
Genomic Analysis Reveals Novel Genes and Adaptive Mechanisms for Artificial Diet Utilization in the Silkworm Strain Guican No.5

 , , and
, , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Silkworm Strain and Sample Collection
2.2. DNA Extraction and Whole-Genome Sequencing
2.3. Bioinformatic Analysis
3. Results
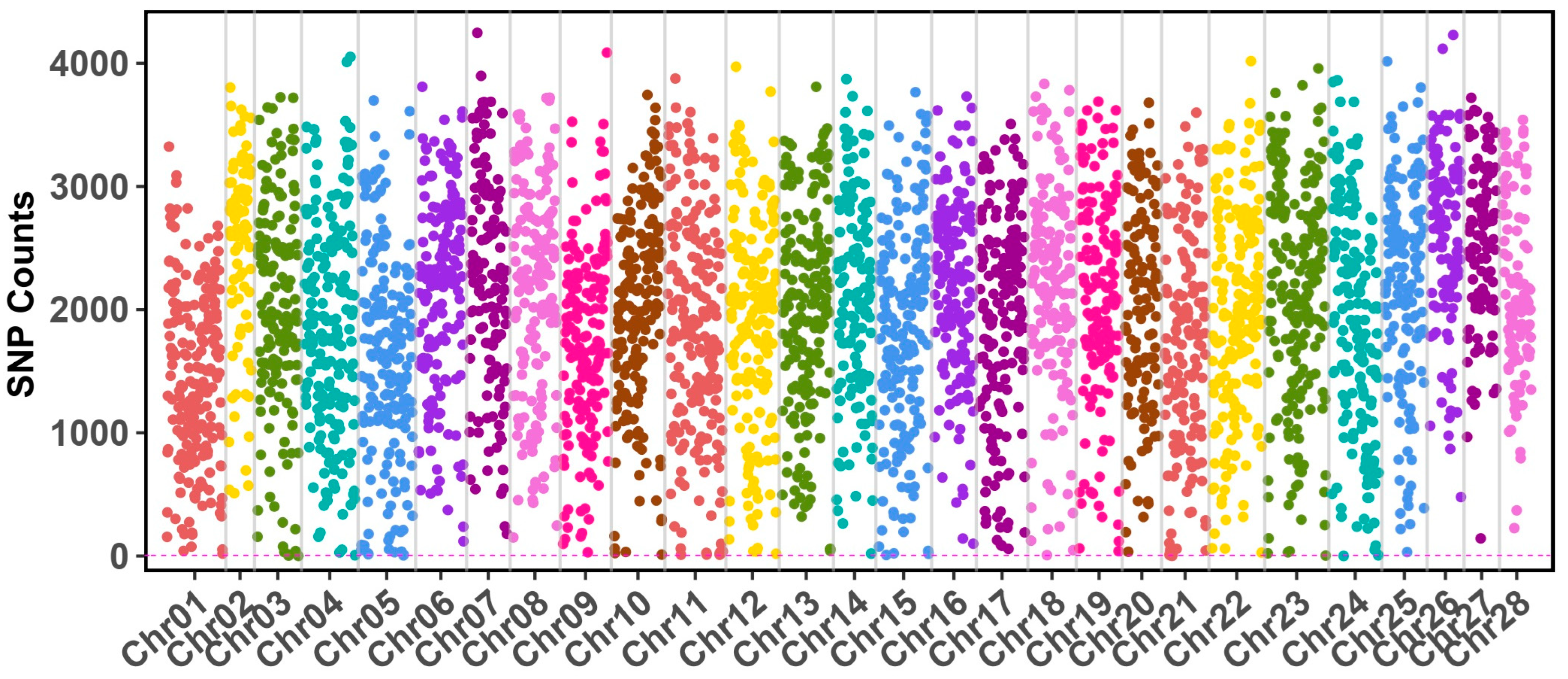
3.1. Genome-Wide SNP Calling
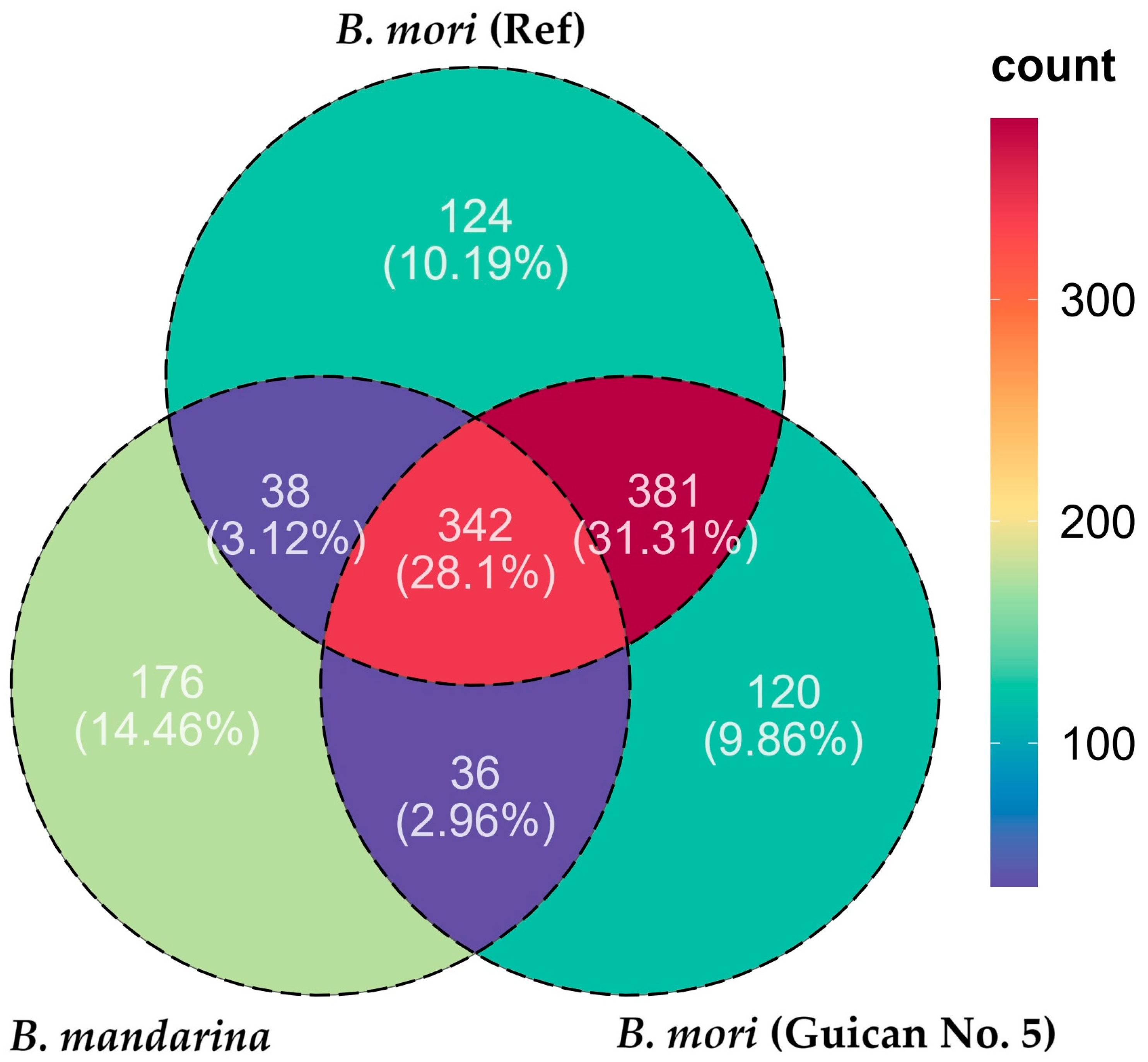
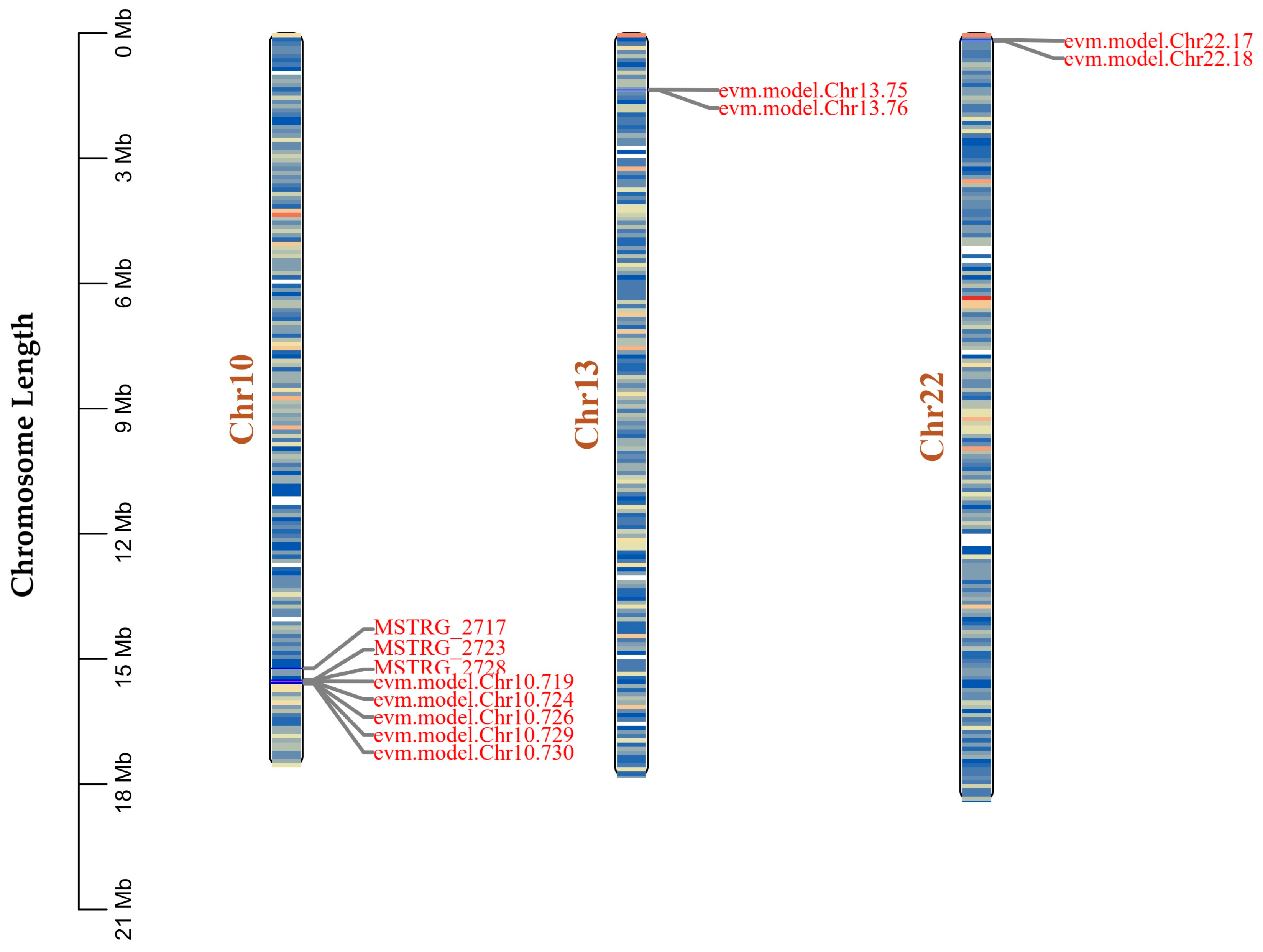
3.2. Consensus Genome Assembly and Structural Analysis
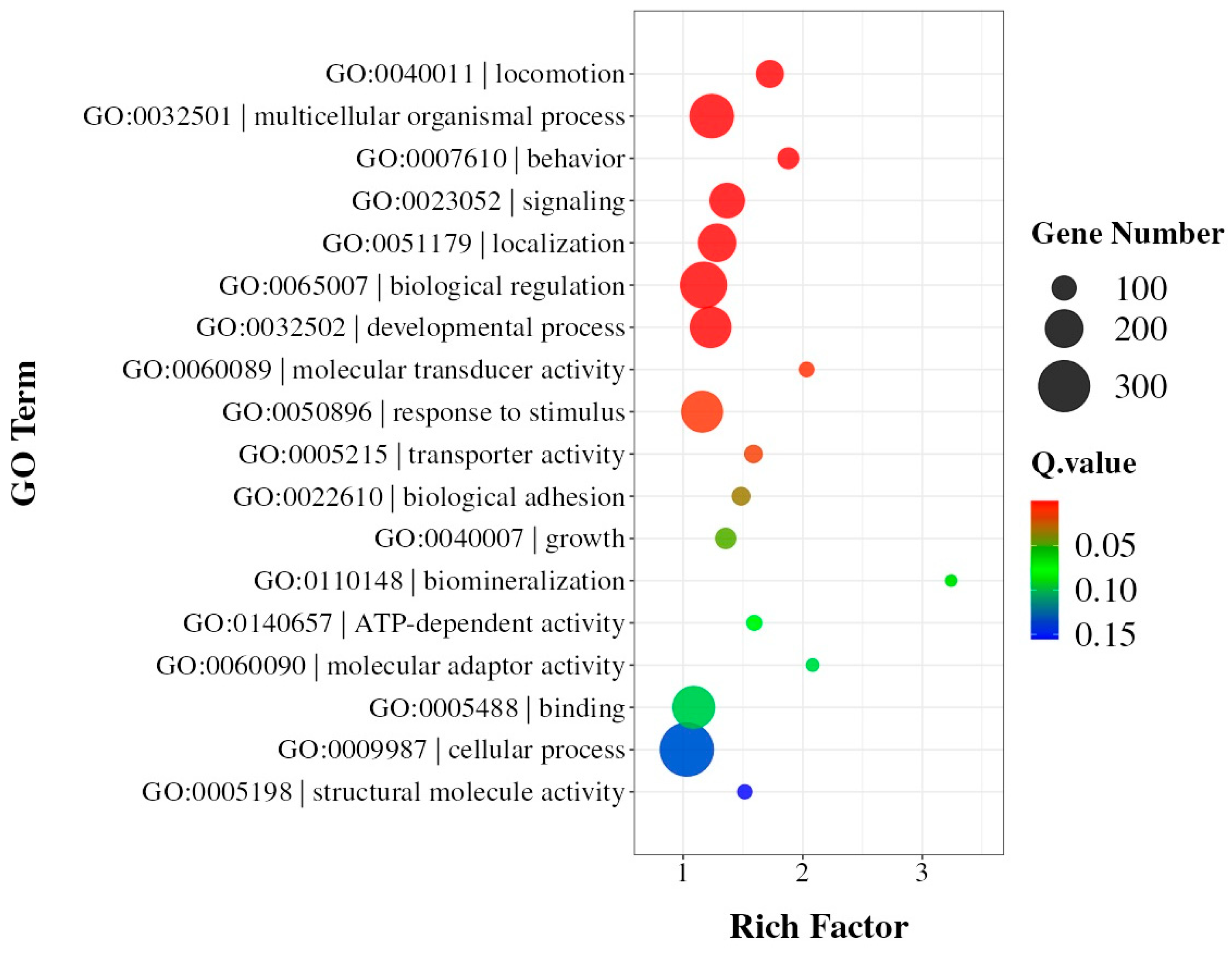
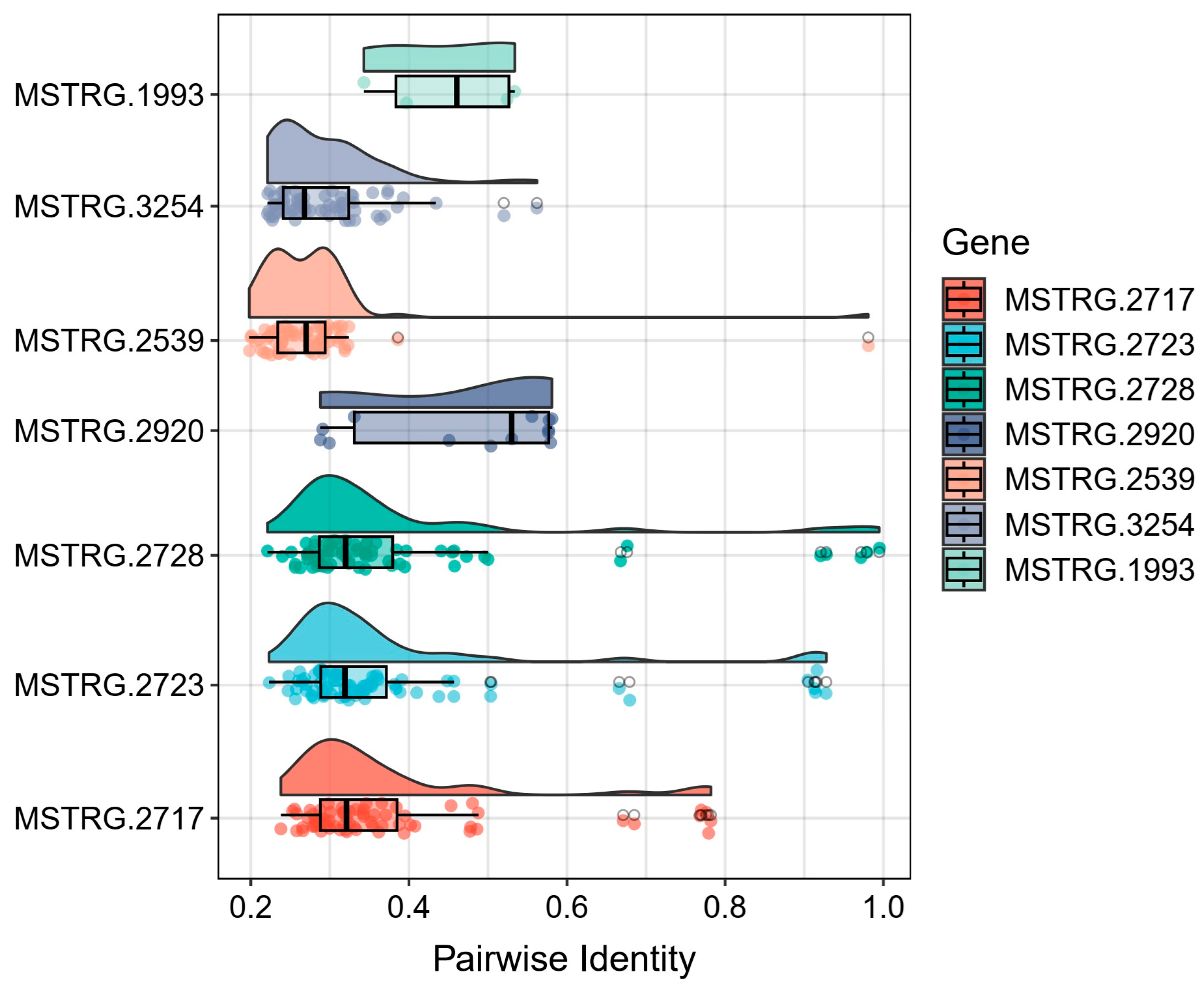
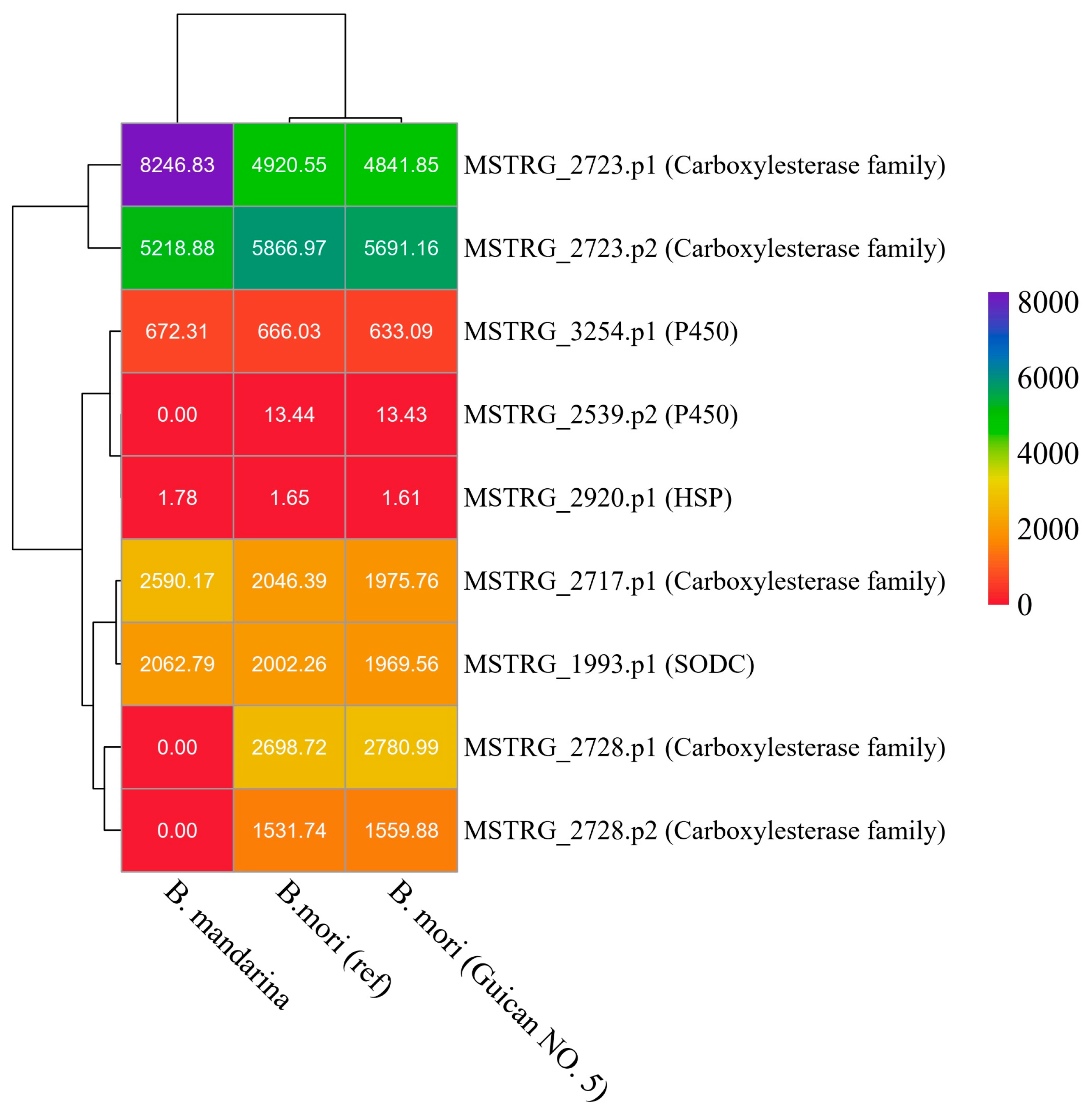
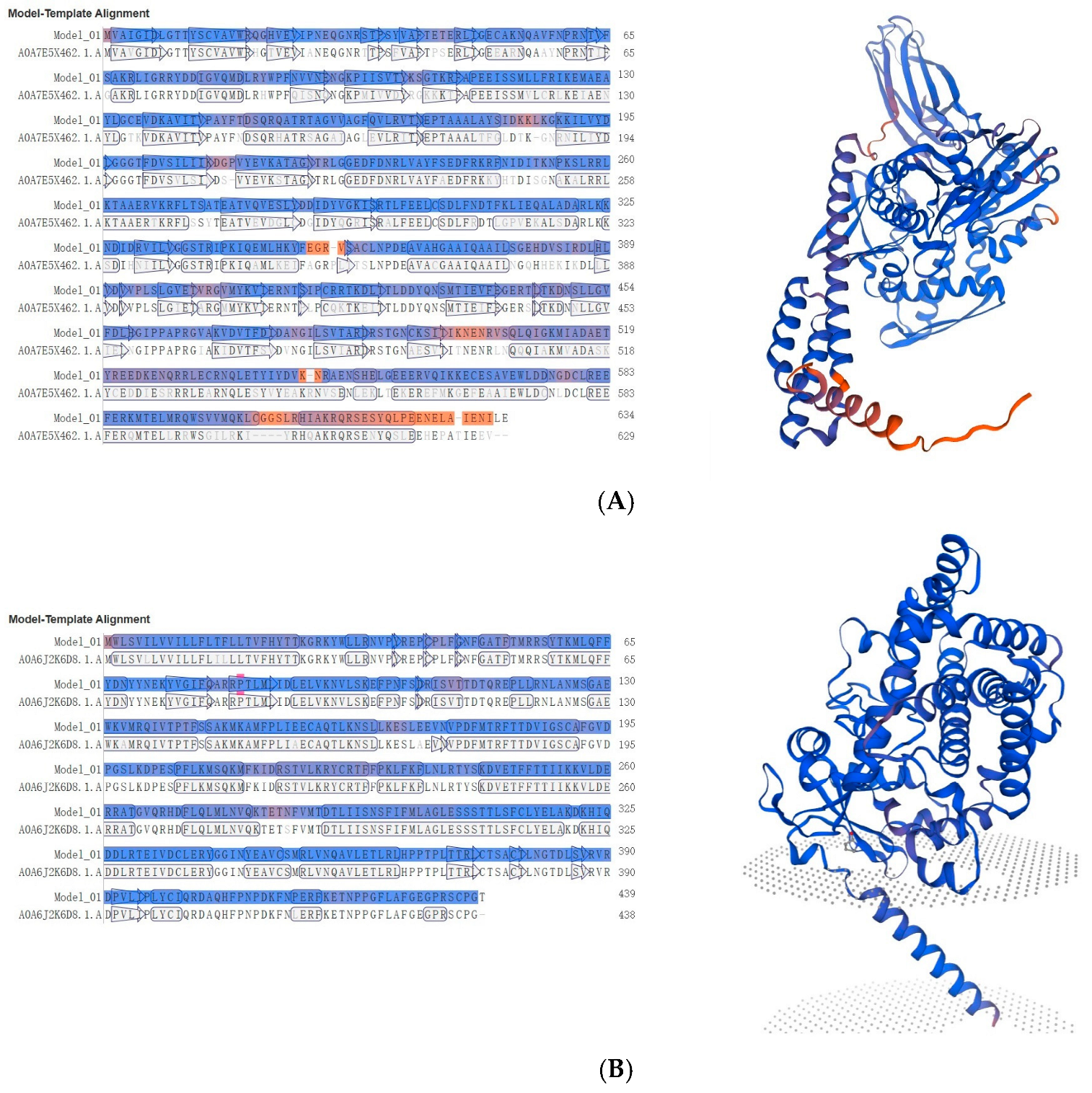
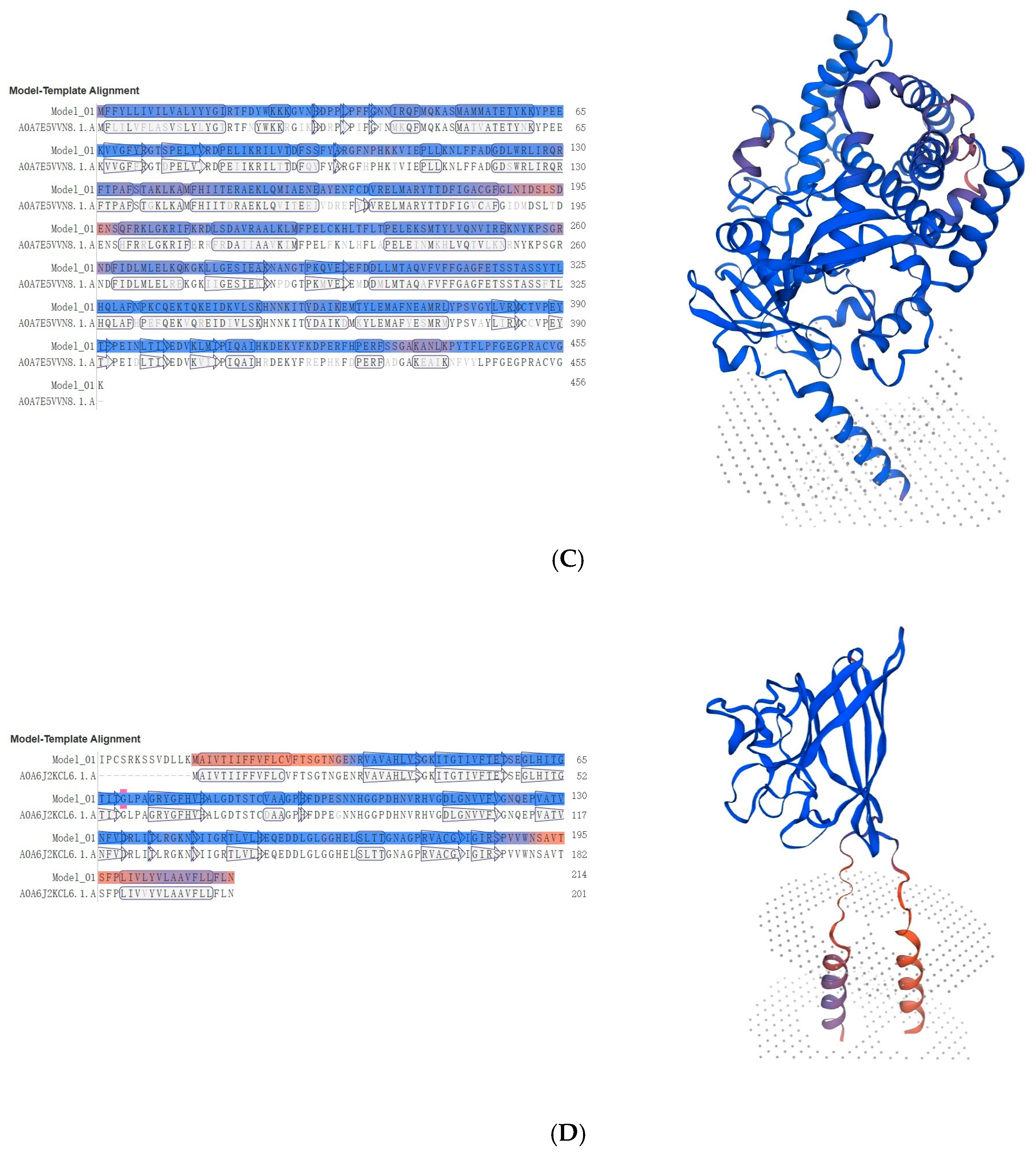
3.3. Identification of Key Coding Domain Sequences Related to Digestion and Detoxification
4. Discussion
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Correction Statement
References
- Shu, Q.; Wang, Y.; Gu, H.; Zhu, Q.; Liu, W.; Dai, Y.; Li, F.; Li, B. Effects of artificial diet breeding on intestinal microbial populations at the young stage of silkworm (Bombyx mori). Arch. Insect Biochem. Physiol. 2023, 113, e22019. [Google Scholar] [CrossRef]
- Lamberti, C.; Gai, F.; Cirrincione, S.; Giribaldi, M.; Purrotti, M.; Manfredi, M.; Marengo, E.; Sicuro, B.; Saviane, A.; Cappellozza, S.; et al. Investigation of the protein profile of silkworm (Bombyx mori) pupae reared on a well-calibrated artificial diet compared to mulberry leaf diet. PeerJ 2019, 7, e6723. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Huang, T.; Liu, Q.; Zhong, S.; Shen, D.; Chen, A.; Zhao, Q. Transcriptome analysis of anorexic and preferred silkworms (Bombyx mori) on artificial diet. Comp. Biochem. Physiol. Part D Genom. Proteom. 2023, 46, 101086. [Google Scholar] [CrossRef] [PubMed]
- Pan, M.; Jiang, K.; Jin, Y.; Mao, Y.; Lu, W.; Jiang, W.; Chen, W. Study on the Structure and Properties of Silk Fibers Obtained from Factory All-Age Artificial Diets. Int. J. Mol. Sci. 2024, 25, 6129. [Google Scholar] [CrossRef] [PubMed]
- Xin, L.; Chen, Y.; Rong, W.; Qin, Y.; Li, X.; Guan, D. Gut Microbiota Analysis in Silkworms (Bombyx mori) Provides Insights into Identifying Key Bacterials for Inclusion in Artificial Diet Formulations. Animals 2024, 14, 1261. [Google Scholar] [CrossRef]
- Li, J.; Deng, J.; Deng, X.; Liu, L.; Zha, X. Metabonomic Analysis of Silkworm Midgut Reveals Differences between the Physiological Effects of an Artificial and Mulberry Leaf Diet. Insects 2023, 14, 347. [Google Scholar] [CrossRef] [PubMed]
- Dong, H.L.; Zhang, S.X.; Chen, Z.H.; Tao, H.; Li, X.; Qiu, J.F.; Cui, W.Z.; Sima, Y.H.; Cui, W.Z.; Xu, S.Q. Differences in gut microbiota between silkworms (Bombyx mori) reared on fresh mulberry (Morus alba var. multicaulis) leaves or an artificial diet. RSC Adv. 2018, 8, 26188–26200. [Google Scholar] [CrossRef]
- Tong, X.; Han, M.J.; Lu, K.; Tai, S.; Liang, S.; Liu, Y.; Hu, H.; Shen, J.; Long, A.; Zhan, C.; et al. High-resolution silkworm pan-genome provides genetic insights into artificial selection and ecological adaptation. Nat. Commun. 2022, 13, 5619. [Google Scholar] [CrossRef]
- Zhang, X.; Nie, M.; Zhao, Q.; Wu, Y.; Wang, G.; Xia, Q. Genome-wide patterns of genetic variation among silkworms. Mol. Genet. Genom. MGG 2015, 290, 1575–1587. [Google Scholar] [CrossRef]
- Xin, S.; Zhang, W. Construction and analysis of the protein-protein interaction network for the detoxification enzymes of the silkworm, Bombyx mori. Arch. Insect Biochem. Physiol. 2021, 108, e21850. [Google Scholar] [CrossRef]
- Rong, W.; Chen, Y.; Lu, J.; Huang, S.; Xin, L.; Guan, D.; Li, X. Effects of Chromium Exposure on the Gene Expression of the Midgut in Silkworms, Bombyx mori. Genes 2023, 14, 1616. [Google Scholar] [CrossRef]
- Chen, Y.Z.; Rong, W.T.; Qin, Y.C.; Lu, L.Y.; Liu, J.; Li, M.J.; Xin, L.; Li, X.D.; Guan, D.L. Integrative analysis of microbiota and metabolomics in chromium-exposed silkworm (Bombyx mori) midguts based on 16S rDNA sequencing and LC/MS metabolomics. Front. Microbiol. 2023, 14, 1278271. [Google Scholar] [CrossRef]
- Yin, X.; Zhang, Y.; Yu, D.; Li, G.; Wang, X.; Wei, Y.; He, C.; Liu, Y.; Li, Y.; Xu, K.; et al. Effects of artificial diet rearing during all instars on silk secretion and gene transcription in Bombyx mori (Lepidoptera: Bombycidae). J. Econ. Entomol. 2023, 116, 1379–1390. [Google Scholar] [CrossRef]
- Liu, L.; Zhao, D.; Wang, G.; He, Q.; Song, Y.; Jiang, Y.; Xia, Q.; Zhao, P. Adaptive Changes in Detoxification Metabolism and Transmembrane Transport of Bombyx mori Malpighian Tubules to Artificial Diet. Int. J. Mol. Sci. 2023, 24, 9949. [Google Scholar] [CrossRef]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinform. 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Lu, F.; Wei, Z.; Luo, Y.; Guo, H.; Zhang, G.; Xia, Q.; Wang, Y. SilkDB 3.0: Visualizing and exploring multiple levels of data for silkworm. Nucleic Acids Res. 2020, 48, D749–D755. [Google Scholar] [CrossRef] [PubMed]
- Jo, H.; Koh, G. Faster single-end alignment generation utilizing multi-thread for BWA. Bio-Med. Mater. Eng. 2015, 26 (Suppl. S1), S1791–S1796. [Google Scholar] [CrossRef]
- Danecek, P.; Bonfield, J.K.; Liddle, J.; Marshall, J.; Ohan, V.; Pollard, M.O.; Whitwham, A.; Keane, T.; McCarthy, S.A.; Davies, R.M.; et al. Twelve years of SAMtools and BCFtools. GigaScience 2021, 10, giab008. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Paggi, J.M.; Park, C.; Bennett, C.; Salzberg, S.L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 2019, 37, 907–915. [Google Scholar] [CrossRef] [PubMed]
- Thakur, V. RNA-Seq Data Analysis for Differential Gene Expression Using HISAT2-StringTie-Ballgown Pipeline. Methods Mol. Biol. 2024, 2812, 101–113. [Google Scholar] [CrossRef]
- Pertea, G.; Pertea, M. GFF Utilities: GffRead and GffCompare. F1000Research 2020, 9, 304. [Google Scholar] [CrossRef]
- Kang, Y.J.; Yang, D.C.; Kong, L.; Hou, M.; Meng, Y.Q.; Wei, L.; Gao, G. CPC2: A fast and accurate coding potential calculator based on sequence intrinsic features. Nucleic Acids Res. 2017, 45, W12–W16. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Song, X.; Wang, K. lncScore: Alignment-free identification of long noncoding RNA from assembled novel transcripts. Sci. Rep. 2016, 6, 34838. [Google Scholar] [CrossRef]
- Cantalapiedra, C.P.; Hernández-Plaza, A.; Letunic, I.; Bork, P.; Huerta-Cepas, J. eggNOG-mapper v2: Functional Annotation, Orthology Assignments, and Domain Prediction at the Metagenomic Scale. Mol. Biol. Evol. 2021, 38, 5825–5829. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.; Hu, E.; Xu, S.; Chen, M.; Guo, P.; Dai, Z.; Feng, T.; Zhou, L.; Tang, W.; Zhan, L.; et al. clusterProfiler 4.0: A universal enrichment tool for interpreting omics data. Innovation 2021, 2, 100141. [Google Scholar] [CrossRef]
- Nunes-Alves, A.; Merz, K. AlphaFold2 in Molecular Discovery. J. Chem. Inf. Model. 2023, 63, 5947–5949. [Google Scholar] [CrossRef] [PubMed]
- Duvaud, S.; Gabella, C.; Lisacek, F.; Stockinger, H.; Ioannidis, V.; Durinx, C. Expasy, the Swiss Bioinformatics Resource Portal, as designed by its users. Nucleic Acids Res. 2021, 49, W216–W227. [Google Scholar] [CrossRef]
- Lyu, F.; Han, F.; Ge, C.; Mao, W.; Chen, L.; Hu, H.; Chen, G.; Lang, Q.; Fang, C. OmicStudio: A composable bioinformatics cloud platform with real-time feedback that can generate high-quality graphs for publication. iMeta 2023, 2, e85. [Google Scholar] [CrossRef] [PubMed]
- Waizumi, R.; Tsubota, T.; Jouraku, A.; Kuwazaki, S.; Yokoi, K.; Iizuka, T.; Yamamoto, K.; Sezutsu, H. Highly accurate genome assembly of an improved high-yielding silkworm strain, Nichi01. G3: Genes Genomes Genet. 2023, 13, jkad044. [Google Scholar] [CrossRef]
- Ma, S.Y.; Smagghe, G.; Xia, Q.Y. Genome editing in Bombyx mori: New opportunities for silkworm functional genomics and the sericulture industry. Insect Sci. 2019, 26, 964–972. [Google Scholar] [CrossRef]
- Kawamoto, M.; Jouraku, A.; Toyoda, A.; Yokoi, K.; Minakuchi, Y.; Katsuma, S.; Fujiyama, A.; Kiuchi, T.; Yamamoto, K.; Shimada, T. High-quality genome assembly of the silkworm, Bombyx mori. Insect Biochem. Mol. Biol. 2019, 107, 53–62. [Google Scholar] [CrossRef]
- Bian, D.; Ren, Y.; Ye, W.; Dai, M.; Li, F.; Wei, J.; Sun, H.; Li, B. Evaluation of tolerance to λ-cyhalothrin and response of detoxification enzymes in silkworms reared on artificial diet. Ecotoxicol. Environ. Saf. 2022, 232, 113232. [Google Scholar] [CrossRef]
- Zhang, X.; Zhao, M.; McCarty, D.R.; Lisch, D. Transposable elements employ distinct integration strategies with respect to transcriptional landscapes in eukaryotic genomes. Nucleic Acids Res. 2020, 48, 6685–6698. [Google Scholar] [CrossRef]
- Li, Y.; Yao, J.; Sang, H.; Wang, Q.; Su, L.; Zhao, X.; Xia, Z.; Wang, F.; Wang, K.; Lou, D.; et al. Pan-genome analysis highlights the role of structural variation in the evolution and environmental adaptation of Asian honeybees. Mol. Ecol. Resour. 2024, 24, e13905. [Google Scholar] [CrossRef] [PubMed]
- Fujimoto, S.; Kawamoto, M.; Shoji, K.; Suzuki, Y.; Katsuma, S.; Iwanaga, M. Whole-genome sequencing and comparative transcriptome analysis of Bombyx mori nucleopolyhedrovirus La strain. Virus Genes 2020, 56, 249–259. [Google Scholar] [CrossRef]
- Li, J.; Chen, C.; Zha, X. Midgut and Head Transcriptomic Analysis of Silkworms Reveals the Physiological Effects of Artificial Diets. Insects 2022, 13, 291. [Google Scholar] [CrossRef] [PubMed]
- Yuan, S.F.; Yue, X.J.; Hu, W.F.; Wang, Y.; Li, Y.Z. Genome-wide analysis of lipolytic enzymes and characterization of a high-tolerant carboxylesterase from Sorangium cellulosum. Front. Microbiol. 2023, 14, 1304233. [Google Scholar] [CrossRef]
- Guerrero-Cruz, S.; Cremers, G.; van Alen, T.A.; Op den Camp, H.J.M.; Jetten, M.S.M.; Rasigraf, O.; Vaksmaa, A. Response of the Anaerobic Methanotroph “Candidatus Methanoperedens nitroreducens” to Oxygen Stress. Appl. Environ. Microbiol. 2018, 84, e01832-18. [Google Scholar] [CrossRef] [PubMed]
- Chertemps, T.; Le Goff, G.; Maïbèche, M.; Hilliou, F. Detoxification gene families in Phylloxera: Endogenous functions and roles in response to the environment. Comp. Biochem. Physiol. Part D Genom. Proteom. 2021, 40, 100867. [Google Scholar] [CrossRef]
- Nauen, R.; Zimmer, C.T.; Vontas, J. Heterologous expression of insect P450 enzymes that metabolize xenobiotics. Curr. Opin. Insect Sci. 2021, 43, 78–84. [Google Scholar] [CrossRef]
- Lu, K.; Song, Y.; Zeng, R. The role of cytochrome P450-mediated detoxification in insect adaptation to xenobiotics. Curr. Opin. Insect Sci. 2021, 43, 103–107. [Google Scholar] [CrossRef]
- Liu, L.; Qian, X.; Chao, M.; Zhao, Y.; Huang, J.; Wang, T.; Sun, F.; Ling, E.; Song, H. Aluminum toxicity related to SOD and expression of presenilin and CREB in Bombyx mori. Arch. Insect Biochem. Physiol. 2018, 99, e21480. [Google Scholar] [CrossRef] [PubMed]
- Peng, Z.; Hu, W.; Yang, X.; Liu, Q.; Shi, X.; Tang, X.; Zhao, P.; Xia, Q. Overexpression of bond-forming active protein for efficient production of silk with structural changes and properties enhanced in silkworm. Int. J. Biol. Macromol. 2024, 264, 129780. [Google Scholar] [CrossRef]
- Kausar, S.; Abbas, M.N.; Yang, L.; Cui, H. Biotic and abiotic stress induces the expression of Hsp70/90 organizing protein gene in silkworm, Bombyx mori. Int. J. Biol. Macromol. 2020, 143, 610–618. [Google Scholar] [CrossRef]
- Zhu, K.; Chen, Y.; Chen, L.; Xiang, H. Comparative Silk Transcriptomics Illuminates Distinctive Impact of Artificial Selection in Silkworm Modern Breeding. Insects 2022, 13, 1163. [Google Scholar] [CrossRef]
- Wan, L.; Zhou, A.; Xiao, W.; Zou, B.; Jiang, Y.; Xiao, J.; Deng, C.; Zhang, Y. Cytochrome P450 monooxygenase genes in the wild silkworm, Bombyx mandarina. PeerJ 2021, 9, e10818. [Google Scholar] [CrossRef] [PubMed]
- Li, J.Y.; Cai, F.; Ye, X.G.; Liang, J.S.; Li, J.K.; Wu, M.Y.; Zhao, D.; Jiang, Z.D.; You, Z.Y.; Zhong, B.X. Comparative Proteomic Analysis of Posterior Silk Glands of Wild and Domesticated Silkworms Reveals Functional Evolution during Domestication. J. Proteome Res. 2017, 16, 2495–2507. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xin, L.; Guan, D.; Wei, N.; Zhang, X.; Deng, W.; Li, X.; Song, J. Genomic Analysis Reveals Novel Genes and Adaptive Mechanisms for Artificial Diet Utilization in the Silkworm Strain Guican No.5. Insects 2024, 15, 1010. https://doi.org/10.3390/insects15121010
Xin L, Guan D, Wei N, Zhang X, Deng W, Li X, Song J. Genomic Analysis Reveals Novel Genes and Adaptive Mechanisms for Artificial Diet Utilization in the Silkworm Strain Guican No.5. Insects. 2024; 15(12):1010. https://doi.org/10.3390/insects15121010
Chicago/Turabian StyleXin, Lei, Delong Guan, Nan Wei, Xiaoyan Zhang, Weian Deng, Xiaodong Li, and Jing Song. 2024. "Genomic Analysis Reveals Novel Genes and Adaptive Mechanisms for Artificial Diet Utilization in the Silkworm Strain Guican No.5" Insects 15, no. 12: 1010. https://doi.org/10.3390/insects15121010
APA StyleXin, L., Guan, D., Wei, N., Zhang, X., Deng, W., Li, X., & Song, J. (2024). Genomic Analysis Reveals Novel Genes and Adaptive Mechanisms for Artificial Diet Utilization in the Silkworm Strain Guican No.5. Insects, 15(12), 1010. https://doi.org/10.3390/insects15121010