 6(2), 524-537. https://doi.org/10.3390/insects6020524