
Tracing the Origin of Korean Invasive Populations of the Spotted Lanternfly, Lycorma delicatula (Hemiptera: Fulgoridae)

 ,
,  ,
,
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. SLF Samples
2.2. Microsatellite Genotyping
2.3. Data Analysis
3. Results
3.1. Genetic Diversity within Populations
3.2. Genetic Variation, Differentiation between Populations, and Bottleneck
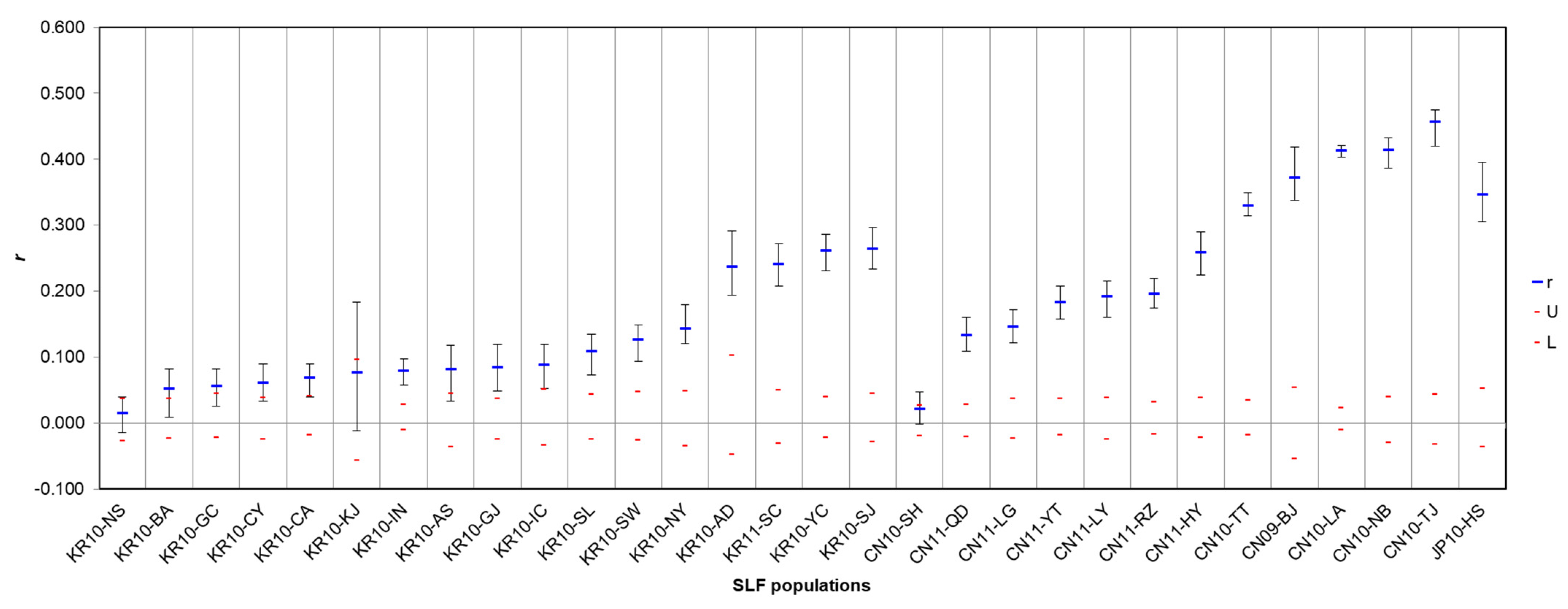
3.3. Spatial Autocorrelation and Isolation-by-Distance
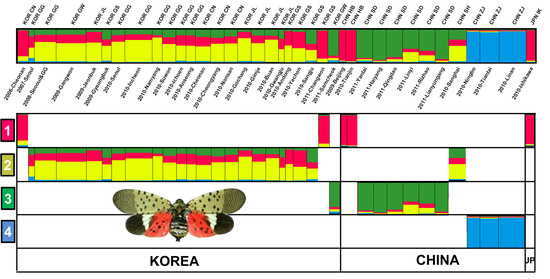
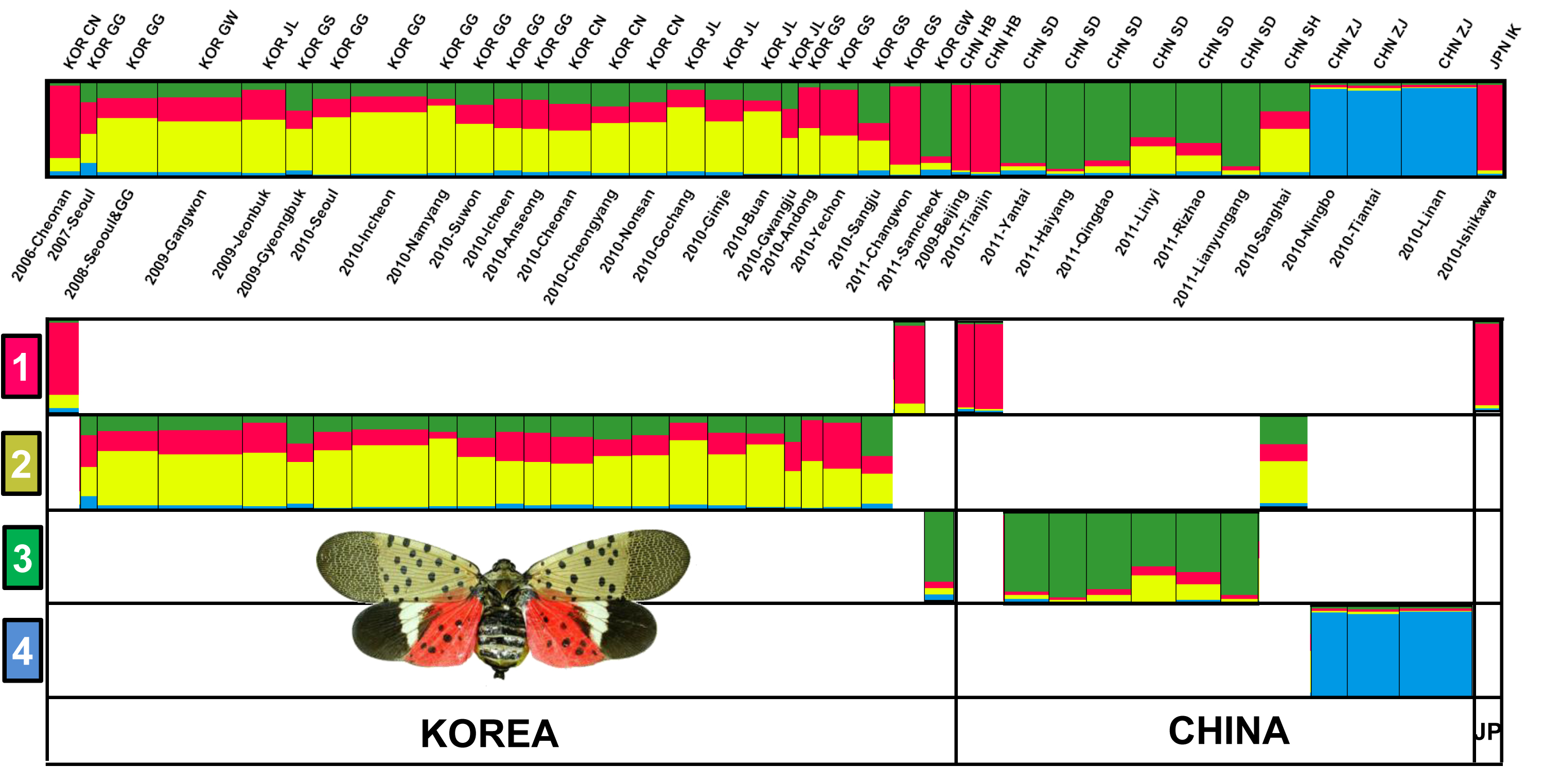
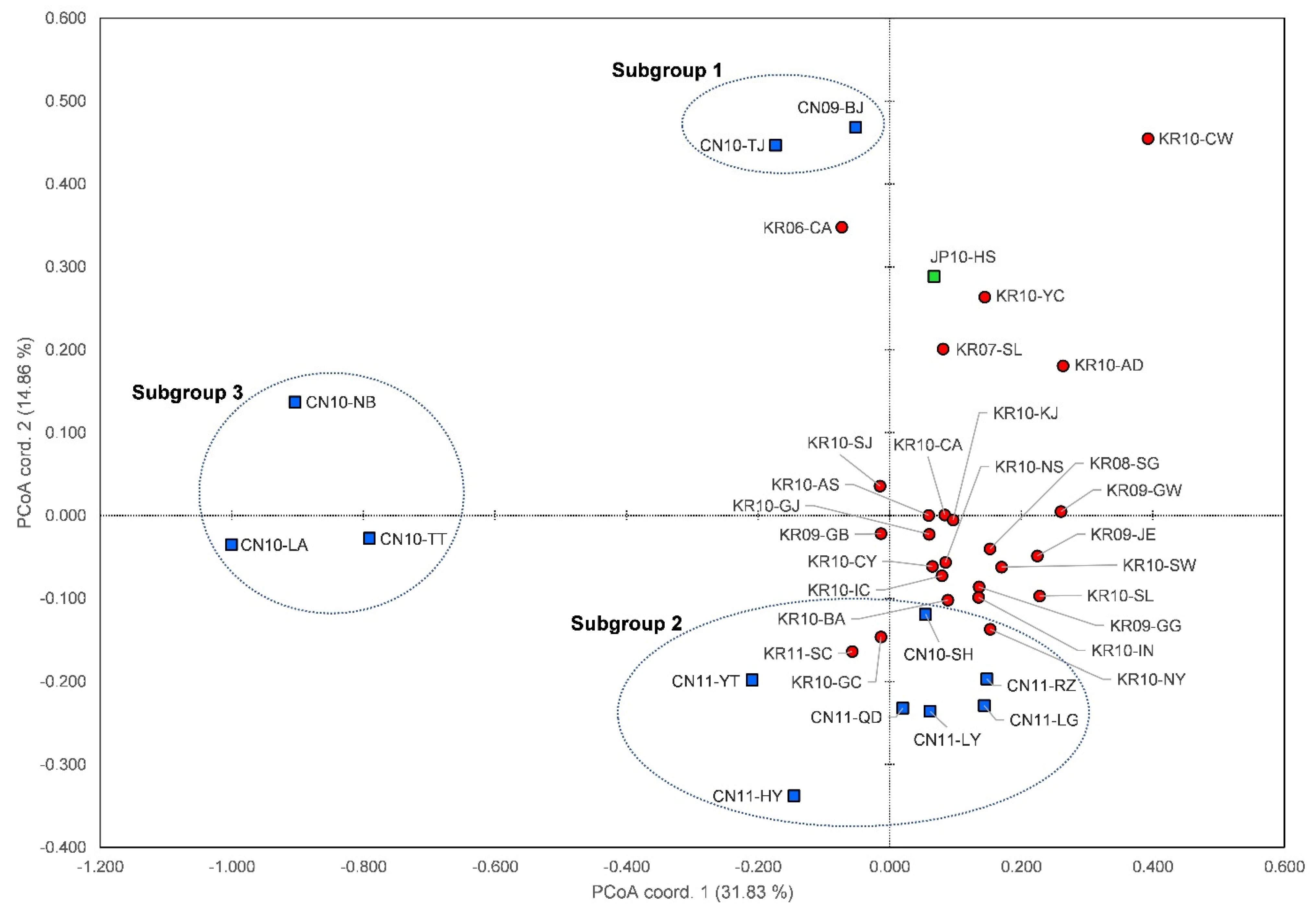
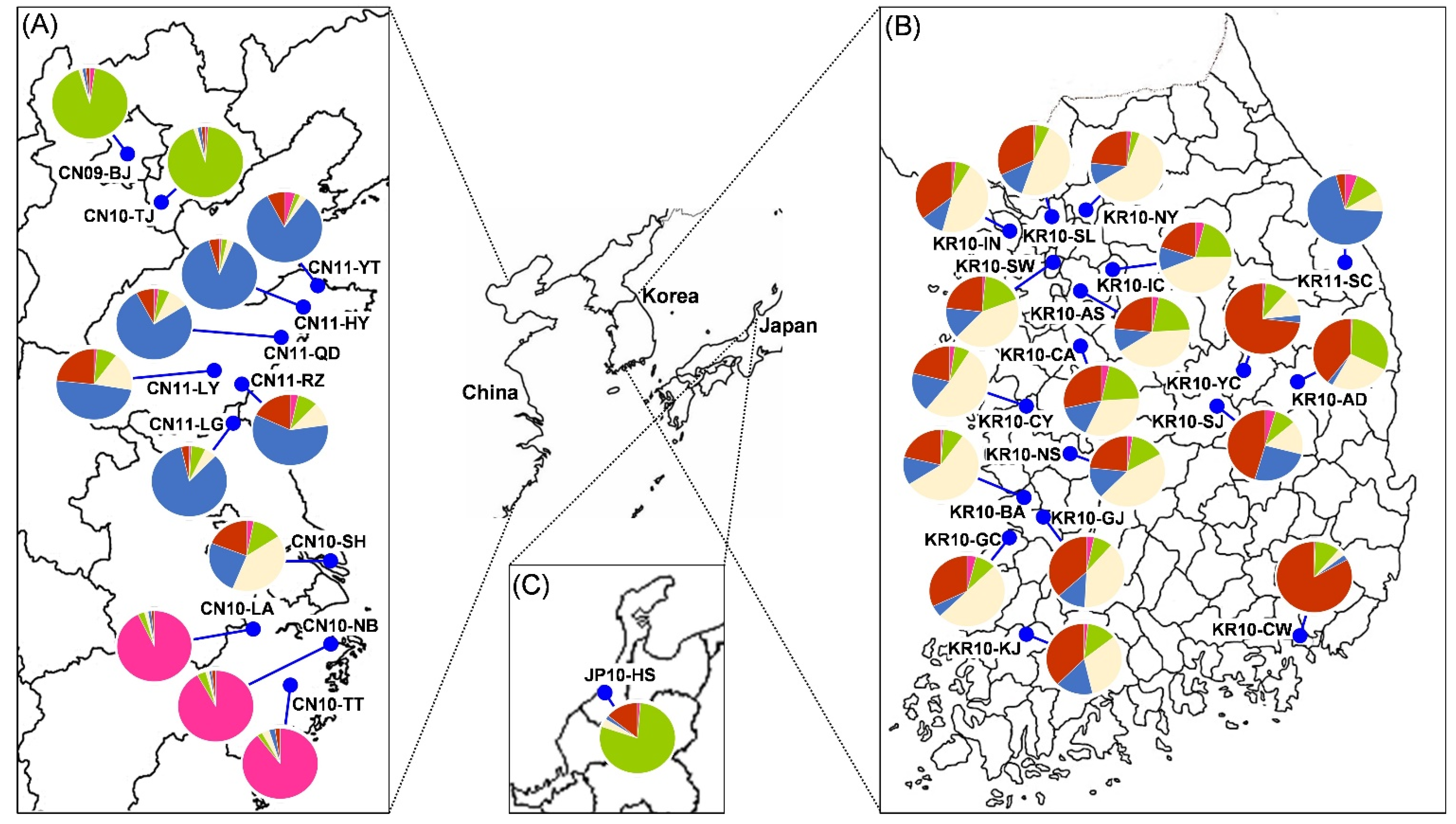
3.4. Genetic Structure and Assignment
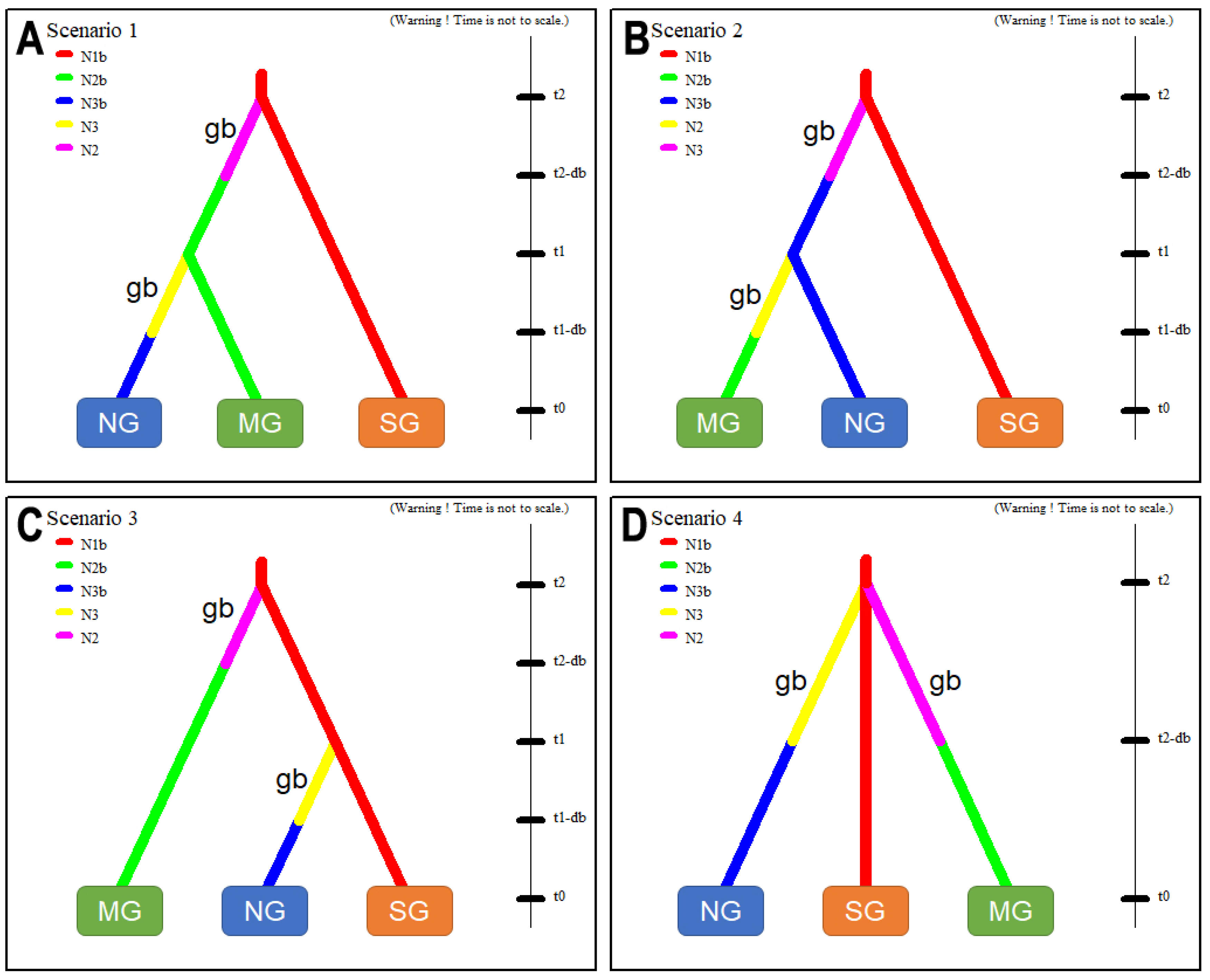
3.5. Inferring an Invasive Route by Testing Hypothetical Scenarios by ABC Analysis
4. Discussion
4.1. Genetic Structures of SLF in the Native and Invasive Areas
4.2. Spatial Distribution of SLF between Origin and Invasion Areas
4.3. Introduction Process of SLF from China
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Hulme, P. Trade, transport and trouble: Managing invasive species pathways in an era of globalization. J. Appl. Ecol. 2009, 46, 10–18. [Google Scholar] [CrossRef]
- Roques, A. Alien forest insects in a warmer world and a globalised economy: Impacts of changes in trade, tourism and climate on forest biosecurity. N. Z. J. For. Sci. 2010, 40, 77–94. [Google Scholar]
- Seebens, H.; Blackburn, T.M.; Dyer, E.E.; Genovesi, P.; Hulme, P.E.; Jeschke, J.M.; Pagad, S.; Pyšek, P.; Winter, M.; Arianoutsou, M.; et al. No saturation in the accumulation of alien species worldwide. Nat. Commun. 2017, 8, 14435. [Google Scholar] [CrossRef]
- Simberloff, D. Invasive Species: What Everyone Needs to Know; Oxford University Press: Oxford, UK, 2013. [Google Scholar]
- Pyšek, P.; Richardson, D.M. Invasive species, environmental change and management, and health. Annu. Rev. Environ. Resour. 2010, 35, 25–55. [Google Scholar] [CrossRef]
- Estoup, A.; Guillemaud, T. Reconstructing routes of invasion using genetic data: Why, how and so what? Mol. Ecol. 2010, 19, 4113–4130. [Google Scholar] [CrossRef] [PubMed]
- Torchin, M.E.; Lafferty, K.D.; Dobson, A.P.; McKenzie, V.J.; Kuris, A.M. Introduced species and their missing parasites. Nature 2003, 421, 628–630. [Google Scholar] [CrossRef] [PubMed]
- Xiao, G. Forest Insect of China, 2nd ed.; Forest Research Institute: Beijing, China, 1992. [Google Scholar]
- Hua, L.-Z. List of Chinese Insects, Vol. I; Zhongshan University Press: Guangzhou, China, 2000; Volume 448. [Google Scholar]
- Kim, H.; Kim, M.; Kwon, D.H.; Park, S.; Lee, Y.; Huang, J.; Kai, S.; Lee, H.-S.; Hong, K.-J.; Jang, Y.; et al. Molecular comparison of Lycorma delicatula (Hemiptera: Fulgoridae) isolates in Korea, China, and Japan. J. Asia-Pac. Entomol. 2013, 16, 503–506. [Google Scholar] [CrossRef]
- Barringer, L.E.; Donovall, L.R.; Spichiger, S.-E.; Lynch, D.; Henry, D. The first new world record of Lycorma delicatula (Insecta: Hemiptera: Fulgoridae). Entomol. News 2015, 125, 20–23. [Google Scholar] [CrossRef]
- Barringer, L.; Ciafré, C.M. Worldwide feeding host plants of spotted lanternfly, with significant additions from North America. Environ. Entomol. 2020, 49, 999–1011. [Google Scholar] [CrossRef]
- Lee, J.E.; Moon, S.R.; Ahn, H.G.; Cho, S.R.; Yang, J.O.; Yoon, C.; Kim, G.H. Feeding Behavior of Lycorma delicatula (Hemiptera: Fulgoridae) and Response on Feeding Stimulants of Some Plants. Korean J. Appl. Entomol. 2009, 48, 467–477. [Google Scholar] [CrossRef]
- Urban, J.M. Perspective: Shedding light on spotted lanternfly impacts in the USA. Pest. Manag. Sci. 2019, 76, 10–17. [Google Scholar] [CrossRef] [PubMed]
- Han, J.M.; Kim, H.; Lim, E.J.; Lee, S.; Kwon, Y.J.; Cho, S. Lycorma delicatula (Hemiptera: Auchenorrhyncha: Fulgoridae: Aphaeninae) finally, but suddenly arrived in Korea. Entomol. Res. 2008, 38, 281–286. [Google Scholar] [CrossRef]
- Park, J.; Kim, M.; Lee, S.; Shin, S.; Kim, J.; Park, I. Biological Characteristics of Lycorma delicatula and the Control Effects of Some Insecticides. Korean J. Appl. Entomol. 2009, 48, 53–57. [Google Scholar] [CrossRef]
- Lee, J.S.; Kim, I.K.; Koh, S.H.; Cho, S.J.; Jang, S.J.; Pyo, S.H.; Choi, W.I. Impact of minimum winter temperature on Lycorma delicatula (Hemiptera: Fulgoridae) egg mortality. J. Asia-Pac. Entomol. 2011, 14, 123–125. [Google Scholar] [CrossRef]
- Namgung, H.; Kim, M.-J.; Baek, S.; Lee, J.-H.; Kim, H. Predicting potential current distribution of Lycorma delicatula (Hemiptera: Fulgoridae) using MaxEnt model in South Korea. J. Asia-Pac. Entomol. 2020, 23, 291–297. [Google Scholar] [CrossRef]
- Kim, H. Morphometric Analysis of Wing Variation of Lantern Fly, Lycorma delicatula from Northeast Asia. Korean J. Appl. Entomol. 2013, 52, 265–271. [Google Scholar] [CrossRef]
- Kim, J.G.; Lee, E.H.; Seo, Y.M.; Kim, N.Y. Cyclic Behavior of Lycorma delicatula (Insecta: Hemiptera: Fulgoridae) on Host Plants. J. Insect Behav. 2011, 24, 423–435. [Google Scholar] [CrossRef]
- Wolfin, M.S.; Binyameen, M.; Wang, Y.; Urban, J.M.; Roberts, D.C.; Baker, T.C. Flight Dispersal Capabilities of Female Spotted Lanternflies (Lycorma delicatula) Related to Size and Mating Status. J. Insect Behav. 2019, 32, 188–200. [Google Scholar] [CrossRef]
- Leach, A.; Leach, H. Characterizing the spatial distributions of spotted lanternfly (Hemiptera: Fulgoridae) in Pennsylvania vineyards. Sci. Rep. 2020, 10, 20588. [Google Scholar] [CrossRef]
- Tadauchi, O.; Inoue, H. On MOKUROKU file based on “A Check List of Japanese Insects” on INTERNET. ESAKIA 2000, 40, 81–84. [Google Scholar]
- Zhang, L.; Zhao, W.; Wang, F.-P.; Qin, D. Genetic Diversity and Population Structure of Natural Lycorma delicatula (White) (Hemiptera: Fulgoridea) Populations in China as Revealed by Microsatellite and Mitochondrial Markers. Insects 2019, 10, 312. [Google Scholar] [CrossRef]
- Park, M.; Kim, K.-S.; Lee, J.-H. Genetic structure of Lycorma delicatula (Hemiptera: Fulgoridae) populations in Korea: Implication for invasion processes in heterogeneous landscapes. Bull. Entomol. Res. 2013, 103, 414–424. [Google Scholar] [CrossRef]
- Kim, H.; Kim, M.; Kwon, D.H.; Park, S.; Lee, Y.; Jang, H.; Lee, S.; Lee, S.H.; Huang, J.; Hong, K.J.; et al. Development and characterization of 15 microsatellite loci from Lycorma delicatula (Hemiptera: Fulgoridae). Anim. Cells Syst. 2011, 15, 295–300. [Google Scholar] [CrossRef]
- Peakall, R.; Smouse, P.E. GenAlEx 6.5: Genetic analysis in Excel. Population genetic software for teaching and research--an update. Bioinformatics 2012, 28, 2537–2539. [Google Scholar] [CrossRef]
- Raymond, M.; Rousset, F. Genepop (Version-1.2)—Population-Genetics Software for Exact Tests and Ecumenicism. J. Hered. 1995, 86, 248–249. [Google Scholar] [CrossRef]
- Rice, W.R. Analyzing Tables of Statistical Tests. Evolution 1989, 43, 223–225. [Google Scholar] [CrossRef]
- Oosterhout, C.V.; Hutchinson, W.F.; Wills, D.P.M.; Shipley, P. Micro-checker: Software for identifying and correcting genotyping errors in microsatellite data. Mol. Ecol. Notes 2004, 4, 535–538. [Google Scholar] [CrossRef]
- Brookfield, J.F.Y. A simple new method for estimating null allele frequency from heterozygote deficiency. Mol. Ecol. 1996, 5, 453–455. [Google Scholar] [CrossRef] [PubMed]
- Excoffier, L.; Smouse, P.E.; Quattro, J.M. Analysis of molecular variance inferred from metric distances among DNA haplotypes: Application to human mitochondrial DNA restriction data. Genetics 1992, 131, 479–491. [Google Scholar] [CrossRef]
- Excoffier, L.; Laval, G.; Schneider, S. Arlequin (version 3.0): An integrated software package for population genetics data analysis. Evol. Bioinform. 2005, 1, 47–50. [Google Scholar] [CrossRef]
- Weir, B.S.; Cockerham, C.C. Estimating F-Statistics for the Analysis of Population Structure. Evolution 1984, 38, 1358–1370. [Google Scholar] [CrossRef] [PubMed]
- Peakall, R.; Smouse, P.E.; Huff, D.R. Evolutionary implications of allozyme and RAPD variation in diploid populations of dioecious buffalograss Buchloë dactyloides. Mol. Ecol. 1995, 4, 135–148. [Google Scholar] [CrossRef]
- Smouse, P.E.; Peakall, R. Spatial autocorrelation analysis of individual multiallele and multilocus genetic structure. Heredity 1999, 82, 561–573. [Google Scholar] [CrossRef]
- Cornuet, J.M.; Luikart, G. Description and power analysis of two tests for detecting recent population bottlenecks from allele frequency data. Genetics 1996, 144, 2001–2014. [Google Scholar] [CrossRef]
- Piry, S.; Luikart, G.; Cornuet, J.M. BOTTLENECK: A computer program for detecting recent reductions in the effective population size using allele frequency data. J. Hered. 1999, 90, 502–503. [Google Scholar] [CrossRef]
- Double, M.C.; Peakall, R.; Beck, N.R.; Cockburn, A. Dispersal, philopatry, and infidelity: Dissecting local genetic structure in superb fairy-wrens (Malurus cyaneus). Evolution 2005, 59, 625–635. [Google Scholar] [CrossRef] [PubMed]
- Peakall, R.; Ruibal, M.; Lindenmayer, D.B. Spatial autocorrelation analysis offers new insights into gene flow in the Australian bush rat, Rattus fuscipes. Evolution 2003, 57, 1182–1195. [Google Scholar] [CrossRef]
- Mantel, N. The detection of disease clustering and a generalized regression approach. Cancer Res. 1967, 27, 209–220. [Google Scholar]
- Pritchard, J.K.; Stephens, M.; Donnelly, P. Inference of population structure using multilocus genotype data. Genetics 2000, 155, 945–959. [Google Scholar] [CrossRef] [PubMed]
- Evanno, G.; Regnaut, S.; Goudet, J. Detecting the number of clusters of individuals using the software STRUCTURE: A simulation study. Mol. Ecol. 2005, 14, 2611–2620. [Google Scholar] [CrossRef]
- Earl, D.A. STRUCTURE HARVESTER: A website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv. Genet. Resour. 2012, 4, 359–361. [Google Scholar] [CrossRef]
- Rosenberg, N.A. DISTRUCT: A program for the graphical display of population structure. Mol. Ecol. Resour. 2004, 4, 137–138. [Google Scholar] [CrossRef]
- Piry, S.; Alapetite, A.; Cornuet, J.-M.; Paetkau, D.; Baudouin, L.; Estoup, A. GENECLASS2: A software for genetic assignment and first-generation migrant detection. J. Hered. 2004, 95, 536–539. [Google Scholar] [CrossRef]
- Rannala, B.; Mountain, J.L. Detecting immigration by using multilocus genotypes. Proc. Natl. Acad. of Sci. USA 1997, 94, 9197–9201. [Google Scholar] [CrossRef]
- Paetkau, D.; Slade, R.; Burden, M.; Estoup, A. Genetic assignment methods for the direct, real-time estimation of migration rate: A simulation-based exploration of accuracy and power. Mol. Ecol. 2004, 13, 55–65. [Google Scholar] [CrossRef] [PubMed]
- Cornuet, J.M.; Santos, F.; Beaumont, M.A.; Robert, C.P.; Marin, J.M.; Balding, D.J.; Guillemaud, T.; Estoup, A. Inferring population history with DIY ABC: A user-friendly approach to approximate Bayesian computation. Bioinformatics 2008, 24, 2713–2719. [Google Scholar] [CrossRef]
- Cornuet, J.M.; Ravigne, V.; Estoup, A. Inference on population history and model checking using DNA sequence and microsatellite data with the software DIYABC (v1.0). BMC Bioinform. 2010, 11, 401. [Google Scholar] [CrossRef]
- Lozier, J.D.; Roderick, G.K.; Mills, N.J. Tracing the invasion history of mealy plum aphid, Hyalopterus pruni (Hemiptera: Aphididae), in North America: A population genetics approach. Biol. Invasions 2009, 11, 299–314. [Google Scholar] [CrossRef]
- Kim, H.; Hoelmer, K.A.; Lee, S. Population genetics of the soybean aphid in North America and East Asia: Test for introduction between native and introduced populations. Biol. Invasions 2017, 19, 597–614. [Google Scholar] [CrossRef]
- Zepeda-Paulo, F.A.; Simon, J.C.; Ramirez, C.C.; Fuentes-Contreras, E.; Margaritopoulos, J.T.; Wilson, A.C.C.; Sorenson, C.E.; Briones, L.M.; Azevedo, R.; Ohashi, D.V.; et al. The invasion route for an insect pest species: The tobacco aphid in the New World. Mol. Ecol. 2010, 19, 4738–4752. [Google Scholar] [CrossRef] [PubMed]
- Kim, I.K.; Koh, S.H.; Lee, J.S.; Il Choi, W.; Shin, S.C. Discovery of an egg parasitoid of Lycorma delicatula (Hemiptera: Fulgoridae) an invasive species in South Korea. J. Asia-Pac. Entomol. 2011, 14, 213–215. [Google Scholar] [CrossRef]
- Kim, K.S.; Sappington, T.W.; Allen, C.T. Genetic profiling to determine potential origins of boll weevils (Coleoptera: Curculionidae) captured in a Texas eradication zone: Endemicity, immigration, or sabotage? J. Econ. Entomol. 2008, 101, 1729–1736. [Google Scholar] [CrossRef] [PubMed]
- Ascunce, M.S.; Yang, C.-C.; Oakey, J.; Calcaterra, L.; Wu, W.-J.; Shih, C.-J.; Goudet, J.; Ross, K.G.; Shoemaker, D. Global Invasion History of the Fire Ant Solenopsis invicta. Science 2011, 331, 1066–1068. [Google Scholar] [CrossRef] [PubMed]
- Keller, J.A.; Johnson, A.E.; Uyi, O.; Wurzbacher, S.; Long, D.; Hoover, K. Dispersal of Lycorma delicatula (Hemiptera: Fulgoridae) Nymphs Through Contiguous, Deciduous Forest. Environ. Entomol. 2020, 49, 1012–1018. [Google Scholar] [CrossRef]
- Gippet, J.M.W.; Liebhold, A.M.; Fenn-Moltu, G.; Bertelsmeier, C. Human-mediated dispersal in insects. Curr. Opin. Insect Sci. 2019, 35, 96–102. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pop. ID | Country | Collection Site | No. | GPS-N | GPS-E | Date |
|---|---|---|---|---|---|---|
| KR06-CA | Korea | Cheonan, CN | 16 | 36.44.01.6 | 127.15.08.1 | 15-Sep-2006 |
| KR07-SL | Korea | Seoul | 9 | 37.33.44.1 | 127.05.12.1 | 26-Sep-2007 |
| KR08-SG | Korea | Seoul and Gyeonggi | 32 | 37.23.52.1 | 126.57.26.1 | 20-Aug-2008 |
| KR09-GG | Korea | Gyeonggi | 44 | 37.38.37.1 | 127.14.10.1 | 30-Jul-2009 |
| KR09-JE | Korea | Jeongeub, JB | 9 | 35.28.55.1 | 126.53.12.1 | 23-Sep-2009 |
| KR09-GB | Korea | Gyeongbuk | 5 | 35.20.22.1 | 128.47.19.1 | 20-Jul-2009 |
| KR09-GW | Korea | Gangwon | 23 | 37.21.29.1 | 127.54.12.1 | 24-Aug-2009 |
| KR10-SL | Korea | Seoul | 20 | 37.33.42.6 | 126.56.39.3 | 28-Jun-2010 |
| KR10-IN | Korea | Incheon, GG | 40 | 37.47.29.9 | 126.16.14.4 | 3-Jul-2010 |
| KR10-NY | Korea | Namyangju, GG | 15 | 37.37.02.6 | 127.13.29.9 | 22-Aug-2010 |
| KR10-SW | Korea | Suwon, GG | 20 | 37.16.07.7 | 126.59.06.7 | 17-Aug-2010 |
| KR10-IC | Korea | Icheon, GG | 15 | 37.20.12.9 | 127.26.29.5 | 14-Jul-2010 |
| KR10-AS | Korea | Anseong, GG | 14 | 37.04.33.1 | 127.07.57.2 | 30-Sep-2010 |
| KR10-CA | Korea | Cheonan, CN | 22 | 36.44.01.6 | 127.15.08.1 | 19-Aug-2010 |
| KR10-CY | Korea | Cheongyang, CN | 20 | 36.26.15.8 | 126.46.05.0 | 14-Sep-2010 |
| KR10-NS | Korea | Nonsan, CN | 20 | 36.13.20.6 | 127.00.55.1 | 29-Sep-2010 |
| KR10-GC | Korea | Gochang, JB | 20 | 35.25.50.8 | 126.43.09.7 | 20-Aug-2010 |
| KR10-GJ | Korea | Gimje, JB | 20 | 35.48.28.7 | 126.59.40.7 | 20-Aug-2010 |
| KR10-BA | Korea | Buan, JB | 20 | 35.40.36.9 | 126.44.24.8 | 20-Aug-2010 |
| KR10-KJ | Korea | Gwangju, JN | 9 | 35.08.06.8 | 126.55.52.3 | 4-Sep-2010 |
| KR10-AD | Korea | Andong, GB | 11 | 36.32.31.1 | 128.47.49.4 | 31-Jul-2010 |
| KR10-YC | Korea | Yechoen, GB | 20 | 36.39.56.0 | 128.31.12.0 | 6-Aug-2010 |
| KR10-SJ | Korea | Sangju, GB | 17 | 36.22.35.3 | 128.08.30.7 | 4-Sep-2010 |
| KR11-CW | Korea | Changwon, GN | 16 | 37.19.41.1 | 127.56.47.1 | 5-Aug-2011 |
| KR11-SC | Korea | Samcheok, GW | 16 | 37.14.28.1 | 129.00.46.2 | 29-Jun-2011 |
| CN09-BJ | China | Beijing | 10 | 39.54.16.8 | 116.24.29.5 | 22-Jul-2009 |
| CN10-TJ | China | Tianjin | 16 | 39.07.15.1 | 117.12.54.1 | 5-Jul-2010 |
| CN11-YT | China | Yantai, SD | 24 | 37.31.40.3 | 121.21.27.9 | 15-Aug-2011 |
| CN11-HY | China | Haiyang, SD | 20 | 36.56.59.5 | 121.03.28.8 | 16-Aug-2011 |
| CN11-QD | China | Qingdao, SD | 24 | 36.19.21.6 | 120.23.36.9 | 15-Aug-2011 |
| CN11-LY | China | Linyi, SD | 20 | 35.43.25.9 | 118.32.47.1 | 17-Aug-2011 |
| CN11-RZ | China | Rihzhao, SD | 24 | 35.28.44.2 | 119.28.15.6 | 16-Aug-2011 |
| CN11-LG | China | Lianyungang, JS | 24 | 34.35.53.1 | 119.10.46.9 | 18-Aug-2011 |
| CN10-SH | China | Shanghai | 26 | 31.37.23.4 | 121.23.50.2 | 4-Sep-2010 |
| CN10-NB | China | Ningbo, ZJ | 20 | 29.47.42.4 | 121.47.37.4 | 5-Sep-2010 |
| CN10-TT | China | Tiantai, ZJ | 28 | 29.03.45.7 | 121.02.45.1 | 6-Sep-2010 |
| CN10-LA | China | Linan, ZJ | 40 | 30.14.01.9 | 119.43.29.0 | 7-Sep-2010 |
| JP10-HS | Japan | Hakusan, IK | 13 | 36.35.40.8 | 136.37.32.1 | 15-Sep-2010 |
| Pop. ID | MLG † | Ho (±s.d.) | He (±s.d.) | HWE | HS | NA | RS | FIS § |
|---|---|---|---|---|---|---|---|---|
| KR06-CA | 16 | 0.443 (0.073) | 0.495 (0.059) | ns | 0.50 | 3.67 | 2.73 | 0.109 |
| KR07-SL | 9 | 0.389 (0.077) | 0.471 (0.069) | ns | 0.48 | 3.33 | 2.81 | 0.182 |
| KR08-SG | 32 | 0.388 (0.061) | 0.445 (0.058) | ns | 0.45 | 4.42 | 2.68 | 0.130 |
| KR09-GG | 44 | 0.445 (0.054) | 0.474 (0.044) | ns | 0.47 | 5.33 | 2.79 | 0.062 |
| KR09-JE | 9 | 0.398 (0.069) | 0.404 (0.066) | ns | 0.40 | 3.17 | 2.59 | 0.014 |
| KR09-GB | 5 | 0.400 (0.070) | 0.513 (0.058) | ns | 0.53 | 2.75 | 2.75 | 0.241 |
| KR09-GW | 23 | 0.322 (0.066) | 0.401 (0.074) | * deficit | 0.40 | 3.75 | 2.55 | 0.199 |
| KR10-SL | 20 | 0.417 (0.061) | 0.436 (0.052) | ns | 0.44 | 3.92 | 2.68 | 0.046 |
| KR10-IN | 40 | 0.440 (0.065) | 0.468 (0.055) | ns | 0.47 | 4.58 | 2.80 | 0.062 |
| KR10-NY | 15 | 0.450 (0.079) | 0.434 (0.065) | ns | 0.43 | 4.17 | 2.82 | −0.039 |
| KR10-SW | 20 | 0.467 (0.073) | 0.445 (0.068) | ns | 0.44 | 4.25 | 2.77 | −0.049 |
| KR10-IC | 15 | 0.467 (0.051) | 0.480 (0.050) | ns | 0.48 | 4.00 | 2.85 | 0.029 |
| KR10-AS | 14 | 0.500 (0.080) | 0.463 (0.061) | ns | 0.46 | 3.42 | 2.68 | −0.083 |
| KR10-CA | 22 | 0.485 (0.071) | 0.487 (0.046) | ns | 0.49 | 4.08 | 2.81 | 0.004 |
| KR10-CY | 20 | 0.467 (0.061) | 0.468 (0.046) | ns | 0.47 | 4.00 | 2.80 | 0.002 |
| KR10-NS | 20 | 0.433 (0.052) | 0.481 (0.042) | ns | 0.48 | 4.58 | 2.88 | 0.101 |
| KR10-GC | 20 | 0.463 (0.082) | 0.505 (0.062) | ns | 0.51 | 4.67 | 3.01 | 0.087 |
| KR10-GJ | 20 | 0.407 (0.063) | 0.437 (0.063) | ns | 0.49 | 3.83 | 2.70 | 0.019 |
| KR10-BA | 20 | 0.442 (0.060) | 0.453 (0.050) | ns | 0.45 | 4.00 | 2.62 | 0.025 |
| KR10-KJ | 9 | 0.407 (0.063) | 0.437 (0.063) | ns | 0.44 | 3.08 | 2.59 | 0.071 |
| KR10-AD | 11 | 0.333 (0.075) | 0.378 (0.069) | ns | 0.38 | 2.50 | 2.21 | 0.124 |
| KR10-YC | 20 | 0.438 (0.053) | 0.432 (0.046) | ns | 0.43 | 3.25 | 2.45 | −0.014 |
| KR10-SJ | 17 | 0.456 (0.076) | 0.444 (0.048) | ns | 0.44 | 2.42 | 2.20 | −0.028 |
| KR10-CW | 16 | 0.172 (0.080) | 0.171 (0.070) | ns | 0.17 | 1.58 | 1.46 | −0.006 |
| KR11-SC | 16 | 0.453 (0.066) | 0.447 (0.063) | ns | 0.45 | 3.50 | 2.63 | −0.014 |
| CN09-BJ | 10 | 0.525 (0.072) | 0.493 (0.062) | ns | 0.49 | 3.58 | 2.88 | −0.068 |
| CN10-TJ | 16 | 0.432 (0.085) | 0.423 (0.069) | ns | 0.42 | 3.00 | 2.40 | −0.024 |
| CN11-YT | 24 | 0.542 (0.052) | 0.558 (0.028) | ns | 0.56 | 3.92 | 2.80 | 0.030 |
| CN11-HY | 20 | 0.463 (0.072) | 0.497 (0.057) | ns | 0.50 | 3.42 | 2.56 | 0.070 |
| CN11-QD | 24 | 0.448 (0.048) | 0.500 (0.036) | ns | 0.50 | 4.08 | 2.73 | 0.106 |
| CN11-LY | 20 | 0.504 (0.052) | 0.489 (0.050) | ns | 0.49 | 3.25 | 2.62 | −0.033 |
| CN11-RZ | 24 | 0.479 (0.084) | 0.457 (0.076) | ns | 0.46 | 4.17 | 2.75 | −0.050 |
| CN11-LG | 24 | 0.424 (0.051) | 0.456 (0.051) | ns | 0.46 | 3.92 | 2.67 | 0.073 |
| CN10-SH | 26 | 0.413 (0.041) | 0.477 (0.048) | ns | 0.48 | 4.67 | 2.77 | 0.136 |
| CN10-NB | 20 | 0.479 (0.065) | 0.547 (0.068) | ns | 0.55 | 5.08 | 3.31 | 0.127 |
| CN10-TT | 28 | 0.548 (0.050) | 0.580 (0.060) | ns | 0.58 | 4.83 | 3.20 | 0.058 |
| CN10-LA | 40 | 0.542 (0.065) | 0.608 (0.060) | * deficit | 0.61 | 7.25 | 3.76 | 0.110 |
| JP10-HS | 13 | 0.436 (0.060) | 0.432 (0.044) | ns | 0.43 | 2.25 | 2.12 | −0.011 |
| Among Groups | Among Populations within Groups | Within Populations | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Case | Va | Percentage | P | Vb | Percentage | p | Vc | Percentage | p |
| 1 | 0.02 | 0.48 | 0.3123 | 0.40 | 12.23 | <0.0001 | 2.84 | 87.29 | <0.0001 |
| 2 | 0.14 | 4.35 | 0.0067 | 0.34 | 10.10 | <0.0001 | 2.88 | 85.54 | <0.0001 |
| 3 | 0.20 | 6.01 | 0.0001 | 0.25 | 7.64 | <0.0001 | 2.84 | 86.35 | <0.0001 |
| 4 | 0.33 | 9.90 | <0.0001 | 0.19 | 5.63 | <0.0001 | 2.84 | 84.47 | <0.0001 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, H.; Kim, S.; Lee, Y.; Lee, H.-S.; Lee, S.-J.; Lee, J.-H. Tracing the Origin of Korean Invasive Populations of the Spotted Lanternfly, Lycorma delicatula (Hemiptera: Fulgoridae). Insects 2021, 12, 539. https://doi.org/10.3390/insects12060539
Kim H, Kim S, Lee Y, Lee H-S, Lee S-J, Lee J-H. Tracing the Origin of Korean Invasive Populations of the Spotted Lanternfly, Lycorma delicatula (Hemiptera: Fulgoridae). Insects. 2021; 12(6):539. https://doi.org/10.3390/insects12060539
Chicago/Turabian StyleKim, Hyojoong, Sohee Kim, Yerim Lee, Heung-Sik Lee, Seong-Jin Lee, and Jong-Ho Lee. 2021. "Tracing the Origin of Korean Invasive Populations of the Spotted Lanternfly, Lycorma delicatula (Hemiptera: Fulgoridae)" Insects 12, no. 6: 539. https://doi.org/10.3390/insects12060539
APA StyleKim, H., Kim, S., Lee, Y., Lee, H.-S., Lee, S.-J., & Lee, J.-H. (2021). Tracing the Origin of Korean Invasive Populations of the Spotted Lanternfly, Lycorma delicatula (Hemiptera: Fulgoridae). Insects, 12(6), 539. https://doi.org/10.3390/insects12060539