
The USDA-ARS Ag100Pest Initiative: High-Quality Genome Assemblies for Agricultural Pest Arthropod Research

 , , , , , , , and
, , , , , , , and
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Species Prioritization
2.2. Sample Collection and Extraction
2.3. Library Preparation, Sequencing, and Assembly
2.4. Mitochondrial and Contaminant Screening
2.5. Genome Annotation
2.5.1. Structural and Functional Annotation
2.5.2. Manual Annotation
2.6. Data Management
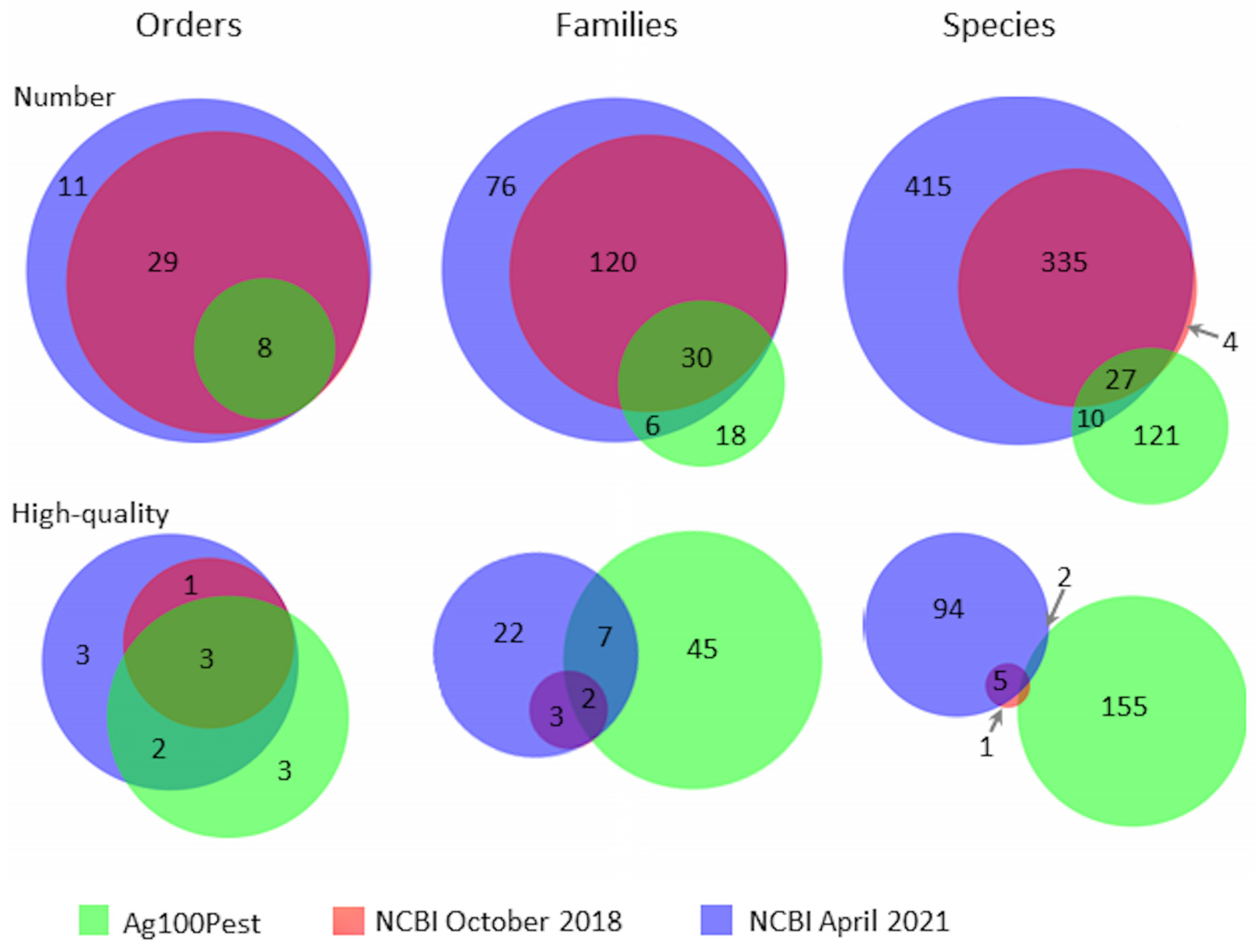
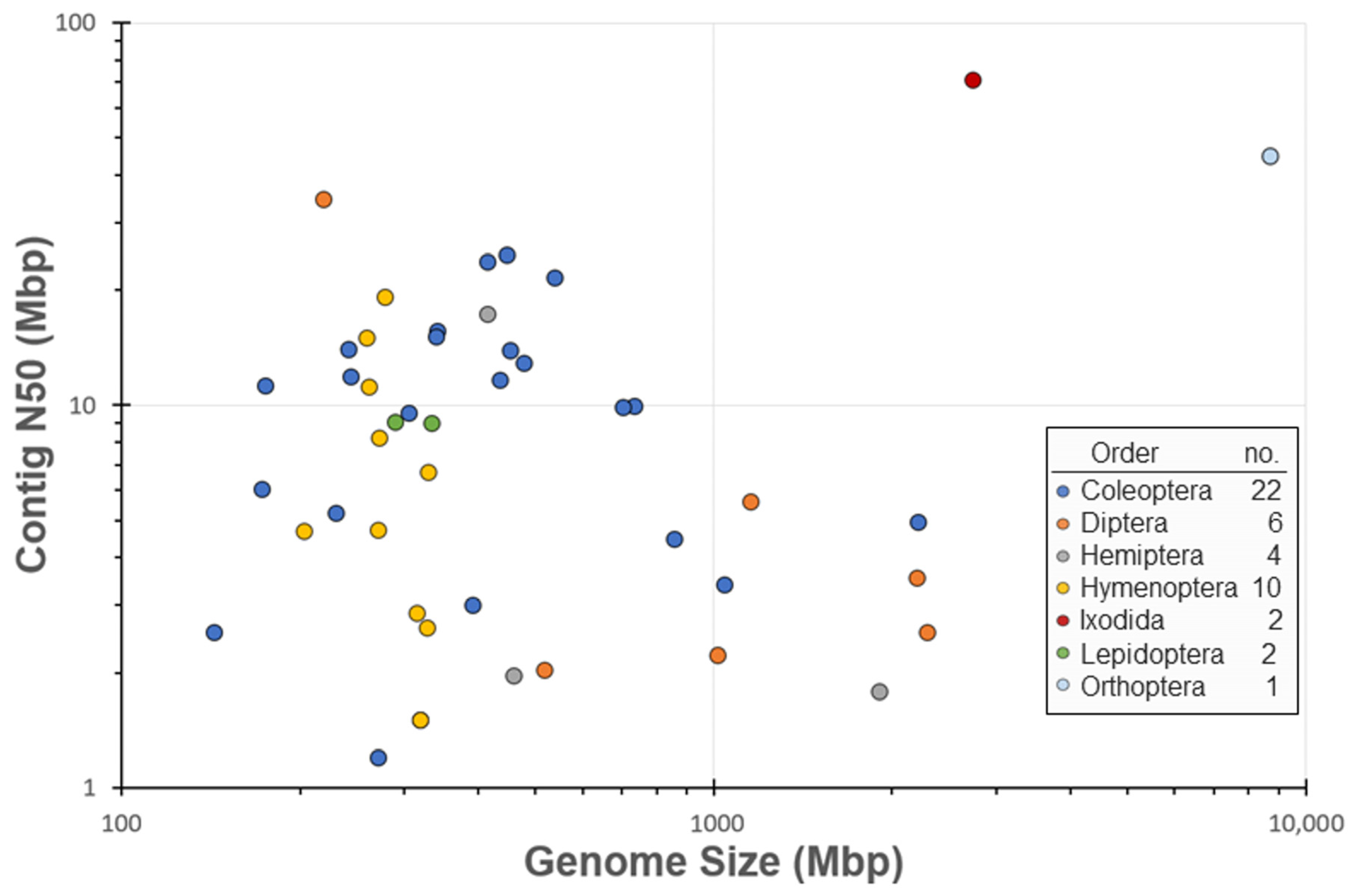
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Davidsson, M. The Financial Implications of a Well-Hidden and Ignored Chronic Lyme Disease Pandemic. Healthcare 2018, 6, 16. [Google Scholar] [CrossRef]
- Deutsch, C.A.; Tewksbury, J.J.; Tigchelaar, M.; Battisti, D.S.; Merrill, S.C.; Huey, R.B.; Naylor, R.L. Increase in crop losses to insect pests in a warming climate. Science 2018, 361, 916–919. [Google Scholar] [CrossRef] [PubMed]
- USDA-National Agricultural Statistics Service-Statistics by Subject. Available online: https://www.nass.usda.gov/Statistics_by_Subject/index.php?sector=CROPS (accessed on 20 April 2021).
- Wechsler, S.; Smith, D. Has Resistance Taken Root in U.S. Corn Fields? Demand for Insect Control. Am. J. Agric. Econ. 2018, 100, 1136–1150. [Google Scholar] [CrossRef]
- Hunter, M.C.; Smith, R.G.; Schipanski, M.E.; Atwood, L.W.; Mortensen, D.A. Agriculture in 2050: Recalibrating Targets for Sustainable Intensification. Bioscience 2017, 67, 386–391. [Google Scholar] [CrossRef]
- Isman, M.B. Challenges of Pest Management in the Twenty First Century: New Tools and Strategies to Combat Old and New Foes Alike. Front. Agron. 2019, 1, 2. [Google Scholar] [CrossRef]
- Sparks, T.C.; Crossthwaite, A.J.; Nauen, R.; Banba, S.; Cordova, D.; Earley, F.; Ebbinghaus-Kintscher, U.; Fujioka, S.; Hirao, A.; Karmon, D.; et al. Insecticides, biologics and nematicides: Updates to IRAC’s mode of action classification-a tool for resistance management. Pestic. Biochem. Physiol. 2020, 167, 104587. [Google Scholar] [CrossRef]
- E Tabashnik, B.; Carrière, Y. Surge in insect resistance to transgenic crops and prospects for sustainability. Nat. Biotechnol. 2017, 35, 926–935. [Google Scholar] [CrossRef] [PubMed]
- Bale, J.S.; Masters, G.J.; Hodkinson, I.D.; Awmack, C.; Bezemer, M.; Brown, V.K.; Butterfield, J.; Buse, A.; Coulson, J.C.; Farrar, J.; et al. Herbivory in global climate change research: Direct effects of rising temperature on insect herbivores. Glob. Chang. Biol. 2002, 8, 1–16. [Google Scholar] [CrossRef]
- Paini, D.R.; Sheppard, A.; Cook, D.C.; De Barro, P.J.; Worner, S.P.; Thomas, M.B. Global threat to agriculture from invasive species. Proc. Natl. Acad. Sci. USA 2016, 113, 7575–7579. [Google Scholar] [CrossRef]
- Kellis, M.; Wold, B.; Snyder, M.P.; Bernstein, B.E.; Kundaje, A.; Marinov, G.K.; Ward, L.; Birney, E.; Crawford, G.E.; Dekker, J.; et al. Defining functional DNA elements in the human genome. Proc. Natl. Acad. Sci. USA 2014, 111, 6131–6138. [Google Scholar] [CrossRef]
- Dimitrieva, S.; Bucher, P. Genomic context analysis reveals dense interaction network between vertebrate ultraconserved non-coding elements. Bioinformatics 2012, 28, i395–i401. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Dance, A. Inner Workings: Researchers peek into chromosomes’ 3D structure in unprecedented detail. Proc. Natl. Acad. Sci. USA 2020, 117, 25186–25189. [Google Scholar] [CrossRef] [PubMed]
- Ou, H.D.; Phan, S.; Deerinck, T.J.; Thor, A.; Ellisman, M.H.; O’shea, C.C. ChromEMT: Visualizing 3D chromatin struc-ture and compaction in interphase and mitotic cells. Science 2017, 357. [Google Scholar] [CrossRef] [PubMed]
- Dekker, J.; Belmont, A.S.; Guttman, M.; Leshyk, V.O.; English, B.; Lomvardas, S.; Mirny, L.A.; O’Shea, C.C.; Park, P.J.; Ren, B.; et al. The 4D nucleome project. Nat. Cell Biol. 2017, 549, 219–226. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.B.; Celniker, S.E. Lessons from modENCODE. Annu. Rev. Genom. Hum. Genet. 2015, 16, 31–53. [Google Scholar] [CrossRef] [PubMed]
- Metzger, B.P.; Wittkopp, P.J.; Coolon, J.D. Evolutionary dynamics of regulatory changes underlying gene expression di-vergence among Saccharomyces species. Genome Biol. Evol. 2017, 9, 843–854. [Google Scholar] [CrossRef] [PubMed]
- Pagani, F.; Baralle, F.E. Genomic variants in exons and introns: Identifying the splicing spoilers. Nat. Rev. Genet. 2004, 5, 389–396. [Google Scholar] [CrossRef]
- Scotti, M.M.; Swanson, M.S. RNA mis-splicing in disease. Nat. Rev. Genet. 2016, 17, 19–32. [Google Scholar] [CrossRef]
- Djuranovic, S.; Nahvi, A.; Green, R. MiRNA-mediated gene silencing by translational repression followed by mRNA deadenylation and decay. Science 2012, 336, 237–240. [Google Scholar] [CrossRef]
- Wang, K.C.; Chang, H.Y. Molecular Mechanisms of Long Noncoding RNAs. Mol. Cell 2011, 43, 904–914. [Google Scholar] [CrossRef] [PubMed]
- Hindorff, L.A.; Sethupathy, P.; Junkins, H.A.; Ramos, E.M.; Mehta, J.P.; Collins, F.S.; Manolio, T.A. Potential etiologic and functional implications of genome-wide association loci for human diseases and traits. Proc. Natl. Acad. Sci. USA 2009, 106, 9362–9367. [Google Scholar] [CrossRef]
- i5K Consortium. The i5K Initiative: Advancing Arthropod Genomics for Knowledge, Human Health, Agriculture, and the Environment. J. Hered. 2013, 104, 595–600. [Google Scholar] [CrossRef]
- About the I5k Initiative. Available online: http://i5k.github.io/about (accessed on 26 May 2021).
- Richards, S.; Murali, S.C. Best practices in insect genome sequencing: What works and what doesn’t. Curr. Opin. Insect Sci. 2015, 7, 1–7. [Google Scholar] [CrossRef]
- Lewin, H.A.; Robinson, G.E.; Kress, W.J.; Baker, W.J.; Coddington, J.; Crandall, K.A.; Durbin, R.; Edwards, S.V.; Forest, F.; Gilbert, M.; et al. Earth BioGenome Project: Sequencing life for the future of life. Proc. Natl. Acad. Sci. USA 2018, 115, 4325–4333. [Google Scholar] [CrossRef] [PubMed]
- Ag100Pest Initiative. Available online: http://i5k.github.io/ag100pest (accessed on 26 May 2021).
- Coates, B.S.; Poelchau, M.; Childers, C.; Evans, J.D.; Handler, A.; Guerrero, F.; Skoda, S.; Hopper, K.; Wintermantel, W.M.; Ling, K.-S.; et al. Arthropod genomics research in the United States Department of Agriculture-Agricultural Research Service: Current impacts and future prospects. Trends Entomol. 2015, 11, 1–27. [Google Scholar]
- Gundersen-Rindal, D.; Adrianos, S.; Allen, M.; Becnel, J.; Chen, Y.; Choi, M.; Estep, A.; Evans, J.; Garczynski, S.; Geib, S.; et al. Arthropod genomics research in the United States Department of Agriculture, Agricultural Research Service: Applications of RNA interference and CRISPR gene-editing technologies in pest control. Trends Entomol. 2017, 13, 109–137. [Google Scholar]
- Welcome to ITAP|Federal Interagency Committee on Invasive Terrestrial Animals and Pathogens. Available online: https://www.itap.gov/ (accessed on 26 May 2021).
- CAPS Program Resource and Collaboration Site|CAPS. Available online: http://caps.ceris.purdue.edu/ (accessed on 26 May 2021).
- BioSample Packages-BioSample-NCBI. Available online: https://www.ncbi.nlm.nih.gov/biosample/docs/packages/ (accessed on 26 May 2021).
- Wenger, A.M.; Peluso, P.; Rowell, W.J.; Chang, P.-C.; Hall, R.J.; Concepcion, G.T.; Ebler, J.; Fungtammasan, A.; Kolesnikov, A.; Olson, N.D.; et al. Accurate circular consensus long-read sequencing improves variant detection and assembly of a human genome. Nat. Biotechnol. 2019, 37, 1155–1162. [Google Scholar] [CrossRef] [PubMed]
- Sim, S.B. HiFiAdapterFilt v1.0.0. Available online: https://github.com/sheinasim/HiFiAdapterFilt (accessed on 23 April 2021).
- Cheng, H.; Concepcion, G.T.; Feng, X.; Zhang, H.; Li, H. Haplotype-resolved de novo assembly using phased assembly graphs with hifiasm. Nat. Methods 2021, 18, 170–175. [Google Scholar] [CrossRef] [PubMed]
- Lieberman-Aiden, E.; Van Berkum, N.L.; Williams, L.; Imakaev, M.; Ragoczy, T.; Telling, A.; Amit, I.; Lajoie, B.R.; Sabo, P.J.; Dorschner, M.O.; et al. Comprehensive Mapping of Long-Range Interactions Reveals Folding Principles of the Human Genome. Science 2009, 326, 289–293. [Google Scholar] [CrossRef]
- Zhang, H.; Emerson, D.J.; Gilgenast, T.G.; Titus, K.R.; Lan, Y.; Huang, P.; Zhang, D.; Wang, H.; Keller, C.A.; Giardine, B.; et al. Chromatin structure dynamics during the mitosis-to-G1 phase transition. Nat. Cell Biol. 2019, 576, 158–162. [Google Scholar] [CrossRef]
- Durand, N.C.; Robinson, J.T.; Shamim, M.S.; Machol, I.; Mesirov, J.P.; Lander, E.S.; Aiden, E.L. Juicebox provides a visu-alization system for Hi-C contact maps with unlimited zoom. Cell Syst. 2016, 3, 99–101. [Google Scholar] [CrossRef]
- Uliano-Silva, M. MitoHiFi. Available online: https://github.com/marcelauliano/MitoHiFi (accessed on 30 January 2021).
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.S.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef]
- Allio, R.; Schomaker-Bastos, A.; Romiguier, J.; Prosdocimi, F.; Nabholz, B.; Delsuc, F. MitoFinder: Efficient automated large-scale extraction of mitogenomic data in target enrichment phylogenomics. Mol. Ecol. Resour. 2020, 20, 892–905. [Google Scholar] [CrossRef]
- Jühling, F.; Pütz, J.; Bernt, M.; Donath, A.; Middendorf, M.; Florentz, C.; Stadler, P.F. Improved systematic tRNA gene annotation allows new insights into the evolution of mitochondrial tRNA structures and into the mechanisms of mitochondrial genome rearrangements. Nucleic Acids Res. 2011, 40, 2833–2845. [Google Scholar] [CrossRef] [PubMed]
- Challis, R.; Richards, E.; Rajan, J.; Cochrane, G.; Blaxter, M. BlobToolKit–Interactive Quality Assessment of Genome Assemblies. G3 Genes Genomes Genet. 2020, 10, 1361–1374. [Google Scholar] [CrossRef]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 2015, 12, 59–60. [Google Scholar] [CrossRef] [PubMed]
- Simão, F.A.; Waterhouse, R.M.; Ioannidis, P.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 2015, 31, 3210–3212. [Google Scholar] [CrossRef]
- Waterhouse, R.M.; Seppey, M.; Simão, F.A.; Manni, M.; Ioannidis, P.; Klioutchnikov, G.; Kriventseva, E.V.; Zdobnov, E. BUSCO Applications from Quality Assessments to Gene Prediction and Phylogenomics. Mol. Biol. Evol. 2018, 35, 543–548. [Google Scholar] [CrossRef]
- Keller, O.; Kollmar, M.; Stanke, M.; Waack, S. A novel hybrid gene prediction method employing protein multiple sequence alignments. Bioinformatics 2011, 27, 757–763. [Google Scholar] [CrossRef] [PubMed]
- Eddy, S.R. Accelerated Profile HMM Searches. PLoS Comput. Biol. 2011, 7, e1002195. [Google Scholar] [CrossRef]
- Yandell, M.; Ence, D. A beginner’s guide to eukaryotic genome annotation. Nat. Rev. Genet. 2012, 13, 329–342. [Google Scholar] [CrossRef]
- Ashburner, M.; Ball, C.A.; Blake, J.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene Ontology: Tool for the unification of biology. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef]
- Thibaud-Nissen, F.; DiCuccio, M.; Hlavina, W.; Kimchi, A.; Kitts, P.A.; Murphy, T.D.; Pruitt, K.D.; Souvorov, A. P8008 The NCBI Eukaryotic Genome Annotation Pipeline. J. Anim. Sci. 2016, 94, 184. [Google Scholar] [CrossRef]
- Souvorov, A.; Kapustin, Y.; Kiryutin, B.; Chetvernin, V.; Tatusova, T.; Lipman, D. Gnomon–NCBI Eukaryotic Gene Prediction Tool. Available online: http://www.ncbi.nlm.nih.gov/core/assets/genome/files/Gnomon-description.pdf (accessed on 15 August 2015).
- The UniProt Consortium. UniProt: The universal protein knowledgebase in 2021. Nucleic Acids Res. 2021, 49, D480–D489. [Google Scholar] [CrossRef] [PubMed]
- The NCBI Eukaryotic Genome Annotation Pipeline. Available online: https://www.ncbi.nlm.nih.gov/genome/annotation_euk/process/#naming (accessed on 26 May 2021).
- NCBI Eukaryotic Genome Annotation Policy on Which Genomes Are Annotated. Available online: https://www.ncbi.nlm.nih.gov/genome/annotation_euk/policy/ (accessed on 26 May 2021).
- AgBase. Available online: https://github.com/AgBase (accessed on 26 May 2021).
- McCarthy, F.M.; Wang, N.; Magee, G.B.; Nanduri, B.; Lawrence, M.L.; Camon, E.B.; Barrell, D.G.; Hill, D.P.; E Dolan, M.; Williams, W.P.; et al. AgBase: A functional genomics resource for agriculture. BMC Genom. 2006, 7, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Blum, M.; Chang, H.-Y.; Chuguransky, S.; Grego, T.; Kandasaamy, S.; Mitchell, A.; Nuka, G.; Paysan-Lafosse, T.; Qureshi, M.; Raj, S.; et al. The InterPro protein families and domains database: 20 years on. Nucleic Acids Res. 2021, 49, D344–D354. [Google Scholar] [CrossRef] [PubMed]
- Xie, C.; Mao, X.; Huang, J.; Ding, Y.; Wu, J.; Dong, S.; Kong, L.; Gao, G.; Li, C.-Y.; Wei, L. KOBAS 2.0: A web server for annotation and identification of enriched pathways and diseases. Nucleic Acids Res. 2011, 39, W316–W322. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef]
- Poelchau, M.; Childers, C.; Moore, G.; Tsavatapalli, V.; Evans, J.; Lee, C.-Y.; Lin, H.; Lin, J.-W.; Hackett, K. The i5k Workspace@NAL—Enabling genomic data access, visualization and curation of arthropod genomes. Nucleic Acids Res. 2015, 43, D714–D719. [Google Scholar] [CrossRef]
- Dunn, N.A.; Unni, D.R.; Diesh, C.; Munoz-Torres, M.; Harris, N.L.; Yao, E.; Rasche, H.; Holmes, I.H.; Elsik, C.G.; Lewis, S.E. Apollo: Democratizing genome annotation. PLoS Comput. Biol. 2019, 15, e1006790. [Google Scholar] [CrossRef]
- ID 555319-BioProject-NCBI. Available online: https://www.ncbi.nlm.nih.gov/bioproject/555319 (accessed on 26 May 2021).
- Invertebrate; Version 1.0 Package-BioSample-NCBI. Available online: https://www.ncbi.nlm.nih.gov/biosample/docs/packages/Invertebrate.1.0/ (accessed on 26 May 2021).
- Rhie, A.; McCarthy, S.; Fedrigo, O.; Damas, J.; Formenti, G.; Koren, S.; Uliano-Silva, M.; Chow, W.; Fungtammasan, A.; Kim, J.; et al. Towards complete and error-free genome assemblies of all vertebrate species. Nat. Cell Biol. 2021, 592, 737–746. [Google Scholar] [CrossRef]
- Kingan, S.B.; Heaton, H.; Cudini, J.; Lambert, C.C.; Baybayan, P.; Galvin, B.D.; Durbin, R.; Korlach, J.; Lawniczak, M.K.N. A High-Quality De novo Genome Assembly from a Single Mosquito Using PacBio Sequencing. Genes 2019, 10, 62. [Google Scholar] [CrossRef] [PubMed]
- Schappach, B.L.; Krell, R.K.; Hornbostel, V.L.; Connally, N.P. Exotic Haemaphysalis longicornis (Acari: Ixodidae) in the United States: Biology, Ecology, and Strategies for Management. J. Integr. Pest Manag. 2020, 11, 21. [Google Scholar] [CrossRef]
- Guan, D.; A McCarthy, S.; Wood, J.; Howe, K.; Wang, Y.; Durbin, R. Identifying and removing haplotypic duplication in primary genome assemblies. Bioinformatics 2020, 36, 2896–2898. [Google Scholar] [CrossRef]
- Amarasinghe, S.L.; Su, S.; Dong, X.; Zappia, L.; Ritchie, M.E.; Gouil, Q. Opportunities and challenges in long-read se-quencing data analysis. Genome Biol. 2020, 21, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Schneider, C.; Woehle, C.; Greve, C.; A D’Haese, C.; Wolf, M.; Hiller, M.; Janke, A.; Bálint, M.; Huettel, B. Two high-quality de novo genomes from single ethanol-preserved specimens of tiny metazoans (Collembola). GigaScience 2021, 10, 35. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Order | NCBI Oct 2018 | NCBI Apr 2021 | Ag100Pest |
|---|---|---|---|
| Coleoptera | 16 (0) | 54 (0) | 50 |
| Diptera | 119 (3) | 186 (31) | 25 |
| Hemiptera | 27 (0) | 51 (4) | 37 |
| Hymenoptera | 73 (1) | 169 (6) | 15 |
| Ixodida | 3 (0) | 11 (1) | 10 |
| Lepidoptera | 53 (1) | 149 (52) | 16 |
| Orthoptera | 3 (0) | 5 (0) | 4 |
| Thysanoptera | 1 (0) | 3 (0) | 1 |
| Order | Family | Scientific Name | TaxID | Common Name | NCBI Representative Assembly | NCBI Assembly Date | NCBI Assembly Length (Mbp) | NCBI Contig N50 (Mbp) | Ag100Pest Assembly Length (Mbp) | Ag100Pest Contig N50 (Mbp) |
|---|---|---|---|---|---|---|---|---|---|---|
| Coleoptera | Silvanidae | Oryzaephilus surinamensis | 41112 | saw-toothed grain beetle | GCA_004796505.1 | 16 April 2019 | 104.01 | 0.019 | 173.49 | 5.98 |
| Coleoptera | Tenebrionidae | Tribolium castaneum | 7070 | red flour beetle | GCF_000002335.3 | 10 March 2016 | 165.94 | 0.073 | 242.40 | 13.86 |
| Diptera | Muscidae | Stomoxys calcitrans | 35570 | stable fly; biting house fly | GCF_001015335.1 | 31 May 2015 | 971.19 | 0.011 | 1159.87 | 5.56 |
| Hemiptera | Aphididae | Aphis gossypii | 80765 | cotton aphid; melon aphid | GCF_004010815.1 | 10 January 2019 | 294.28 | 0.077 | 416.81 | 17.16 |
| Hymenoptera | Diprionidae | Neodiprion lecontei | 441921 | redheaded pine sawfly | GCA_001263575.2 | 21 June 2018 | 239.78 | 0.087 | 273.27 | 8.16 |
| Hymenoptera | Diprionidae | Neodiprion pinetum | 441929 | white pine sawfly | GCA_004916985.1 | 26 April 2019 | 269.78 | 0.016 | 272.19 | 4.68 |
| Hymenoptera | Formicidae | Wasmannia auropunctata | 64793 | little fire ant | GCF_000956235.1 | 17 March 2015 | 324.12 | 0.038 | 320.50 | 1.49 |
| Hymenoptera | Vespidae | Vespula pensylvanica | 30213 | western yellowjacket | GCA_014466175.1 | 9 September 2020 | 179.37 | 0.097 | 204.70 | 4.64 |
| Ixodida | Ixodidae | Haemaphysalis longicornis | 44386 | longhorned tick | GCA_013339765.1 | 16 June 2020 | 2554.97 | 0.740 | 5576.40 | 0.88 |
| Lepidoptera | Pyralidae | Plodia interpunctella | 58824 | Indianmeal moth | GCA_900182495.1 | 6 May 2017 | 382.24 | 0.312 | 291.43 | 8.96 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Childers, A.K.; Geib, S.M.; Sim, S.B.; Poelchau, M.F.; Coates, B.S.; Simmonds, T.J.; Scully, E.D.; Smith, T.P.L.; Childers, C.P.; Corpuz, R.L.; et al. The USDA-ARS Ag100Pest Initiative: High-Quality Genome Assemblies for Agricultural Pest Arthropod Research. Insects 2021, 12, 626. https://doi.org/10.3390/insects12070626
Childers AK, Geib SM, Sim SB, Poelchau MF, Coates BS, Simmonds TJ, Scully ED, Smith TPL, Childers CP, Corpuz RL, et al. The USDA-ARS Ag100Pest Initiative: High-Quality Genome Assemblies for Agricultural Pest Arthropod Research. Insects. 2021; 12(7):626. https://doi.org/10.3390/insects12070626
Chicago/Turabian StyleChilders, Anna K., Scott M. Geib, Sheina B. Sim, Monica F. Poelchau, Brad S. Coates, Tyler J. Simmonds, Erin D. Scully, Timothy P. L. Smith, Christopher P. Childers, Renee L. Corpuz, and et al. 2021. "The USDA-ARS Ag100Pest Initiative: High-Quality Genome Assemblies for Agricultural Pest Arthropod Research" Insects 12, no. 7: 626. https://doi.org/10.3390/insects12070626
APA StyleChilders, A. K., Geib, S. M., Sim, S. B., Poelchau, M. F., Coates, B. S., Simmonds, T. J., Scully, E. D., Smith, T. P. L., Childers, C. P., Corpuz, R. L., Hackett, K., & Scheffler, B. (2021). The USDA-ARS Ag100Pest Initiative: High-Quality Genome Assemblies for Agricultural Pest Arthropod Research. Insects, 12(7), 626. https://doi.org/10.3390/insects12070626