


Novel Microsatellite Loci, Cross-Species Validation of Multiplex Assays, and By-Catch Mitochondrial Genomes on Ochthebius Beetles from Supratidal Rockpools
 , , ,
, , ,  , and
, and
Abstract
Simple Summary
Abstract
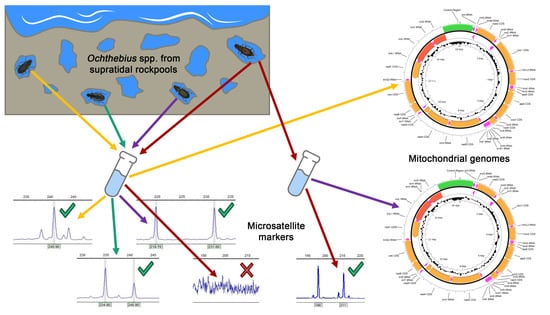
1. Introduction
2. Materials and Methods
2.1. Microsatellite Design
2.2. Multiplex Optimization
2.3. By-Catch Shotgun-Sequencing Data
3. Results
3.1. Isolation and Characterization of Microsatellite Loci
3.2. Multiplex Optimization and Cross-Species Amplification
3.3. Mitochondrial Genomes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hayward, M.D.; Hamilton, N.R.S. Genetic Diversity, Population Structure and Conservation. In Biotechnology and Plant Genetic Resources—Conservation and Use; Callow, J.A., Ford-Lloyd, B.V., Newbury, H.J., Eds.; Biotechnology in Agriculture Series; CAB International: Wallingford, UK, 1997; pp. 49–76. ISBN 0851991424. [Google Scholar]
- Frankham, R. Genetics and Extinction. Biol. Conserv. 2005, 126, 131–140. [Google Scholar] [CrossRef]
- Kajungiro, R.A.; Palaiokostas, C.; Pinto, F.A.L.; Mmochi, A.J.; Mtolera, M.; Houston, R.D.; De Koning, D.J. Population Structure and Genetic Diversity of Nile Tilapia (Oreochromis niloticus) Strains Cultured in Tanzania. Front. Genet. 2019, 10, 1269. [Google Scholar] [CrossRef]
- Lande, R.; Barrowclough, G.F. Effective Population Size, Genetic Variation, and Their Use in Population Management. In Viable Populations for Conservation; Soulé, M.E., Ed.; Cambridge University Press: Cambridge, UK, 1987; pp. 87–124. ISBN 978-0-521-33657-4. [Google Scholar]
- Újvári, B.; Madsen, T.; Kotenko, T.; Olsson, M.; Shine, R.; Wittzell, H. Low Genetic Diversity Threatens Imminent Extinction for the Hungarian Meadow Viper (Vipera ursinii rakosiensis). Biol. Conserv. 2002, 105, 127–130. [Google Scholar] [CrossRef]
- Gaston, K.J.; Lawton, J.H. The Population Ecology of Rare Species. J. Fish Biol. 1990, 37, 97–104. [Google Scholar] [CrossRef]
- Fahrig, L. Effects of Habitat Fragmentation on Biodiversity. Annu. Rev. Ecol. Evol. Syst. 2003, 34, 487–515. [Google Scholar] [CrossRef]
- Ribera, I. Habitat Constraints and the Generation of Diversity in Freshwater Macroinvertebrates. In Aquatic Insects: Challenges to Populations; Lancaster, J., Briers, R.A., Eds.; CABI: Wallingford, UK, 2008; pp. 289–311. ISBN 978-1-84593-396-8. [Google Scholar]
- Maxwell, S.L.; Cazalis, V.; Dudley, N.; Hoffmann, M.; Rodrigues, A.S.L.; Stolton, S.; Visconti, P.; Woodley, S.; Kingston, N.; Lewis, E.; et al. Area-Based Conservation in the Twenty-First Century. Nature 2020, 586, 217–227. [Google Scholar] [CrossRef]
- Sánchez-Bayo, F.; Wyckhuys, K.A.G. Worldwide Decline of the Entomofauna: A Review of Its Drivers. Biol. Conserv. 2019, 232, 8–27. [Google Scholar] [CrossRef]
- Thomas, C.D.; Jones, T.H.; Hartley, S.E. “Insectageddon”: A Call for More Robust Data and Rigorous Analyses. Glob. Chang. Biol. 2019, 25, 1891–1892. [Google Scholar] [CrossRef]
- Cook, L.G.; Edwards, R.D.; Crisp, M.D.; Hardy, N.B. Need Morphology Always Be Required for New Species Descriptions? Invertebr. Syst. 2010, 24, 322. [Google Scholar] [CrossRef]
- Dopheide, A.; Tooman, L.K.; Grosser, S.; Agabiti, B.; Rhode, B.; Xie, D.; Stevens, M.I.; Nelson, N.; Buckley, T.R.; Drummond, A.J.; et al. Estimating the Biodiversity of Terrestrial Invertebrates on a Forested Island Using DNA Barcodes and Metabarcoding Data. Ecol. Appl. 2019, 29, e01877. [Google Scholar] [CrossRef]
- Sagarin, R.D.; Gaines, S.D. Geographical Abundance Distributions of Coastal Invertebrates: Using One-dimensional Ranges to Test Biogeographic Hypotheses. J. Biogeogr. 2002, 29, 985–997. [Google Scholar] [CrossRef]
- Coll, M.; Piroddi, C.; Albouy, C.; Ben Rais Lasram, F.; Cheung, W.W.L.; Christensen, V.; Karpouzi, V.S.; Guilhaumon, F.; Mouillot, D.; Paleczny, M.; et al. The Mediterranean Sea under Siege: Spatial Overlap between Marine Biodiversity, Cumulative Threats and Marine Reserves: The Mediterranean Sea under Siege. Glob. Ecol. Biogeogr. 2012, 21, 465–480. [Google Scholar] [CrossRef]
- García-Meseguer, A.J.; Abellán, P.; Mirón-Gatón, J.M.; Botella-Cruz, M.; Guareschi, S.; Millán, A.; Velasco, J. Fine-Scale Niche Differences Allow the Co-Existence of Congeneric Aquatic Beetles in Supratidal Rockpools. Hydrobiologia 2023. [Google Scholar] [CrossRef]
- Struck, T.H.; Feder, J.L.; Bendiksby, M.; Birkeland, S.; Cerca, J.; Gusarov, V.I.; Kistenich, S.; Larsson, K.-H.; Liow, L.H.; Nowak, M.D.; et al. Finding Evolutionary Processes Hidden in Cryptic Species. Trends Ecol. Evol. 2018, 33, 153–163. [Google Scholar] [CrossRef]
- Bickford, D.; Lohman, D.J.; Sodhi, N.S.; Ng, P.K.L.; Meier, R.; Winker, K.; Ingram, K.K.; Das, I. Cryptic Species as a Window on Diversity and Conservation. Trends Ecol. Evol. 2007, 22, 148–155. [Google Scholar] [CrossRef]
- Pfenninger, M.; Schwenk, K. Cryptic Animal Species Are Homogeneously Distributed among Taxa and Biogeographical Regions. BMC Evol. Biol. 2007, 7, 121. [Google Scholar] [CrossRef]
- Adams, M.; Raadik, T.A.; Burridge, C.P.; Georges, A. Global Biodiversity Assessment and Hyper-Cryptic Species Complexes: More Than One Species of Elephant in the Room? Syst. Biol. 2014, 63, 518–533. [Google Scholar] [CrossRef]
- Whitham, T.G.; Bailey, J.K.; Schweitzer, J.A.; Shuster, S.M.; Bangert, R.K.; LeRoy, C.J.; Lonsdorf, E.V.; Allan, G.J.; DiFazio, S.P.; Potts, B.M.; et al. A Framework for Community and Ecosystem Genetics: From Genes to Ecosystems. Nat. Rev. Genet. 2006, 7, 510–523. [Google Scholar] [CrossRef]
- Moodley, Y.; Masello, J.F.; Cole, T.L.; Calderon, L.; Munimanda, G.K.; Thali, M.R.; Alderman, R.; Cuthbert, R.J.; Marin, M.; Massaro, M.; et al. Evolutionary Factors Affecting the Cross-species Utility of Newly Developed Microsatellite Markers in Seabirds. Mol. Ecol. Resour. 2015, 15, 1046–1058. [Google Scholar] [CrossRef]
- Fox, G.; Preziosi, R.F.; Antwis, R.E.; Benavides-Serrato, M.; Combe, F.J.; Harris, W.E.; Hartley, I.R.; Kitchener, A.C.; De Kort, S.R.; Nekaris, A.; et al. Multi-individual Microsatellite Identification: A Multiple Genome Approach to Microsatellite Design (MiMi). Mol. Ecol. Resour. 2019, 19, 1672–1680. [Google Scholar] [CrossRef]
- Selkoe, K.A.; Toonen, R.J. Microsatellites for Ecologists: A Practical Guide to Using and Evaluating Microsatellite Markers. Ecol. Lett. 2006, 9, 615–629. [Google Scholar] [CrossRef] [PubMed]
- Guichoux, E.; Lagache, L.; Wagner, S.; Chaumeil, P.; Léger, P.; Lepais, O.; Lepoittevin, C.; Malausa, T.; Revardel, E.; Salin, F.; et al. Current Trends in Microsatellite Genotyping. Mol. Ecol. Resour. 2011, 11, 591–611. [Google Scholar] [CrossRef]
- Rose, O.; Falush, D. A Threshold Size for Microsatellite Expansion. Mol. Biol. Evol. 1998, 15, 613–615. [Google Scholar] [CrossRef] [PubMed]
- Moore, S.S.; Sargeant, L.L.; King, T.J.; Mattick, J.S.; Georges, M.; Hetzel, D.J.S. The Conservation of Dinucleotide Microsatellites among Mammalian Genomes Allows the Use of Heterologous PCR Primer Pairs in Closely Related Species. Genomics 1991, 10, 654–660. [Google Scholar] [CrossRef] [PubMed]
- Brandes, M.; Albach, D.C.; Vogt, J.C.; Mayland-Quellhorst, E.; Mendieta-Leiva, G.; Golubic, S.; Palinska, K.A. Supratidal Extremophiles—Cyanobacterial Diversity in the Rock Pools of the Croatian Adria. Microb. Ecol. 2015, 70, 876–888. [Google Scholar] [CrossRef]
- Mirón-Gatón, J.M.; Botella-Cruz, M.; García-Meseguer, A.J.; Millán, A.; Velasco, J. Discordant Pattern between Realised and Fundamental Saline Niches in Two Supralittoral Ochthebius Species (Coleoptera: Hydraenidae). Ecol. Entomol. 2023, 48, 284–294. [Google Scholar] [CrossRef]
- Mirón-Gatón, J.M.; Botella-Cruz, M.; García-Meseguer, A.J.; Millán, A.; Velasco, J. Thermal Tolerance Differs between Co-Occurring Congeneric Beetle Species in Marine Supratidal Rockpools. Mar. Ecol. Prog. Ser. 2022, 681, 185–196. [Google Scholar] [CrossRef]
- Hawkins, S.J.; Pack, K.E.; Hyder, K.; Benedetti-Cecchi, L.; Jenkins, S.R. Rocky Shores as Tractable Test Systems for Experimental Ecology. J. Mar. Biol. Ass. 2020, 100, 1017–1041. [Google Scholar] [CrossRef]
- Ranta, E. Animal Communities in Rock Pools. Ann. Zool. Fenn. 1982, 19, 337–347. [Google Scholar]
- Denny, M.W.; Steven, S.D. Encyclopedia of Tidepools and Rocky Shores, 1st ed.; University of California Press: Berkeley, CA, USA, 2007; ISBN 978-0-520-93375-0. [Google Scholar]
- Margalef, R. Sobre la ecología de las larvas del mosquito Aëdes mariae. Publicaciones Inst. Biol. Apl. 1949, 6, 83–101. [Google Scholar]
- Villastrigo, A.; Hernando, C.; Millán, A. The Ochthebius (Coleoptera, Hydraenidae) from Western Palaearctic Supratidal Rockpools. Boln. Asoc. Esp. Ent. 2022, 4, 100–108. [Google Scholar]
- Villastrigo, A.; Bilton, D.T.; Abellán, P.; Millán, A.; Ribera, I.; Velasco, J. Cryptic Lineages, Cryptic Barriers: Historical Seascapes and Oceanic Fronts Drive Genetic Diversity in Supralittoral Rockpool Beetles (Coleoptera: Hydraenidae). Zool. J. Linn. Soc. 2022, 196, 740–756. [Google Scholar] [CrossRef]
- Palmer, J.D. Tidal Rhythms: The Clock Control of the Rhythmic Physiology of Marine Organisms. Biol. Rev. 1973, 48, 377–418. [Google Scholar] [CrossRef]
- Newell, R.C. Adaptations to Intertidal Life. In Adaptation to Environment; Elsevier: Amsterdam, The Netherlands, 1976; pp. 1–82. ISBN 978-0-408-70778-7. [Google Scholar]
- Hanski, I.; Gaggiotti, O. Metapopulation Biology: Past, Present, and Future. In Ecology, Genetics and Evolution of Metapopulations; Elsevier: Amsterdam, The Netherlands, 2004; pp. 3–22. ISBN 978-0-12-323448-3. [Google Scholar]
- Bretman, A.; Rodríguez-Muñoz, R.; Walling, C.; Slate, J.; Tregenza, T. Fine-Scale Population Structure, Inbreeding Risk and Avoidance in a Wild Insect Population. Mol. Ecol. 2011, 20, 3045–3055. [Google Scholar] [CrossRef]
- Sabatelli, S.; Audisio, P.; Antonini, G.; Solano, E.; Martinoli, A.; Trizzino, M. Molecular Ecology and Phylogenetics of the Water Beetle Genus Ochthebius Revealed Multiple Independent Shifts to Marine Rockpools Lifestyle. Zool. Scr. 2016, 45, 175–186. [Google Scholar] [CrossRef]
- Sabatelli, S.; Ruspantini, P.; Cardoli, P.; Audisio, P. Underestimated Diversity: Cryptic Species and Phylogenetic Relationships in the Subgenus Cobalius (Coleoptera: Hydraenidae) from Marine Rockpools. Mol. Phylogenetics Evol. 2021, 163, 107243. [Google Scholar] [CrossRef]
- Villastrigo, A.; Hernando, C.; Millán, A.; Ribera, I. The Neglected Diversity of the Ochthebius Fauna from Eastern Atlantic and Central and Western Mediterranean Coastal Rockpools (Coleoptera, Hydraenidae). Org. Divers. Evol. 2020, 20, 785–801. [Google Scholar] [CrossRef]
- Mejjad, N.; Rossi, A.; Pavel, A.B. The Coastal Tourism Industry in the Mediterranean: A Critical Review of the Socio-Economic and Environmental Pressures&Impacts. Tour. Manag. Perspect. 2022, 44, 101007. [Google Scholar] [CrossRef]
- Villastrigo, A.; Arribas, P.; Ribera, I. Irreversible Habitat Specialization Does Not Constrain Diversification in Hypersaline Water Beetles. Mol. Ecol. 2020, 29, 3637–3648. [Google Scholar] [CrossRef]
- Andrews, S. FASTQC. A Quality Control Tool for High Throughput Sequence Data. 2010. Available online: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 16 September 2023).
- Edgar, R.C. Search and Clustering Orders of Magnitude Faster than BLAST. Bioinformatics 2010, 26, 2460–2461. [Google Scholar] [CrossRef]
- Benson, G. Tandem Repeats Finder: A Program to Analyze DNA Sequences. Nucleic Acids Res. 1999, 27, 573–580. [Google Scholar] [CrossRef] [PubMed]
- Untergasser, A.; Cutcutache, I.; Koressaar, T.; Ye, J.; Faircloth, B.C.; Remm, M.; Rozen, S.G. Primer3—New Capabilities and Interfaces. Nucleic Acids Res. 2012, 40, e115. [Google Scholar] [CrossRef] [PubMed]
- Peakall, R.; Smouse, P.E. GenAlEx 6.5: Genetic Analysis in Excel. Population Genetic Software for Teaching and Research—An Update. Bioinformatics 2012, 28, 2537–2539. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing. 2023. Available online: https://www.R-project.org (accessed on 16 September 2023).
- Pritchard, J.K.; Stephens, M.; Donnelly, P. Inference of Population Structure Using Multilocus Genotype Data. Genetics 2000, 155, 945–959. [Google Scholar] [CrossRef]
- Earl, D.A.; vonHoldt, B.M. Structure Harvester: A Website and Program for Visualizing Structure Output and Implementing the Evanno Method. Conserv. Genet. Resour. 2012, 4, 359–361. [Google Scholar] [CrossRef]
- Evanno, G.; Regnaut, S.; Goudet, J. Detecting the Number of Clusters of Individuals Using the Software Structure: A Simulation Study. Mol. Ecol. 2005, 14, 2611–2620. [Google Scholar] [CrossRef] [PubMed]
- Jakobsson, M.; Rosenberg, N.A. CLUMPP: A Cluster Matching and Permutation Program for Dealing with Label Switching and Multimodality in Analysis of Population Structure. Bioinformatics 2007, 23, 1801–1806. [Google Scholar] [CrossRef]
- Criscuolo, N.G.; Angelini, C. StructuRly: A Novel Shiny App to Produce Comprehensive, Detailed and Interactive Plots for Population Genetic Analysis. PLoS ONE 2020, 15, e0229330. [Google Scholar] [CrossRef]
- Watts, C.H.S.; Villastrigo, A.; Langille, B.L.; Stringer, D.N.; Bradford, T.M.; Humphreys, W.F.; Austin, A.D.; Balke, M.; Cooper, S.J.B. Phylogenetic Placement and Description of Ngaliadessus humphreysi Gen. et Sp. Nov., Watts&Villastrigo (Coleoptera: Dytiscidae), a Subterranean Diving Beetle from the Ngalia Basin in Central Australia. Austral Entomol. 2023, 62, 300–309. [Google Scholar] [CrossRef]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. Fastp: An Ultra-Fast All-in-One FASTQ Preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Prjibelski, A.; Antipov, D.; Meleshko, D.; Lapidus, A.; Korobeynikov, A. Using SPAdes De Novo Assembler. Curr. Protoc. Bioinform. 2020, 70, e102. [Google Scholar] [CrossRef] [PubMed]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and Applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef] [PubMed]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious Basic: An Integrated and Extendable Desktop Software Platform for the Organization and Analysis of Sequence Data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef]
- Donath, A.; Jühling, F.; Al-Arab, M.; Bernhart, S.H.; Reinhardt, F.; Stadler, P.F.; Middendorf, M.; Bernt, M. Improved Annotation of Protein-Coding Genes Boundaries in Metazoan Mitochondrial Genomes. Nucleic Acids Res. 2019, 47, 10543–10552. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed]
- Rico, C.; Normandeau, E.; Dion-Côté, A.-M.; Rico, M.I.; Côté, G.; Bernatchez, L. Combining Next-Generation Sequencing and Online Databases for Microsatellite Development in Non-Model Organisms. Sci. Rep. 2013, 3, 3376. [Google Scholar] [CrossRef]
- Schoebel, C.N.; Brodbeck, S.; Buehler, D.; Cornejo, C.; Gajurel, J.; Hartikainen, H.; Keller, D.; Leys, M.; Říčanová, Š.; Segelbacher, G.; et al. Lessons Learned from Microsatellite Development for Nonmodel Organisms Using 454 Pyrosequencing. J. Evol. Biol. 2013, 26, 600–611. [Google Scholar] [CrossRef]
- Hu, P.; Huang, C.-L.; Luo, M.-X.; Hsu, Y.-F.; Wang, R.-J. Development and Characterization of Novel Microsatellite Markers in Chestnut Tiger Butterfly Parantica sita (Lepidoptera: Nymphalidae) Using next-Generation Sequencing. Appl. Entomol. Zool. 2020, 55, 281–286. [Google Scholar] [CrossRef]
- Mahalle, R.; Bosamia, T.; Chakravarty, S.; Srivastava, K.; Meena, R.; Kadam, U.; Srivastava, C. De Novo Mining and Validating Novel Microsatellite Markers to Assess Genetic Diversity in Maruca vitrata (F.), a Legume Pod Borer. Genes 2023, 14, 1433. [Google Scholar] [CrossRef] [PubMed]
- Marcy-Quay, B.; Wilson, C.C.; Osborne, C.A.; Marsden, J.E. Optimization of an Amplicon Sequencing-based Microsatellite Panel and Protocol for Stock Identification and Kinship Inference of Lake Trout (Salvelinus namaycush). Ecol. Evol. 2023, 13, e10020. [Google Scholar] [CrossRef]
- Mishra, G.; Meena, R.K.; Kant, R.; Pandey, S.; Ginwal, H.S.; Bhandari, M.S. Genome-Wide Characterization Leading to Simple Sequence Repeat (SSR) Markers Development in Shorea robusta. Funct. Integr. Genom. 2023, 23, 51. [Google Scholar] [CrossRef] [PubMed]
- Oreshkova, N.V.; Bondar, E.I.; Sharov, V.V.; Dhungana, S.P.; Gailing, O.; Krutovsky, K.V. Population Genetic Variation of Microsatellite Markers Developed for Siberian Fir (Abies sibirica Ledeb.) and European Silver Fir (Abies alba Mill.) Using Whole Genome Sequencing Data. Plant Genet. Resour. 2023, 21, 149–158. [Google Scholar] [CrossRef]
- Liljegren, M.M.; De Muinck, E.J.; Trosvik, P. Microsatellite Length Scoring by Single Molecule Real Time Sequencing—Effects of Sequence Structure and PCR Regime. PLoS ONE 2016, 11, e0159232. [Google Scholar] [CrossRef] [PubMed]
- Corner, S.; Yuzbasiyan-Gurkan, V.; Agnew, D.; Venta, P.J. Development of a 12-Plex of New Microsatellite Markers Using a Novel Universal Primer Method to Evaluate the Genetic Diversity of Jaguars (Panthera onca) from North American Zoological Institutions. Conserv. Genet. Resour. 2019, 11, 487–497. [Google Scholar] [CrossRef]
- Kostro-Ambroziak, A.; Siekiera, A.; Czajkowska, M.; Pomorski, J.J.; Panagiotopoulou, H. Development of Microsatellite Loci and Optimization of a Multiplex Assay for Latibulus argiolus (Hymenoptera: Ichneumonidae), the Specialized Parasitoid of Paper Wasps. Sci. Rep. 2020, 10, 16068. [Google Scholar] [CrossRef] [PubMed]
- Chistiakov, D.A.; Hellemans, B.; Volckaert, F.A.M. Microsatellites and Their Genomic Distribution, Evolution, Function and Applications: A Review with Special Reference to Fish Genetics. Aquaculture 2006, 255, 1–29. [Google Scholar] [CrossRef]
- Blondin, L.; Badisco, L.; Pagès, C.; Foucart, A.; Risterucci, A.; Bazelet, C.S.; Vanden Broeck, J.; Song, H.; Ould Ely, S.; Chapuis, M. Characterization and Comparison of Microsatellite Markers Derived from Genomic and Expressed Libraries for the Desert Locust. J. Appl. Entomol. 2013, 137, 673–683. [Google Scholar] [CrossRef]
- Wilson, A.C.C.; Massonnet, B.; Simon, J.-C.; Prunier-Leterme, N.; Dolatti, L.; Llewellyn, K.S.; Figueroa, C.C.; Ramirez, C.C.; Blackman, R.L.; Estoup, A.; et al. Cross-Species Amplification of Microsatellite Loci in Aphids: Assessment and Application: PRIMER NOTE. Mol. Ecol. Notes 2004, 4, 104–109. [Google Scholar] [CrossRef]
- Mikac, K.M. Isolation and Characterization of the First Microsatellite Loci from the Order Psocoptera in the Economically Important Pest Insect Liposcelis Decolor (Pearman) and Cross-Species Amplification: PRIMER NOTE. Mol. Ecol. Notes 2006, 6, 1102–1104. [Google Scholar] [CrossRef]
- Meglécz, E.; Anderson, S.J.; Bourguet, D.; Butcher, R.; Caldas, A.; Cassel-Lundhagen, A.; d′Acier, A.C.; Dawson, D.A.; Faure, N.; Fauvelot, C.; et al. Microsatellite Flanking Region Similarities among Different Loci within Insect Species. Insect Mol. Biol. 2007, 16, 175–185. [Google Scholar] [CrossRef]
- Weng, Y.; Azhaguvel, P.; Michels, G.J.; Rudd, J.C. Cross-species Transferability of Microsatellite Markers from Six Aphid (Hemiptera: Aphididae) Species and Their Use for Evaluating Biotypic Diversity in Two Cereal Aphids. Insect Mol. Biol. 2007, 16, 613–622. [Google Scholar] [CrossRef] [PubMed]
- Galbusera, P. Cross-Species Amplification of Microsatellite Primers in Passerine Birds. Conserv. Genet. 2000, 1, 163–168. [Google Scholar] [CrossRef]
- Hinomoto, N.; Higaki, T.; Noda, T. Development of Microsatellite Markers for the Minute Pirate Bug Orius sauteri (Poppius), and Their Cross-Species Amplification in O. minutus (L.) and O. strigicollis (Poppius) (Heteroptera: Anthocoridae). Appl. Entomol. Zool. 2009, 44, 635–642. [Google Scholar] [CrossRef][Green Version]
- Shikano, T.; Ramadevi, J.; Shimada, Y.; Merilä, J. Utility of Sequenced Genomes for Microsatellite Marker Development in Non-Model Organisms: A Case Study of Functionally Important Genes in Nine-Spined Sticklebacks (Pungitius pungitius). BMC Genom. 2010, 11, 334. [Google Scholar] [CrossRef] [PubMed]
- Villastrigo, A.; Jäch, M.A.; Cardoso, A.; Valladares, L.F.; Ribera, I. A Molecular Phylogeny of the Tribe Ochthebiini (Coleoptera, Hydraenidae, Ochthebiinae). Syst. Entomol. 2019, 44, 273–288. [Google Scholar] [CrossRef]
- Urbanelli, S.; Sallicandro, P.; De Vito, E.; Colonnelli, E.; Bullini, L. Molecular Reexamination of the Taxonomy of Ochthebius (Calobius) (Coleoptera: Hydraenidae) from the Mediterranean and Macaronesian Regions. Ann. Entomol. Soc. Am. 1996, 89, 623–636. [Google Scholar] [CrossRef]
- Audisio, P.; Trizzino, M.; De Biase, A.; Rossetti, G.; Mancini, E.; Antonini, G. Molecular and Morphological Evidence of a New Sibling Species of Calobius (Coleoptera: Hydraenidae) of the C. quadricollis Complex from Peninsular Italy. Ital. J. Zool. 2010, 77, 29–37. [Google Scholar] [CrossRef]
- Urbanelli, S. Genetic Divergence and Reproductive Isolation in the Ochthebius (Calobius) Complex (Coleoptera: Hydraenidae). Heredity 2002, 88, 333–341. [Google Scholar] [CrossRef][Green Version]
- Goudet, J.; Perrin, N.; Waser, P. Tests for Sex-biased Dispersal Using Bi-parentally Inherited Genetic Markers. Mol. Ecol. 2002, 11, 1103–1114. [Google Scholar] [CrossRef]
- Prugnolle, F.; De Meeus, T. Inferring Sex-Biased Dispersal from Population Genetic Tools: A Review. Heredity 2002, 88, 161–165. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| O. lejolisii, O. subinteger and O. celatus | |||||||||
| Locus | Primer Sequences 5′-3′ | Motif | Allele Size Range (bp) | No. of Alleles | Multiplex | Dye | Final Primer Concentration (μM) | Annealing Temp. °C | |
| os_37067 | F: R: | GGGAGCGGTGCATATTGTTG ACAAAGTGATAAAAAGCGAAAAGC | (TCT)7 | 220–241 | 4 | 1 | FAM | 0.3 | 60 |
| os_120990 | F: R: | TCGGAAAGGTGCTACTAACAAAC ATAATTGTCACTTGGACGACAG | (TA)13 | 135–208 | 5 | 1 | ATTO550 | 0.3 | 60 |
| os_890215 | F: R: | CACAGGTCGGGGCTAAAATG TCGAAAACTTTAACCCAAGATTGC | (ATA)8 | 138–144 | 3 | 1 | FAM | 0.3 | 60 |
| os_1099692 | F: R: | TGCCACTTGCTCGAAGAAAC TCTCGTAAATTTTGTAGAGTTGGGG | (TTA)8 | 173–186 | 5 | 1 | ATTO565 | 0.3 | 60 |
| os_1225179 | F: R: | AACAAAAGGCGCTTATGACG AGAACAATTACGTTCTACAATGTGC | (AT)27 | 125–130 | 2 | 1 | ATTO532 | 0.3 | 60 |
| os_70525 | F: R: | ACAACAATCATGGAGGTCCG CGTAGGTCGAAAACTAATGTCCTC | (AAT)9 | 243–261 | 6 | 2 | FAM | 0.3 | 60 |
| os_336684 | F: R: | AGTTTCCTTACTTATCAAATAAAAGCG AGTCTGAAAAGCCCACTTGC | (AT)24 | 146–211 | 5 | 2 | ATTO565 | 0.3 | 60 |
| os_712676 | F: R: | ATTACAGTGCGTCTGAGTGC AGACAACTTATTCCAACGAAGC | (AAT)8 | 85–97 | 4 | 2 | ATTO532 | 0.3 | 60 |
| os_866755 | F: R: | CACCGATTGTATCAGCAGCC TGAACAAATAAAGTGCGCTTCTTC | (TAA)8 | 146–156 | 4 | 2 | ATTO550 | 0.3 | 60 |
| os_1099017 | F: R: | AAATTAAAATTGGGATTTTCAAGTGC GGATGTGTATCAAAAATACTCTCTAGG | (TA)25 | 139–142 | 2 | 2 | FAM | 0.3 | 60 |
| O. quadricollis | |||||||||
| Locus | Primer Sequences 5′-3′ | Motif | Allele Size Range (bp) | No. of Alleles | Multiplex | Dye | Final Primer Concentration (μM) | Annealing Temp. °C | |
| Oq_116301 | F: R: | AGTCATGTTTGGTTAATGGATGTC ACTACAGTGAGTGACGTAAGC | (TTA)9 | 188–200 | 3 | 3 | ATTO550 | 0.3 | 60 |
| Oq_117536 | F: R: | ACTCGGTGTTCCACAGATCG CATCAAGCCTTCTTCAGACCG | (TTA)9 | 207–217 | 3 | 3 | FAM | 0.3 | 60 |
| Oq_1610909 | F: R: | CGACCCTCTTCAATACCAAGC GTCCACCAAAGAACGAGGAC | (ATT)9 | 222–231 | 4 | 3 | ATTO565 | 0.5 | 60 |
| Oq_2015046 | F: R: | TCCGTTTGAGAGTAGCACCC GGGACGGTATATGGGGATGG | (AGT)10 | 201–210 | 4 | 3 | ATTO532 | 0.3 | 60 |
| Oq_2184903 | F: R: | ATGTTTGGACCGCCATTGTG TGTTAGTTTGATGATTTTCTTCGAC | (ATTA)7 | 118–143 | 2 | 3 | ATTO565 | 0.5 | 60 |
| Oq_122989 | F: R: | ATAAATGGTGAGCAAGTAGCG ATATGGTACAACGGAGGCGG | (TAT)8 | 205–212 | 3 | 4 | ATTO565 | 0.3 | 60 |
| Oq_743481 | F: R: | CACTCCAATTTGAACTACAATAAGTCC AGCATCCTCTGGTGATGTCC | (TAT)8 | 232–247 | 6 | 4 | FAM | 0.3 | 60 |
| Oq_1061334 | F: R: | TGTTTCCTAAGTGCTTGTGCG ACTGGTTACATTCAGCAAACTG | (TAA)10 | 170–228 | 5 | 4 | ATTO550 | 0.5 | 60 |
| Oq_1457302 | F: R: | CTACATCCTGATCGGAGCCC CACCATCCAGAACACCAAGC | (ATA)9 | 180–194 | 6 | 4 | FAM | 0.3 | 60 |
| Oq_2353236 | F: R: | AACACTCCTAGTGCTCGCTC ATCTGGAGCTCATATCCGCC | (TTA)8 | 216–225 | 4 | 4 | ATTO532 | 0.3 | 60 |
| O. celatus | O. subinteger | O. lejolisii | Total | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Locus | NA | NE | HO | HE | NA | NE | HO | HE | NA | NE | HO | HE | N | NA | NE | HO | HE |
| os_23931 | 0 | 0.000 | 0.000 | 0.000 | 2 | 1.385 | 0.333 | 0.278 | 0 | 0.000 | 0.000 | 0.000 | 3 | 2 | 1.385 | 0.333 | 0.278 |
| os_37067 * | 2 | 2.000 | 1.000 | 0.500 | 1 | 1.000 | 0.000 | 0.000 | 2 | 2.000 | 1.000 | 0.500 | 10 | 4 | 2.597 | 0.700 | 0.615 |
| os_70525 * | 4 | 2.778 | 0.800 | 0.640 | 2 | 1.385 | 0.333 | 0.278 | 1 | 1.000 | 0.000 | 0.000 | 10 | 6 | 4.545 | 0.500 | 0.780 |
| os_120990 * | 1 | 1.000 | 0.000 | 0.000 | 4 | 3.600 | 1.000 | 0.722 | 0 | 0.000 | 0.000 | 0.000 | 7 | 5 | 2.649 | 0.429 | 0.622 |
| os_163989 | 0 | 0.000 | 0.000 | 0.000 | 2 | 1.385 | 0.333 | 0.278 | 0 | 0.000 | 0.000 | 0.000 | 3 | 2 | 1.385 | 0.333 | 0.278 |
| os_166922 | 0 | 0.000 | 0.000 | 0.000 | 3 | 2.571 | 0.667 | 0.611 | 2 | 1.600 | 0.500 | 0.375 | 5 | 5 | 4.167 | 0.600 | 0.760 |
| os_198491 | 0 | 0.000 | 0.000 | 0.000 | 4 | 3.600 | 0.333 | 0.722 | 0 | 0.000 | 0.000 | 0.000 | 3 | 4 | 3.600 | 0.333 | 0.722 |
| os_258100 | 0 | 0.000 | 0.000 | 0.000 | 4 | 3.000 | 1.000 | 0.667 | 0 | 0.000 | 0.000 | 0.000 | 3 | 4 | 3.000 | 1.000 | 0.667 |
| os_265699 | 0 | 0.000 | 0.000 | 0.000 | 4 | 3.600 | 0.667 | 0.722 | 2 | 1.600 | 0.500 | 0.375 | 5 | 6 | 5.000 | 0.600 | 0.800 |
| os_314710 | 0 | 0.000 | 0.000 | 0.000 | 2 | 1.385 | 0.333 | 0.278 | 0 | 0.000 | 0.000 | 0.000 | 3 | 2 | 1.385 | 0.333 | 0.278 |
| os_336684 * | 1 | 1.000 | 0.000 | 0.000 | 4 | 3.600 | 0.333 | 0.722 | 0 | 0.000 | 0.000 | 0.000 | 6 | 5 | 3.130 | 0.167 | 0.681 |
| os_458175 | 0 | 0.000 | 0.000 | 0.000 | 3 | 2.000 | 0.667 | 0.500 | 0 | 0.000 | 0.000 | 0.000 | 3 | 3 | 2.000 | 0.667 | 0.500 |
| os_532182 | 0 | 0.000 | 0.000 | 0.000 | 3 | 2.571 | 1.000 | 0.611 | 0 | 0.000 | 0.000 | 0.000 | 3 | 3 | 2.571 | 1.000 | 0.611 |
| os_537494 | 0 | 0.000 | 0.000 | 0.000 | 2 | 2.000 | 1.000 | 0.500 | 0 | 0.000 | 0.000 | 0.000 | 3 | 2 | 2.000 | 1.000 | 0.500 |
| os_712676 * | 1 | 1.000 | 0.000 | 0.000 | 2 | 1.800 | 0.667 | 0.444 | 1 | 1.000 | 0.000 | 0.000 | 10 | 3 | 1.852 | 0.200 | 0.460 |
| os_858059 | 0 | 0.000 | 0.000 | 0.000 | 3 | 2.571 | 0.333 | 0.611 | 0 | 0.000 | 0.000 | 0.000 | 3 | 3 | 2.571 | 0.333 | 0.611 |
| os_866755 * | 2 | 1.220 | 0.200 | 0.180 | 2 | 1.385 | 0.333 | 0.278 | 2 | 1.600 | 0.500 | 0.375 | 10 | 4 | 2.740 | 0.300 | 0.635 |
| os_890215 * | 1 | 1.000 | 0.000 | 0.000 | 3 | 2.571 | 0.333 | 0.611 | 0 | 0.000 | 0.000 | 0.000 | 8 | 3 | 1.471 | 0.125 | 0.320 |
| os_897615 | 0 | 0.000 | 0.000 | 0.000 | 2 | 2.000 | 1.000 | 0.500 | 1 | 1.000 | 0.000 | 0.000 | 4 | 2 | 1.882 | 0.750 | 0.469 |
| os_898357 | 0 | 0.000 | 0.000 | 0.000 | 1 | 1.000 | 0.000 | 0.000 | 0 | 0.000 | 0.000 | 0.000 | 3 | 1 | 1.000 | 0.000 | 0.000 |
| os_953020 | 0 | 0.000 | 0.000 | 0.000 | 3 | 2.667 | 1.000 | 0.625 | 1 | 1.000 | 0.000 | 0.000 | 3 | 4 | 3.600 | 0.667 | 0.722 |
| os_1099017 * | 2 | 1.923 | 0.800 | 0.480 | 0 | 0.000 | 0.000 | 0.000 | 0 | 0.000 | 0.000 | 0.000 | 5 | 2 | 1.923 | 0.800 | 0.480 |
| os_1099692 * | 3 | 1.515 | 0.400 | 0.340 | 3 | 3.000 | 1.000 | 0.667 | 1 | 1.000 | 0.000 | 0.000 | 10 | 5 | 3.922 | 0.500 | 0.745 |
| os_1225179 * | 1 | 1.000 | 0.000 | 0.000 | 1 | 1.000 | 0.000 | 0.000 | 1 | 1.000 | 0.000 | 0.000 | 10 | 2 | 1.724 | 0.000 | 0.420 |
| Locus | N | NA | NE | HO | HE |
|---|---|---|---|---|---|
| Oq_116301 * | 7 | 3 | 1.342 | 0.286 | 0.255 |
| Oq_117536 * | 7 | 3 | 1.342 | 0.286 | 0.255 |
| Oq_122989 * | 7 | 3 | 2.800 | 0.714 | 0.643 |
| Oq_129897 | 7 | 5 | 2.227 | 0.714 | 0.551 |
| Oq_222765 | 7 | 3 | 2.000 | 0.714 | 0.500 |
| Oq_235636 | 7 | 4 | 2.970 | 0.571 | 0.663 |
| Oq_349914 | 7 | 2 | 1.508 | 0.429 | 0.337 |
| Oq_467933 | 7 | 2 | 1.324 | 0.286 | 0.245 |
| Oq_475499 | 7 | 6 | 4.083 | 0.857 | 0.755 |
| Oq_556301 | 7 | 2 | 2.000 | 0.714 | 0.500 |
| Oq_558655 | 7 | 2 | 1.690 | 0.000 | 0.408 |
| Oq_673482 | 7 | 2 | 1.153 | 0.143 | 0.133 |
| Oq_730507 | 7 | 5 | 3.379 | 0.571 | 0.704 |
| Oq_743481 * | 7 | 4 | 1.849 | 0.571 | 0.459 |
| Oq_793953 | 7 | 2 | 1.153 | 0.143 | 0.133 |
| Oq_801691 | 7 | 2 | 1.960 | 0.571 | 0.490 |
| Oq_857784 | 7 | 2 | 1.508 | 0.429 | 0.337 |
| Oq_958857 | 7 | 3 | 2.085 | 0.286 | 0.520 |
| Oq_1061334 * | 7 | 5 | 3.920 | 0.857 | 0.745 |
| Oq_1151835 | 7 | 6 | 3.769 | 0.571 | 0.735 |
| Oq_1217367 | 7 | 3 | 1.342 | 0.286 | 0.255 |
| Oq_1227338 | 7 | 6 | 4.900 | 1.000 | 0.796 |
| Oq_1237963 | 7 | 4 | 2.882 | 0.286 | 0.653 |
| Oq_1388647 | 7 | 2 | 1.153 | 0.143 | 0.133 |
| Oq_1457302 * | 7 | 6 | 3.769 | 1.000 | 0.735 |
| Oq_1489818 | 7 | 5 | 2.649 | 0.857 | 0.622 |
| Oq_1493143 | 6 | 6 | 4.800 | 0.167 | 0.792 |
| Oq_1555513 | 7 | 3 | 2.085 | 0.714 | 0.520 |
| Oq_1557685 | 7 | 3 | 2.279 | 0.571 | 0.561 |
| Oq_1610909 * | 7 | 4 | 3.379 | 1.000 | 0.704 |
| Oq_1708927 | 7 | 2 | 1.153 | 0.143 | 0.133 |
| Oq_1714988 | 7 | 2 | 1.153 | 0.143 | 0.133 |
| Oq_1750394 | 7 | 5 | 3.379 | 0.571 | 0.704 |
| Oq_1763578 | 7 | 2 | 1.960 | 0.571 | 0.490 |
| Oq_1821745 | 7 | 2 | 1.153 | 0.143 | 0.133 |
| Oq_1879905 | 7 | 3 | 1.782 | 0.571 | 0.439 |
| Oq_1966394 | 7 | 2 | 1.153 | 0.143 | 0.133 |
| Oq_2006473 | 7 | 2 | 1.960 | 0.286 | 0.490 |
| Oq_2010748 | 7 | 6 | 5.158 | 0.714 | 0.806 |
| Oq_2015046 * | 7 | 4 | 3.161 | 0.857 | 0.684 |
| Oq_2044728 | 7 | 3 | 2.970 | 0.143 | 0.663 |
| Oq_2057906 | 7 | 2 | 1.849 | 0.429 | 0.459 |
| Oq_2144776 | 7 | 4 | 2.970 | 0.286 | 0.663 |
| Oq_2145901 | 7 | 2 | 1.153 | 0.143 | 0.133 |
| Oq_2184903 * | 7 | 2 | 1.508 | 0.143 | 0.337 |
| Oq_2254408 | 7 | 3 | 2.882 | 0.571 | 0.653 |
| Oq_2353236 * | 7 | 4 | 2.882 | 0.714 | 0.653 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
García-Meseguer, A.J.; Villastrigo, A.; Mirón-Gatón, J.M.; Millán, A.; Velasco, J.; Muñoz, I. Novel Microsatellite Loci, Cross-Species Validation of Multiplex Assays, and By-Catch Mitochondrial Genomes on Ochthebius Beetles from Supratidal Rockpools. Insects 2023, 14, 881. https://doi.org/10.3390/insects14110881
García-Meseguer AJ, Villastrigo A, Mirón-Gatón JM, Millán A, Velasco J, Muñoz I. Novel Microsatellite Loci, Cross-Species Validation of Multiplex Assays, and By-Catch Mitochondrial Genomes on Ochthebius Beetles from Supratidal Rockpools. Insects. 2023; 14(11):881. https://doi.org/10.3390/insects14110881
Chicago/Turabian StyleGarcía-Meseguer, Antonio José, Adrián Villastrigo, Juana María Mirón-Gatón, Andrés Millán, Josefa Velasco, and Irene Muñoz. 2023. "Novel Microsatellite Loci, Cross-Species Validation of Multiplex Assays, and By-Catch Mitochondrial Genomes on Ochthebius Beetles from Supratidal Rockpools" Insects 14, no. 11: 881. https://doi.org/10.3390/insects14110881
APA StyleGarcía-Meseguer, A. J., Villastrigo, A., Mirón-Gatón, J. M., Millán, A., Velasco, J., & Muñoz, I. (2023). Novel Microsatellite Loci, Cross-Species Validation of Multiplex Assays, and By-Catch Mitochondrial Genomes on Ochthebius Beetles from Supratidal Rockpools. Insects, 14(11), 881. https://doi.org/10.3390/insects14110881