
Laboratory Test Indirectly Reveals the Unreliability of RNA-Dependent 16S rRNA Amplicon Sequences in Detecting the Gut Bacterial Diversity of Delia antiqua

 and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Insect Rearing and Microbial Culture Media
2.2. Larval Gut Sample Collection
2.3. DNA- and RNA-Dependent 16S rRNA Amplicon Sequence
DNA/RNA Extraction and Amplicon Sequencing
2.4. Isolation and Identification of D. antiqua Gut Bacteria
2.5. SynComs Constructed According to DNA/RNA-Dependent Amplicon Sequencing
2.6. Effects of SynComs on Larval Survival and Growth
2.7. Data Analysis
3. Results
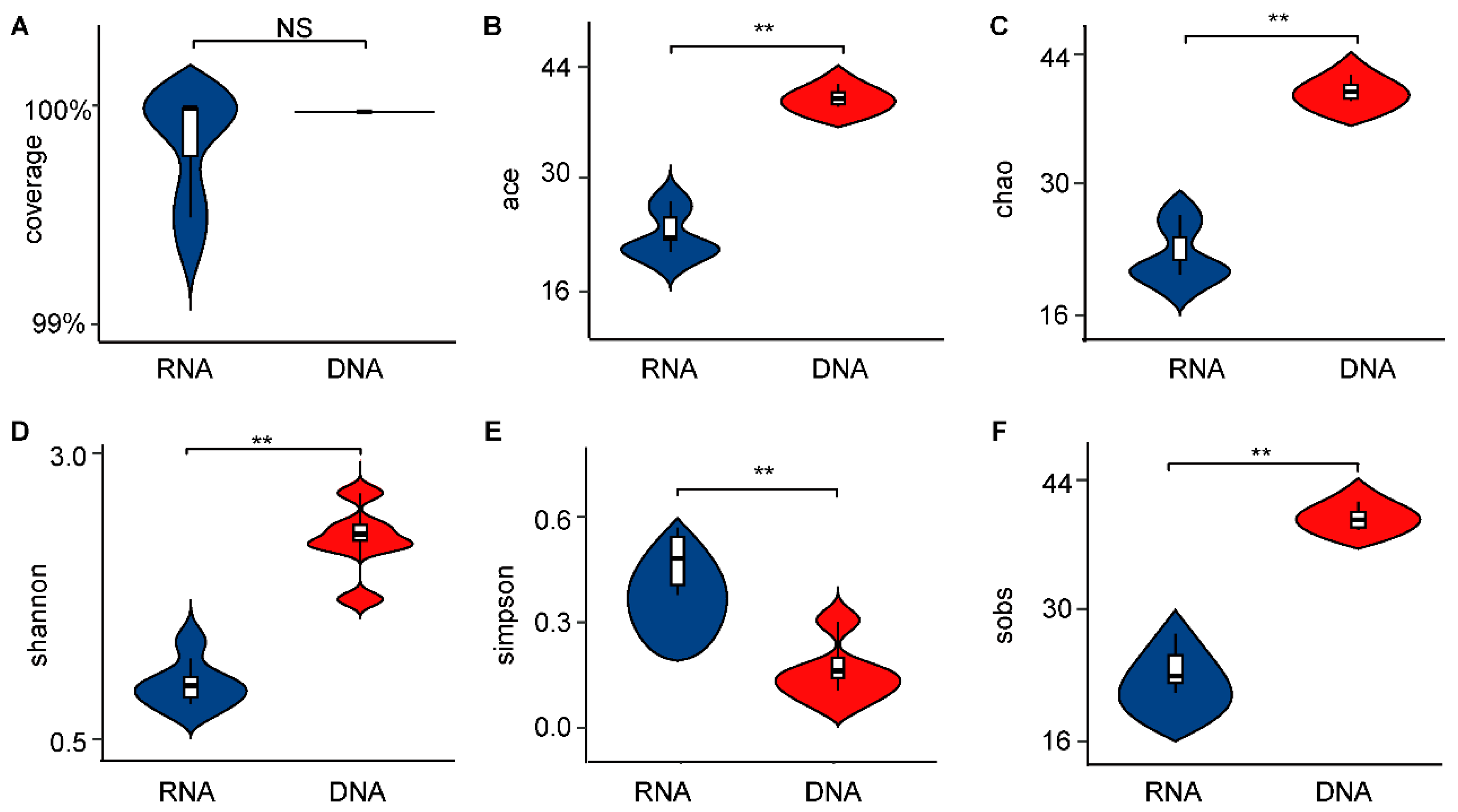
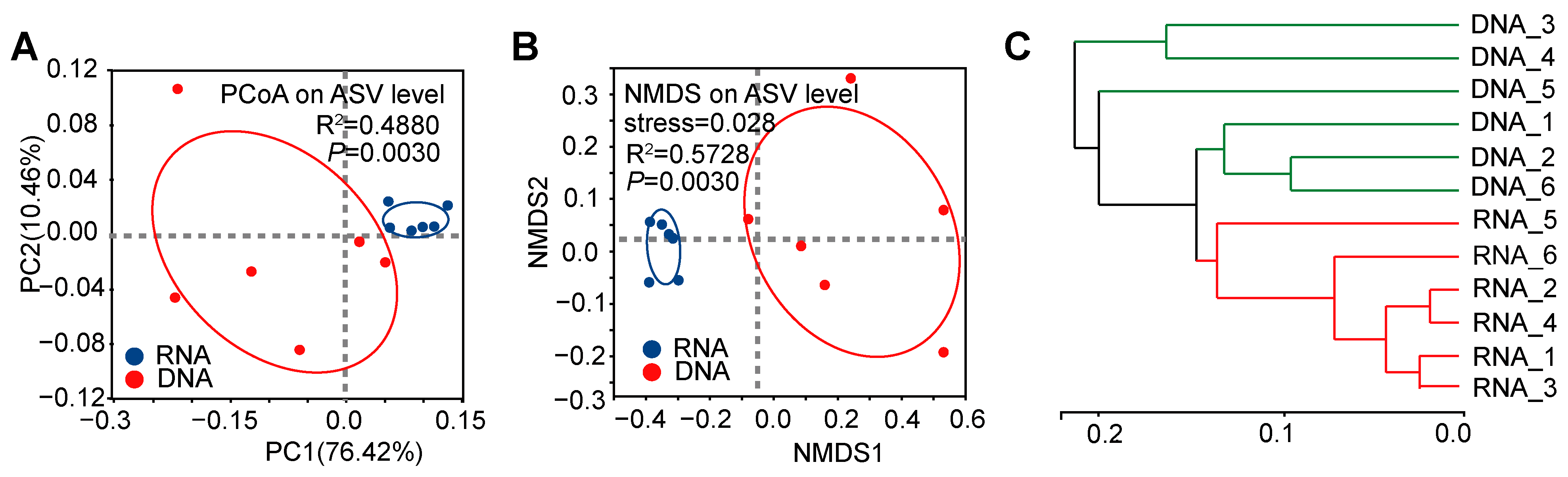
3.1. Significant Differences in α-Diversity and β-Diversity of Bacterial Communities Between DNA- and RNA-Dependent Sequence Samples
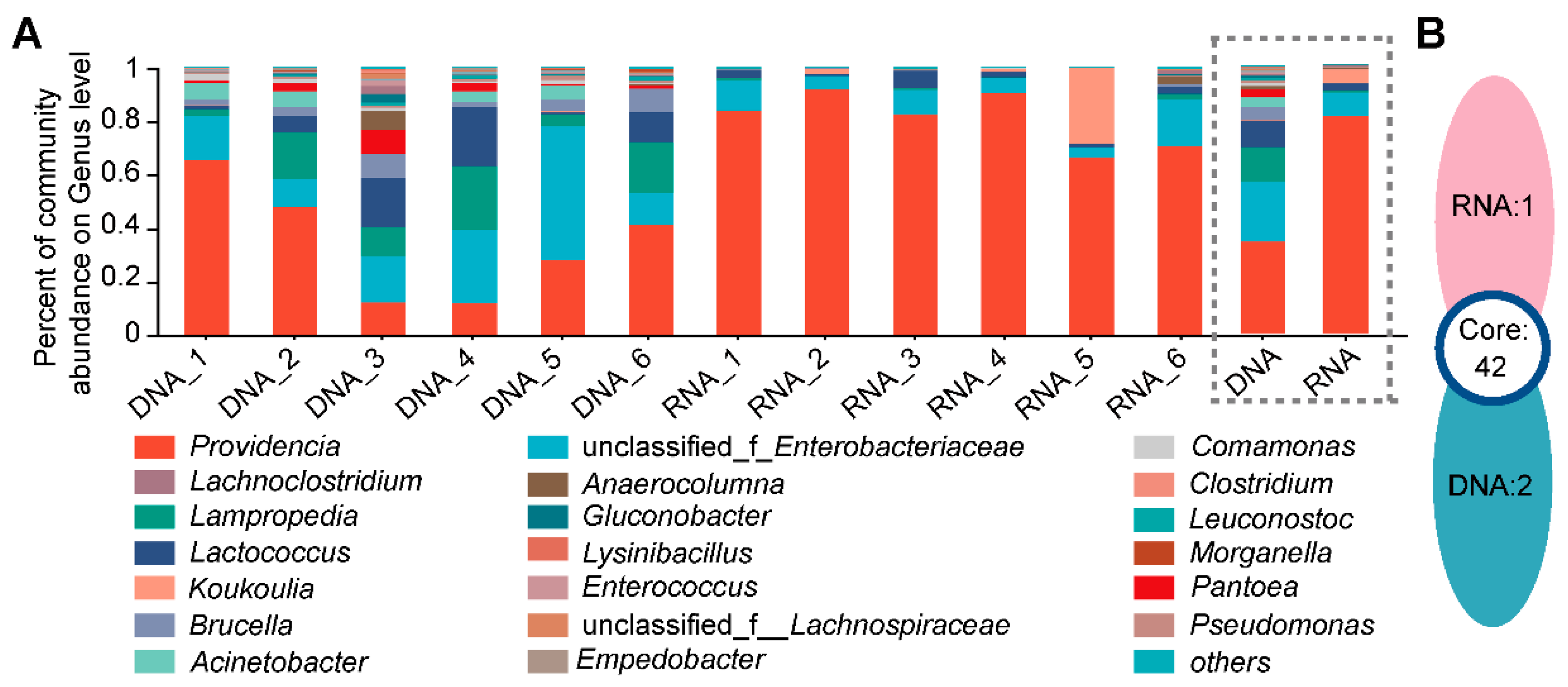
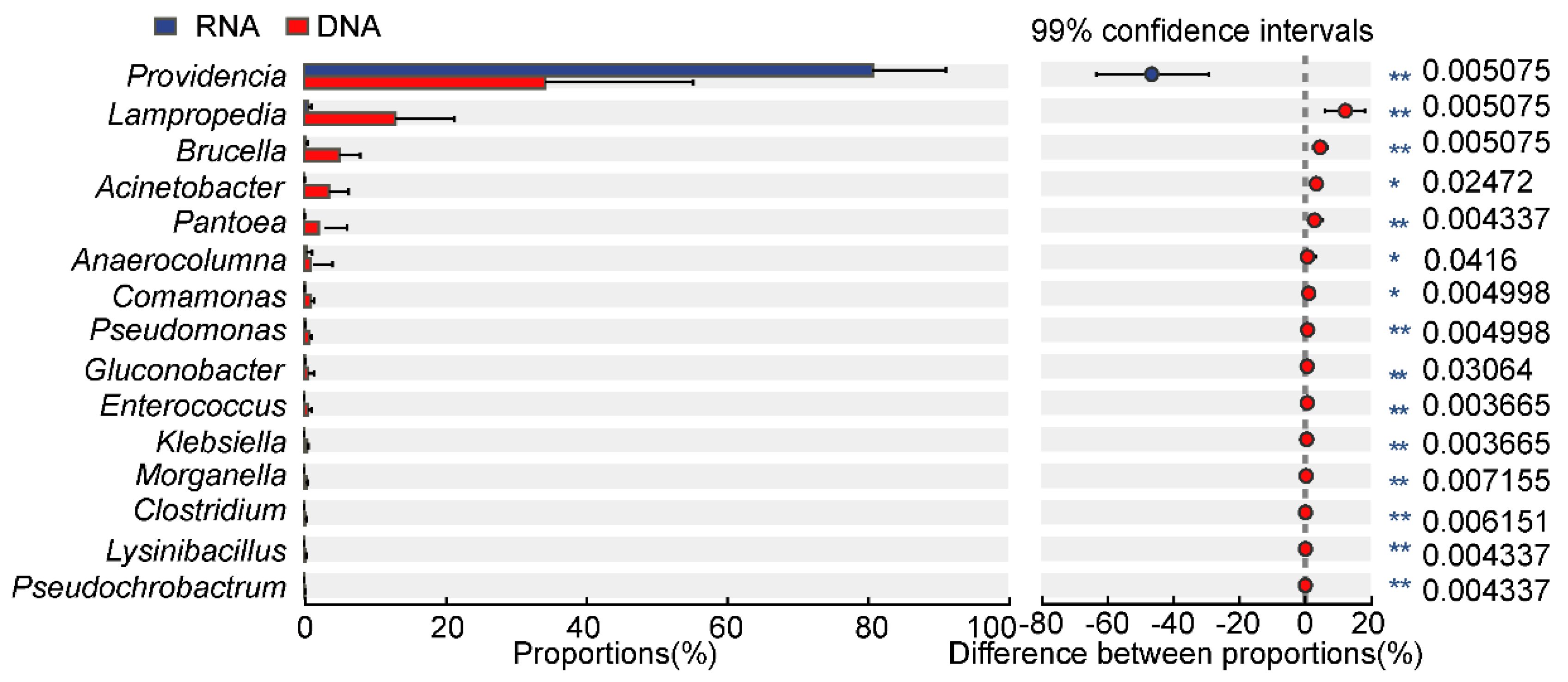
3.2. Different Bacterial Species Composition and Genus Abundance in the DNA- and RNA-Dependent Sequence Samples
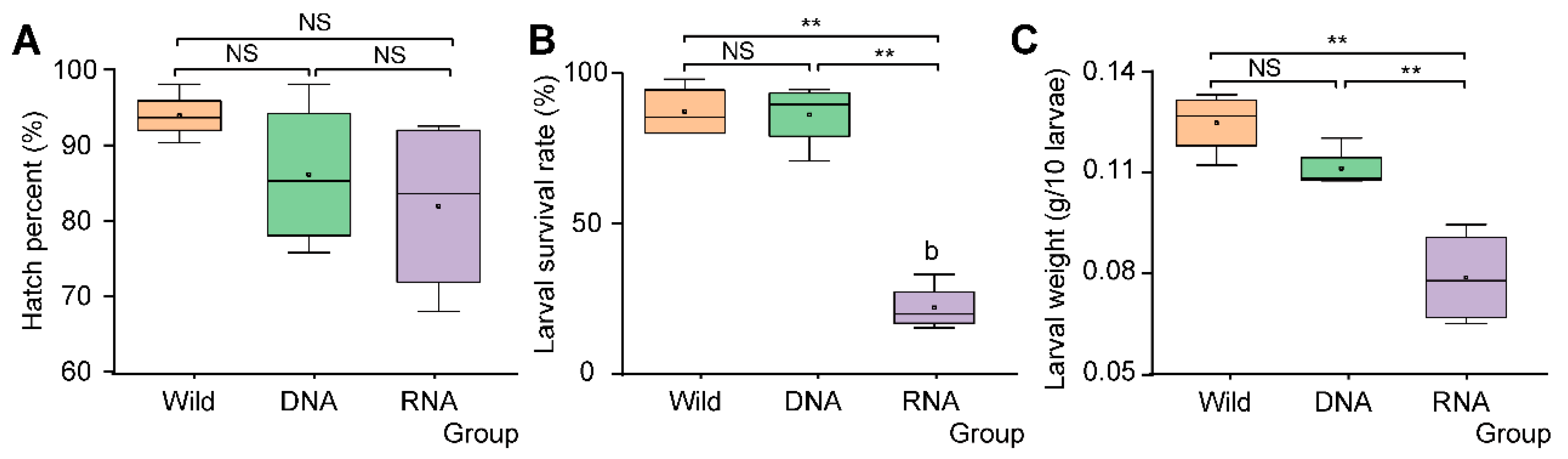
3.3. SynComs Constructed According to RNA-Dependent 16S rRNA Amplicon Sequencing Showed Inhibition Effects on D. antiqua Larvae
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Douglas, A.E. Multiorganismal insects: Diversity and function of resident microorganisms. Annu. Rev. Entomol. 2015, 60, 17–34. [Google Scholar] [CrossRef] [PubMed]
- Nahar, K.; Sultana, S.; Akter, T.; Begum, S. Biological traits and susceptibility of Delia antiqua (Meigen, 1826) (Diptera: Anthomyiidae) in onion. Bangladesh J. Zool. 2019, 47, 325–332. [Google Scholar] [CrossRef]
- Erica, M.; Kyle, W.; Brian, N. Environmental factors and crop management that affect Delia antiqua damage in onion fields. Agric. Ecosyst. Environ. 2021, 314, 107420. [Google Scholar] [CrossRef]
- Alejandro, T.M.; Guillermo, R.A.; Patricio, I.C.; Agustín, H. Incidence of Delia platura (Meigen) (Diptera: Anthomyiidae) in onion and scallion crops in Mexico. Fla. Entomol. 2022, 105, 319–320. [Google Scholar] [CrossRef]
- Akman, L.; Yamashita, A.; Watanabe, H.; Oshima, K.; Shiba, T.; Hattori, M.; Aksoy, S. Genome sequence of the endocellular obligate symbiont of tsetse flies, Wigglesworthia glossinidia. Nat. Genet. 2002, 32, 402–407. [Google Scholar] [CrossRef]
- Zhou, F.; Xu, L.; Wu, X.; Zhao, X.; Liu, M.; Zhang, X. Symbiotic bacterium-derived organic acids protect Delia antiqua larvae from entomopathogenic fungal Infection. mSystems 2020, 5, e00778-20. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Yan, Y.; Thompson, K.N.; Bae, S.; Accorsi, E.K.; Zhang, Y.; Shen, J.; Vlamakis, H.; Hartmann, E.M.; Huttenhower, C. Whole microbial community viability is not quantitatively reflected by propidium monoazide sequencing approach. Microbiome 2021, 9, 17. [Google Scholar] [CrossRef]
- Shen, J.; McFarland, A.G.; Young, V.B.; Hayden, M.K.; Hartmann, E.M. Toward accurate and robust environmental surveillance using metagenomics. Front. Genet. 2021, 12, 600111. [Google Scholar] [CrossRef]
- Rackaityte, E.; Halkias, J.; Fukui, E.M.; Mendoza, V.F.; Hayzelden, C.; Crawford, E.D.; Fujimura, K.E.; Burt, T.D.; Lynch, S.V. Viable bacterial colonization is highly limited in the human intestine in utero. Nat. Med. 2020, 26, 599–607. [Google Scholar] [CrossRef]
- Ting, Y.; Hyder, Q.I.; Jinhao, L.; Peijia, Y.; Hongyu, Y.; Xueyin, Z.; Weili, L.; Jiping, L. Analysis of changes in bacterial diversity in healthy and bacterial wilt mulberry samples using metagenomic sequencing and culture-dependent approaches. Front. Plant Sci. 2023, 14, 1206691. [Google Scholar] [CrossRef]
- Bowsher, A.W.; Kearns, P.J.; Shade, A. 16S rRNA/rRNA gene ratios and cell activity staining reveal consistent patterns of microbial activity in plant-associated soil. mSystems 2019, 4, e00003-19. [Google Scholar] [CrossRef] [PubMed]
- Schostag, M.; Stibal, M.; Jacobsen, C.S.; Bælum, J.; Taş, N.; Elberling, B.; Jansson, J.K.; Semenchuk, P.; Priemé, A. Distinct summer and winter bacterial communities in the active layer of Svalbard permafrost revealed by DNA- and RNA-based analyses. Front. Microbiol. 2015, 6, 399. [Google Scholar] [CrossRef] [PubMed]
- Wutkowska, M.; Vader, A.; Mundra, S.; Cooper, E.J.; Eidesen, P.B. Dead or Alive; or does it really matter? level of congruency between trophic modes in total and active fungal communities in high arctic soil. Front. Microbiol. 2018, 9, 3243. [Google Scholar] [CrossRef]
- Blazewicz, S.J.; Barnard, R.L.; Daly, R.A.; Firestone, M.K. Evaluating rRNA as an indicator of microbial activity in environmental communities: Limitations and uses. ISME J. 2013, 7, 2061–2068. [Google Scholar] [CrossRef]
- Ishikawa, Y.; Yamashita, T.; Nomura, M. Characteristics of summer diapause in the onion maggot, Delia antiqua (Diptera: Anthomyiidae). J. Insect Physiol. 2000, 46, 161–167. [Google Scholar] [CrossRef]
- Xin, C.; Liang, Q.; Li, M.; Wu, X.; Fan, S.; Zhang, X.; Zhou, F.; Zhao, Z. Rearing axenic Delia antiqua with half-fermented sterile diets. J. Vis. Exp. 2023, 202, e66259. [Google Scholar] [CrossRef]
- Zhou, F.Y.; Wu, X.Q.; Fan, S.S.; Zhao, X.; Li, M.; Song, F.; Huang, Y.; Zhang, X. Detoxification of phoxim by a gut bacterium of Delia antiqua. Sci. Total Environ. 2024, 943, 173866. [Google Scholar] [CrossRef]
- Fangyuan, Z.; Qingxia, L.; Xiaoyan, Z.; Xiaoqing, W.; Susu, F.; Xinjian, Z. Comparative metaproteomics reveal co-contribution of onion maggot and its gut microbiota to phoxim resistance. Ecotoxicol. Environ. Saf. 2023, 267, 115649. [Google Scholar] [CrossRef]
- Fangyuan, Z.; Yunxiao, G.; Mei, L.; Letian, X.; Xiaoqing, W.; Xiaoyan, Z.; Xinjian, Z. Bacterial Inhibition on beauveria bassiana contributes to microbiota stability in Delia antiqua. Front. Microbiol. 2021, 12, 710800. [Google Scholar] [CrossRef]
- Fangyuan, Z.; Xiaoqing, W.; Letian, X.; Shuhai, G.; Guanhong, C.; Xinjian, Z. Repressed beauveria bassiana Infections in Delia antiqua due to associated microbiota. Pest Manag. Sci. 2018, 75, 170–179. [Google Scholar] [CrossRef]
- Cao, X.; Li, M.; Wu, X.; Fan, S.; Lin, L.; Xu, L.; Zhang, X.; Zhou, F. Gut fungal diversity across different life stages of the onion fly Delia antiqua. Microb. Ecol. 2024, 87, 115. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Zhao, X.; Li, X.; Wu, X.; Zhou, H.; Gao, Y.; Zhang, X.; Zhou, F. Antagonistic effects of Delia antiqua (Diptera: Anthomyiidae)-associated bacteria against four phytopathogens. J. Econ. Entomol. 2021, 114, 597–610. [Google Scholar] [CrossRef] [PubMed]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef] [PubMed]
- Jingying, Z.; Xin, L.Y.; Xiaoxuan, G.; Yuan, Q.; Ruben, G.O.; Paul, S.L.; Yang, B. High-throughput cultivation and identification of bacteria from the plant root microbiota. Nat. Protoc. 2021, 16, 988–1012. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Stöver, B.C.; Müller, K.F. TreeGraph 2: Combining and visualizing evidence from different phylogenetic analyses. BMC Bioinf. 2010, 11, 7. [Google Scholar] [CrossRef]
- Fakruddin, M.; Mannan, S.B. Methods for analyzing diversity of microbial communities in natural environments. Ceylon J. Sci. Biol. Sci. 2013, 42, 19. [Google Scholar] [CrossRef]
- Rabee, A.E.; Forster, R.J.; Elekwachi, C.O.; Kewan, K.Z.; Sabra, E.; Mahrous, H.A.; Khamiss, O.A.; Shawket, S.M. Composition of bacterial and archaeal communities in the rumen of dromedary camel using cDNA-amplicon sequencing. Int. Microbiol. 2020, 23, 137–148. [Google Scholar] [CrossRef]
- Adams, R.I.; Lymperopoulou, D.S.; Misztal, P.K.; De Cassia Pessotti, R.; Behie, S.W.; Tian, Y.; Goldstein, A.H.; Lindow, S.E.; Nazaroff, W.W.; Taylor, J.W.; et al. Microbes and associated soluble and volatile chemicals on periodically wet household surfaces. Microbiome 2017, 5, 128. [Google Scholar] [CrossRef]
- Kajla, M.; Gupta, K.; Gupta, L.; Kumar, S. A Fine-Tuned management between physiology and immunity maintains the gut microbiota in insects. Biochem. Physiol.Open Access 2015, 4, 1000182. [Google Scholar] [CrossRef]
- Lax, S.; Cardona, C.; Zhao, D.; Winton, V.J.; Goodney, G.; Gao, P.; Gottel, N.; Hartmann, E.M.; Henry, C.; Thomas, P.M.; et al. Microbial and metabolic succession on common building materials under high humidity conditions. Nat. Commun. 2019, 10, 1767. [Google Scholar] [CrossRef] [PubMed]
- Fahimipour, A.K.; Hartmann, E.M.; Siemens, A.; Kline, J.; Levin, D.A.; Wilson, H.; Betancourt-Román, C.M.; Brown, G.Z.; Fretz, M.; Northcutt, D.; et al. Daylight exposure modulates bacterial communities associated with household dust. Microbiome 2018, 6, 175. [Google Scholar] [CrossRef] [PubMed]
- Nkongolo, K.K.; Narendrula-Kotha, R. Advances in monitoring soil microbial community dynamic and function. J. Appl. Genet. 2020, 61, 249–263. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Thompson, K.N.; Yan, Y.; Short, M.I.; Zhang, Y.; Franzosa, E.A.; Shen, J.; Hartmann, E.M.; Huttenhower, C. RNA-based amplicon sequencing is ineffective in measuring metabolic activity in environmental microbial communities. Microbiome 2023, 11, 131. [Google Scholar] [CrossRef]
- Tan, S.C.; Yiap, B.C. DNA, RNA, and protein extraction: The past and the present. BioMed Res. Int. 2009, 2009, 574398. [Google Scholar] [CrossRef]
- Ledeker, B.M.; De Long, S.K. The effect of multiple primer-template mismatches on quantitative PCR accuracy and development of a multi-primer set assay for accurate quantification of pcrA gene sequence variants. J. Microbiol. Methods 2013, 94, 224–231. [Google Scholar] [CrossRef]
- Gaby, J.C.; Buckley, D.H. The use of degenerate primers in qPCR analysis of functional genes can cause dramatic quantification bias as revealed by Investigation of nifH primer performance. Microb. Ecol. 2017, 74, 701–708. [Google Scholar] [CrossRef]
- Reid, N.M.; Addison, S.L.; Macdonald, L.J.; Lloyd-Jones, G. Biodiversity of active and inactive bacteria in the gut flora of wood-feeding huhu beetle larvae (Prionoplus reticularis). Appl. Environ. Microbiol. 2011, 77, 7000–7006. [Google Scholar] [CrossRef]
- Mugo Kamiri, L.; Querejeta, M.; Raymond, B.; Herniou, E.A. The effect of diet composition on the diversity of active gut bacteria and on the growth of Spodoptera exigua (Lepidoptera: Noctuidae). J. Insect Sci. 2024, 24, 13. [Google Scholar] [CrossRef]
- Zhang, Y.; Ke, Z.; Xu, L.; Yang, Y.; Chang, L.; Zhang, J. A faster killing effect of plastid-mediated RNA interference on a leaf beetle through induced dysbiosis of the gut bacteria. Plant Commun. 2024, 5, 100974. [Google Scholar] [CrossRef]
- Zeng, T.; Fu, Q.Y.; Luo, F.; Dai, J.; Fu, R.; Qi, Y.; Deng, X.; Lu, Y.; Xu, Y. Lactic acid bacteria modulate the CncC pathway to enhance resistance to β-cypermethrin in the oriental fruit fly. ISME J. 2024, 18, wrae058. [Google Scholar] [CrossRef] [PubMed]
- Zhou, F.; Xu, L.; Wang, S.; Wang, B.; Lou, Q.; Lu, M.; Sun, J. Bacterial volatile ammonia regulates the consumption sequence of d-pinitol and d-glucose in a fungus associated with an invasive bark beetle. ISME J. 2017, 11, 2809–2820. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, M.; Cao, X.; Xu, L.; Lin, L.; Wu, X.; Fan, S.; Zhang, X.; Zhou, F. Laboratory Test Indirectly Reveals the Unreliability of RNA-Dependent 16S rRNA Amplicon Sequences in Detecting the Gut Bacterial Diversity of Delia antiqua. Insects 2025, 16, 611. https://doi.org/10.3390/insects16060611
Li M, Cao X, Xu L, Lin L, Wu X, Fan S, Zhang X, Zhou F. Laboratory Test Indirectly Reveals the Unreliability of RNA-Dependent 16S rRNA Amplicon Sequences in Detecting the Gut Bacterial Diversity of Delia antiqua. Insects. 2025; 16(6):611. https://doi.org/10.3390/insects16060611
Chicago/Turabian StyleLi, Miaomiao, Xin Cao, Linfeng Xu, Luyao Lin, Xiaoqing Wu, Susu Fan, Xinjian Zhang, and Fangyuan Zhou. 2025. "Laboratory Test Indirectly Reveals the Unreliability of RNA-Dependent 16S rRNA Amplicon Sequences in Detecting the Gut Bacterial Diversity of Delia antiqua" Insects 16, no. 6: 611. https://doi.org/10.3390/insects16060611
APA StyleLi, M., Cao, X., Xu, L., Lin, L., Wu, X., Fan, S., Zhang, X., & Zhou, F. (2025). Laboratory Test Indirectly Reveals the Unreliability of RNA-Dependent 16S rRNA Amplicon Sequences in Detecting the Gut Bacterial Diversity of Delia antiqua. Insects, 16(6), 611. https://doi.org/10.3390/insects16060611