
Characterization of the Complete Mitochondrial Genome of Eight Diurnal Hawkmoths (Lepidoptera: Sphingidae): New Insights into the Origin and Evolution of Diurnalism in Sphingids
 ,
,  and
and 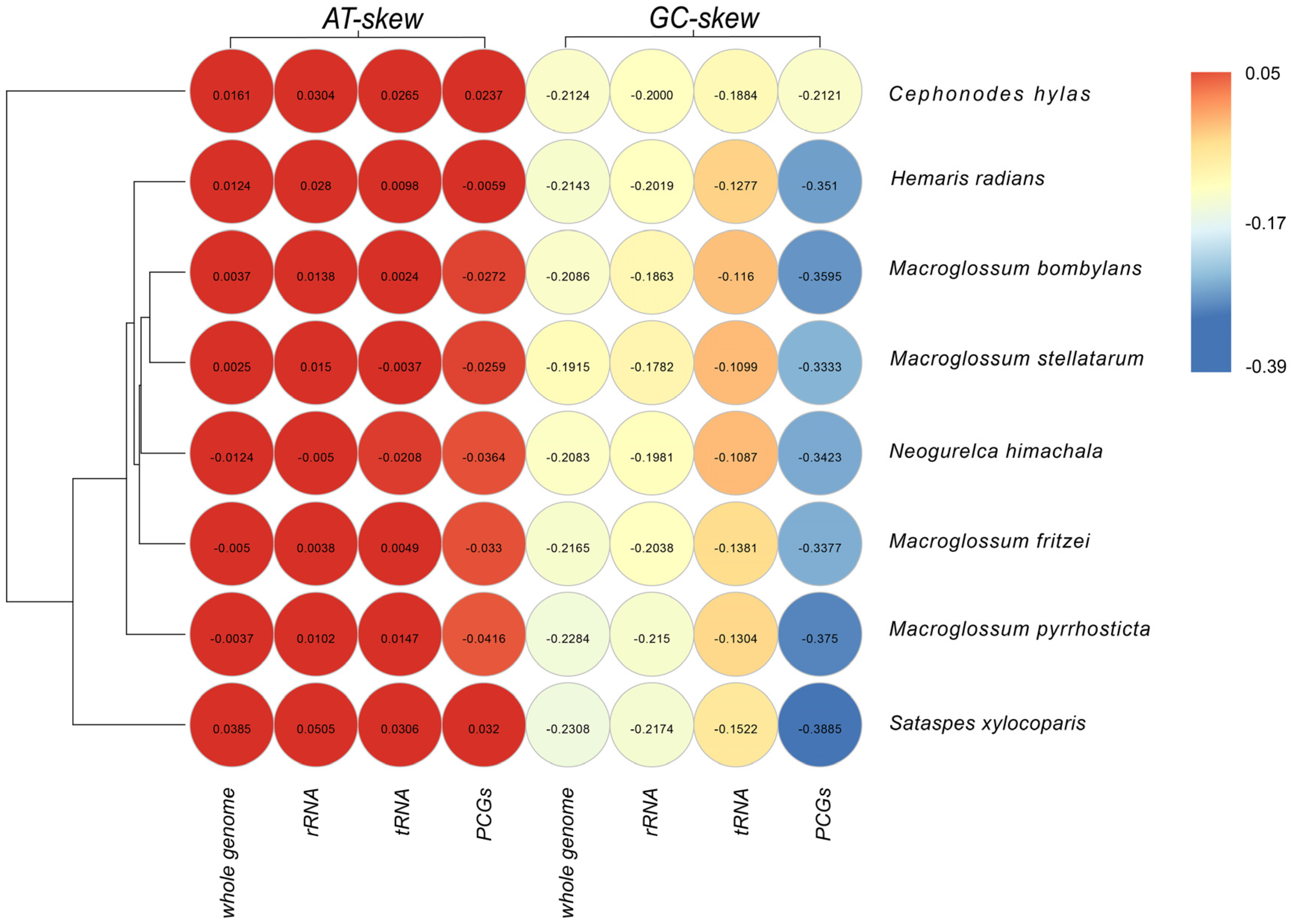{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
2. Material and Methods
2.1. Sampling and DNA Extraction
2.2. Sequencing and Assembly
2.3. Mitochondrial Genome Annotation and Analysis
2.4. Phylogenetic Analysis
2.5. Ancestral Character State Reconstruction
2.6. Divergence Time Estimation
3. Results
3.1. Genome Structure and Organization
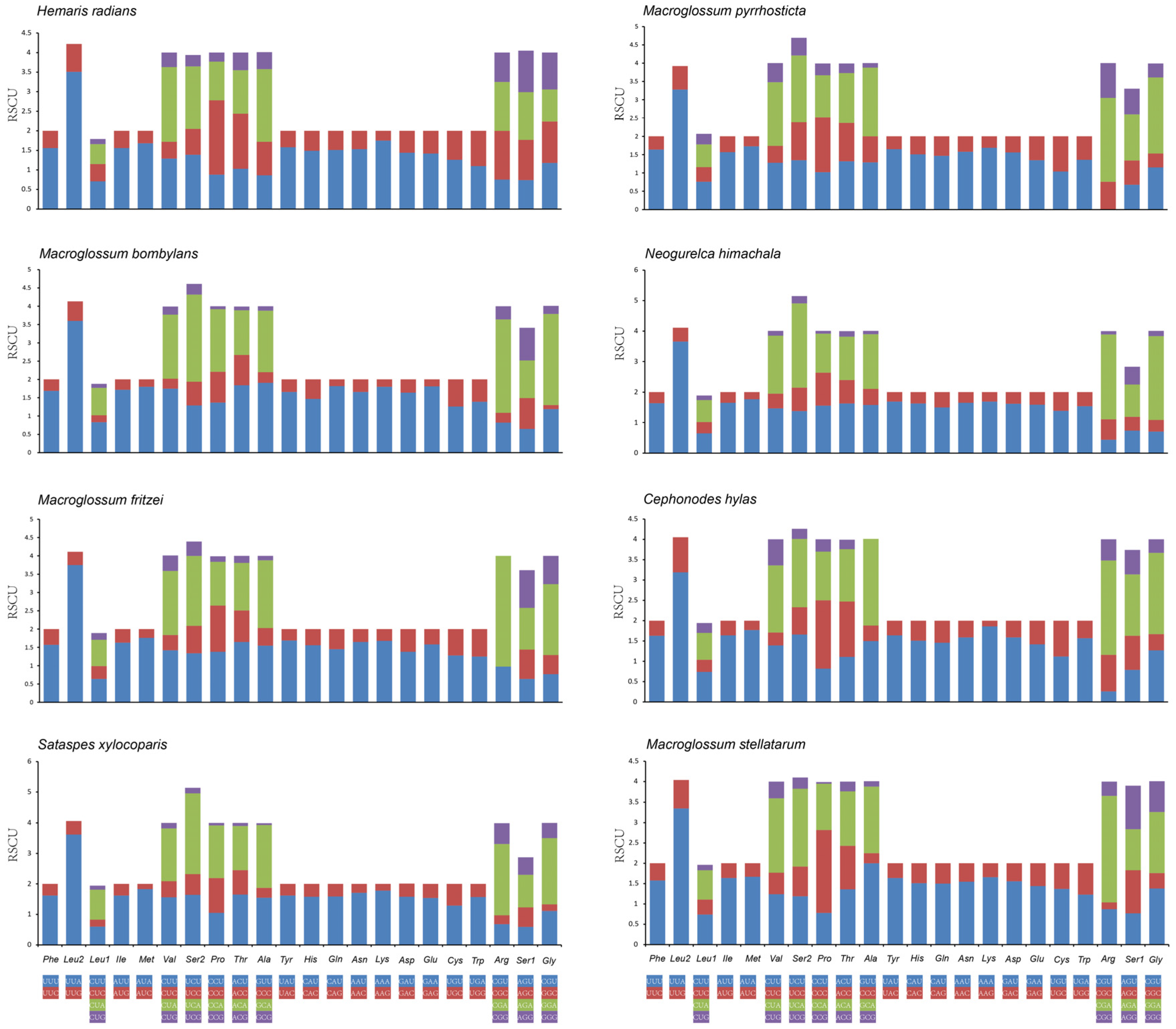
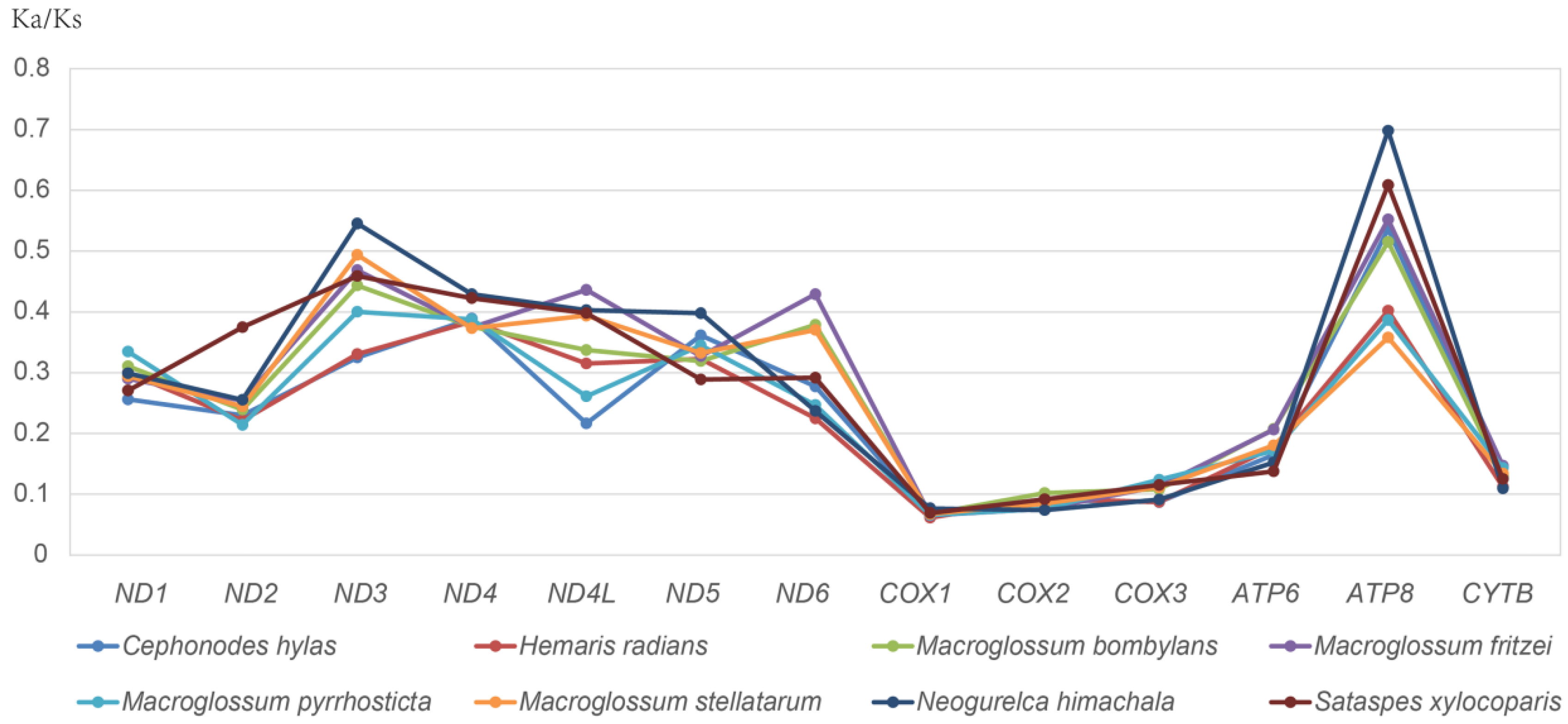
3.2. Protein-Coding Genes (PCGs)
3.3. Transfer and Ribosomal RNA Genes
3.4. Intergenic Spacers and Overlapping Sequences
3.5. Control Region
3.6. Phylogenetic Analyses
3.7. Divergence Time Estimation and Reconstruction of the Origin of Diurnalism
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Keisuke, N.; Keiich, H.; Suzuka, N.; Yoshinori, S. Rearing Theretra oldenlandiae (Lepidoptera: Sphingidae) Larvae on an Artificial Diet. J. Insect Sci. 2019, 19, 1–5. [Google Scholar] [CrossRef]
- Stöckl, A.L.; Kelber, A. Fuelling on the wing: Sensory ecology of hawkmoth foraging. J. Comp. Physiol. A Neuroethol. Sens. Neural Behav. Physiol. 2019, 205, 399–413. [Google Scholar] [CrossRef]
- Kelber, A.; Balkenius, A.; Warrant, E.J. Colour Vision in Diurnal and Nocturnal Hawkmoths. Integr. Comp. Biol. 2003, 43, 571–579. [Google Scholar] [CrossRef]
- Deora, T.; Ahmed, M.A.; Brunton, B.W.; Daniel, T.L. Learning to feed in the dark: How light level influences feeding in the hawkmoth Manduca sexta. Biol. Lett. 2021, 17, 20210320. [Google Scholar] [CrossRef]
- Boore, J.L. Animal mitochondrial genomes. Nucleic Acids Res. 1999, 27, 1767–1780. [Google Scholar] [CrossRef]
- Kim, K.Y.; Lee, S.Y.; Bang, I.C.; Nam, Y.K. Complete mitogenome sequence of an endangered freshwater fish, Iksookimia choii (Teleostei; Cypriniformes; Cobitidae). Mitochondrial DNA 2008, 19, 438–445. [Google Scholar] [CrossRef]
- Zhang, D.X.; Hewitt, G.M. Insect mitochondrial control region: A review of its structure, evolution and usefulness in evolutionary studies. Biochem. Syst. Ecol. Evol. 1997, 25, 99–120. [Google Scholar] [CrossRef]
- Wolstenholme, D.R. Animal Mitochondrial DNA: Structure and Evolution. Int. Rev. Cytol. 1992, 141, 173–216. [Google Scholar] [CrossRef]
- Liu, H.; Beckenbach, A.T. Evolution of the mitochondrial cytochrome oxidase II gene among 10 orders of insects. Mol. Phylogenet. Evol. 1992, 1, 41–52. [Google Scholar] [CrossRef]
- Xu, W.; Hao, Z.; Kitching, I.; Xu, Z.B.; Huang, Y.X. First mitogenome of subfamily Langiinae (Lepidoptera: Sphingidae) with its phylogenetic implications. Gene 2021, 789, 145677. [Google Scholar] [CrossRef]
- Li, J.; Zhang, Y.; Hu, K.; Zhao, Y.; Lin, R.; Li, Y.; Huang, Z.; Zhang, X.; Geng, X.; Ding, J. Mitochondrial genome characteristics of two Sphingidae insects (Psilogramma increta and Macroglossum stellatarum) and implications for their phylogeny. Int. J. Biol. Macromol. 2018, 113, 592–600. [Google Scholar] [CrossRef] [PubMed]
- Shahjahan, R.M.; Hughes, K.J.; Leopold, R.A.; Devault, J.D. Lower incubation temperature increases yield of insect genomic DNA isolated by the CTAB method. Biotechniques 1995, 19, 332–334. [Google Scholar] [CrossRef] [PubMed]
- Andrews, S. FASTQC. Available online: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 10 February 2022).
- Yang, M.S.; Song, L.; Zhou, L.; Shi, Y.X.; Song, N.; Zhang, Y.L. Mitochondrial genomes of four satyrine butterflies and phylogenetic relationships of the family Nymphalidae (Lepidoptera: Papilionoidea). Int. J. Biol. Macromol. 2019, 145, 272–281. [Google Scholar] [CrossRef] [PubMed]
- Schubert, M.; Lindgreen, S.; Orlando, L. AdapterRemoval v2: Rapid adapter trimming, identification, and read merging. BMC Res. Notes 2016, 9, 88. [Google Scholar] [CrossRef] [PubMed]
- Nicolas, D.; Patrick, M.; Smits, G. NOVOPlasty: De novo assembly of organelle genomes from whole genome data. Nucleic Acids Res. 2017, 45, e18. [Google Scholar] [CrossRef]
- Meng, G.; Li, Y.; Yang, C.; Liu, S. MitoZ: A toolkit for animal mitochondrial genome assembly, annotation and visualization. Nucleic Acids Res. 2019, 47, e63. [Google Scholar] [CrossRef]
- Bernt, M.; Donath, A.; Jühling, F.; Externbrink, F.; Florentz, C.; Fritzsch, G.; Pütz, J.; Middendorf, M.; Stadler, P.F. MITOS: Improved de novo metazoan mitochondrial genome annotation. Mol. Phylogenet. Evol. 2013, 69, 313–319. [Google Scholar] [CrossRef]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef]
- Greiner, S.; Lehwark, P.; Bock, R. OrganellarGenomeDRAW (OGDRAW) version 1.3.1: Expanded toolkit for the graphical visualization of organellar genomes. Nucleic Acids Res. 2019, 47, 59–64. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef]
- Benson, G. Tandem repeats finder: A program to analyze DNA sequences. Nucleic Acids Res. 1999, 27, 573–580. [Google Scholar] [CrossRef] [PubMed]
- Benson, G. Tandem Repeats Finder Web Server. Available online: https://tandem.bu.edu/trf/trf.html (accessed on 28 June 2022).
- Felsenstein, J. Evolutionary trees from DNA sequences: A maximum likelihood approach. J. Mol. Evol. 1981, 17, 368–376. [Google Scholar] [CrossRef] [PubMed]
- Hundsdoerfer, A.K.; Kitching, I.J.; Wink, M. A molecular phylogeny of the hawkmoth genus Hyles (Lepidoptera: Sphingidae, Macroglossinae). Mol. Phylogenet. Evol. 2005, 35, 442–458. [Google Scholar] [CrossRef] [PubMed]
- Kazutaka, K.; Standley, D.M. A simple method to control over-alignment in the MAFFT multiple sequence alignment program. Bioinformatics 2016, 32, 1933–1942. [Google Scholar] [CrossRef]
- Xia, X.H. DAMBE5: A Comprehensive Software Package for Data Analysis in Molecular Biology and Evolution. Mol. Biol. Evol. 2013, 30, 1720–1728. [Google Scholar] [CrossRef]
- Lanfear, R.; Calcott, B.; Ho, S.Y.W.; Guindon, S. PartitionFinder: Combined Selection of Partitioning Schemes and Substitution Models for Phylogenetic Analyses. Mol. Biol. Evol. 2012, 29, 1695–1701. [Google Scholar] [CrossRef]
- Minh, B.Q.; Nguyen, M.A.T.; Haeseler, A.V. Ultrafast Approximation for Phylogenetic Bootstrap. Mol. Biol. Evol. 2013, 30, 1188–1195. [Google Scholar] [CrossRef]
- Rambaut, A. FigTree. Available online: http://tree.bio.ed.ac.uk/software/figtree/ (accessed on 3 May 2022).
- Kawahara, A.Y.; Plotkin, D.; Espeland, M.; Meusemann, K.; Toussaint, E.F.A.; Donath, A.; Gimnich, F.; Frandsen, P.B.; Zwick, A.; Reis, M.D.; et al. Phylogenomics reveals the evolutionary timing and pattern of butterflies and moths. Proc. Natl. Acad. Sci. USA 2019, 116, 22657–22663. [Google Scholar] [CrossRef]
- Drummond, A.J.; Rambaut, A. BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol. Biol. 2007, 7, 214. [Google Scholar] [CrossRef]
- Li, J.; Lin, R.-R.; Zhang, Y.-Y.; Hu, K.-J.; Zhao, Y.-Q.; Li, Y.; Huang, Z.-R.; Zhang, X.; Geng, X.-X.; Ding, J.-H. Characterization of the complete mitochondrial DNA of Theretra japonica and its phylogenetic position within the Sphingidae (Lepidoptera, Sphingidae). Zookeys 2018, 754, 127–139. [Google Scholar] [CrossRef]
- Korkmaz, E.M.; Doğan, Ö.; Budak, M.; Başıbüyük, H.H. Two nearly complete mitogenomes of wheat stem borers, Cephus pygmeus (L.) and Cephus sareptanus Dovnar-Zapolskij (Hymenoptera: Cephidae): An unusual elongation of rrnS gene. Gene 2015, 558, 254–264. [Google Scholar] [CrossRef] [PubMed]
- Fenn, J.D.; Cameron, S.L.; Whiting, M.F. The complete mitochondrial genome sequence of the Mormon cricket (Anabrus simplex: Tettigoniidae: Orthoptera) and an analysis of control region variability. Insect Mol. Biol. 2010, 16, 239–252. [Google Scholar] [CrossRef] [PubMed]
- Donath, A.; Jühling, F.; Al-Arab, M.; Bernhart, S.H.; Reinhardt, F.; Stadler, P.F.; Middendorf, M.; Bernt, M. Improved annotation of protein-coding genes boundaries in metazoan mitochondrial genomes. Nucleic Acids Res. 2019, 47, 10543–10552. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Chen, P.Y.; Xue, X.F.; Hua, H.Q.; Li, Y.X.; Zhang, F.; Wei, S.J. Extensive gene rearrangements in the mitochondrial genomes of two egg parasitoids, Trichogramma japonicum and Trichogramma ostriniae (Hymenoptera: Chalcidoidea: Trichogrammatidae). Sci. Rep. 2018, 8, 7034. [Google Scholar] [CrossRef]
- Hershberg, R.; Petrov, D.A. Selection on codon bias. Annu. Rev. Genet. 2008, 42, 287–299. [Google Scholar] [CrossRef]
- Hurst, L.D. The Ka/Ks ratio: Diagnosing the form of sequence evolution. Trends Genet. TIG 2002, 18, 486. [Google Scholar] [CrossRef]
- Yang, M.S.; Zhang, H.F.; Song, L.; Shi, Y.X.; Liu, X.M. The complete mitochondrial genome of Mahanta tanyae compared with other zygaenoid moths (Lepidoptera: Zygaenoidea). J. Asia-Pac. Entomol. 2019, 22, 513–521. [Google Scholar] [CrossRef]
- Song, H.J.; Sheffield, N.C.; Cameron, S.L.; Miller, K.B.; Whiting, M.F. When phylogenetic assumptions are violated: Base compositional heterogeneity and among-site rate variation in beetle mitochondrial phylogenomics. Syst. Entomol. 2010, 35, 429–448. [Google Scholar] [CrossRef]
- Min, J.K.; Kang, A.R.; Jeong, H.C.; Kim, K.G.; Kim, I. Reconstructing intraordinal relationships in Lepidoptera using mitochondrial genome data with the description of two newly sequenced lycaenids, Spindasis takanonis and Protantigius superans (Lepidoptera: Lycaenidae). Mol. Phylogenet. Evol. 2011, 61, 436–445. [Google Scholar] [CrossRef]
- Yang, X.S.; Cameron, S.L.; Lees, D.C.; Xue, D.Y.; Han, H.X. A mitochondrial genome phylogeny of owlet moths (Lepidoptera: Noctuoidea), and examination of the utility of mitochondrial genomes for lepidopteran phylogenetics. Mol. Phylogenet. Evol. 2015, 85, 230–237. [Google Scholar] [CrossRef]
- Kitching, I.J.; Scoble, M.J.; Smith, C.; James, S.; Young, R.; Blagoderov, V. Sphingidae Taxonomic Inventory. Available online: https://sphingidae.myspecies.info/node/20910 (accessed on 4 May 2022).
- Kawahara, A.Y.; Mignault, A.A.; Regier, J.C.; Kitching, I.J.; Mitter, C. Phylogeny and Biogeography of Hawkmoths (Lepidoptera: Sphingidae): Evidence from Five Nuclear Genes. PLoS ONE 2009, 4, e5719. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, M.; Cotton, J.A.; Lapointe, F.-J.; Pisani, D. Properties of supertree methods in the consensus setting. Syst. Biol. 2007, 56, 330–337. [Google Scholar] [CrossRef]
- Matsumoto, Y.; Yanase, T.; Tsuda, T.; Noda, H. Species-specific mitochondrial gene rearrangements in biting midges and vector species identification. Med. Vet. Entomol. 2009, 23, 47–55. [Google Scholar] [CrossRef] [PubMed]
- Iyengar, A.; Diniz, F.M.; Gilbert, T.; Woodfine, T.; Maclean, N. Structure and evolution of the mitochondrial control region in oryx. Mol. Phylogenet. Evol. 2006, 40, 305–314. [Google Scholar] [CrossRef] [PubMed]
- Galtier, N.; Roux, C.; Rousselle, M.; Romiguier, J.; Figuet, E.; Glémin, S.; Bierne, N.; Duret, L. Codon Usage Bias in Animals: Disentangling the Effects of Natural Selection, Effective Population Size, and GC-Biased Gene Conversion. Mol. Biol. Evol. 2018, 35, 1092–1103. [Google Scholar] [CrossRef]
- Schnitzler, C.E.; Pang, K.; Powers, M.L.; Reitzel, A.M.; Ryan, J.F.; Simmons, D.; Tada, T.; Park, M.; Gupta, J.; Brooks, S.Y.; et al. Genomic organization, evolution, and expression of photoprotein and opsin genes in Mnemiopsis leidyi: A new view of ctenophore photocytes. BMC Biol. 2012, 10, 107. [Google Scholar] [CrossRef]
- Prum, R.O.; Berv, J.S.; Dornburg, A.; Field, D.J.; Townsend, J.P.; Lemmon, E.M.; Lemmon, A.R. A comprehensive phylogeny of birds (Aves) using targeted next-generation DNA sequencing. Nature 2015, 526, 569–573. [Google Scholar] [CrossRef]
- Spicer, R.A.; Harris, N.B.W.; Widdowson, M.; Herman, A.B.; Guo, S.; Valdes, P.J.; Wolfe, J.A.; Kelley, S.P. Constant elevation of southern Tibet over the past 15 million years. Nature 2003, 421, 622–624. [Google Scholar] [CrossRef]
- ZhiSheng, A.; Kutzbach, J.E.; Prell, W.L.; Porter, S.C. Evolution of Asian monsoons and phased uplift of the Himalaya-Tibetan plateau since Late Miocene times. Nature 2006, 411, 62–66. [Google Scholar] [CrossRef]
- Ghanavi, H.R.; Twort, V.; Hartman, T.J.; Zahiri, R.; Wahlberg, N. The (non) accuracy of mitochondrial genomes for family level phylogenetics: The case of erebid moths (Lepidoptera; Erebidae). Zool. Scr. 2021. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, Y.-X.; Xing, Z.-P.; Zhang, H.; Xu, Z.-B.; Tao, L.-L.; Hu, H.-Y.; Kitching, I.J.; Wang, X. Characterization of the Complete Mitochondrial Genome of Eight Diurnal Hawkmoths (Lepidoptera: Sphingidae): New Insights into the Origin and Evolution of Diurnalism in Sphingids. Insects 2022, 13, 887. https://doi.org/10.3390/insects13100887
Huang Y-X, Xing Z-P, Zhang H, Xu Z-B, Tao L-L, Hu H-Y, Kitching IJ, Wang X. Characterization of the Complete Mitochondrial Genome of Eight Diurnal Hawkmoths (Lepidoptera: Sphingidae): New Insights into the Origin and Evolution of Diurnalism in Sphingids. Insects. 2022; 13(10):887. https://doi.org/10.3390/insects13100887
Chicago/Turabian StyleHuang, Yi-Xin, Zhi-Ping Xing, Hao Zhang, Zhen-Bang Xu, Li-Long Tao, Hao-Yuan Hu, Ian J. Kitching, and Xu Wang. 2022. "Characterization of the Complete Mitochondrial Genome of Eight Diurnal Hawkmoths (Lepidoptera: Sphingidae): New Insights into the Origin and Evolution of Diurnalism in Sphingids" Insects 13, no. 10: 887. https://doi.org/10.3390/insects13100887
APA StyleHuang, Y.-X., Xing, Z.-P., Zhang, H., Xu, Z.-B., Tao, L.-L., Hu, H.-Y., Kitching, I. J., & Wang, X. (2022). Characterization of the Complete Mitochondrial Genome of Eight Diurnal Hawkmoths (Lepidoptera: Sphingidae): New Insights into the Origin and Evolution of Diurnalism in Sphingids. Insects, 13(10), 887. https://doi.org/10.3390/insects13100887