
Reference Transcriptome Data in Silkworm Bombyx mori

 , , ,
, , , 
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
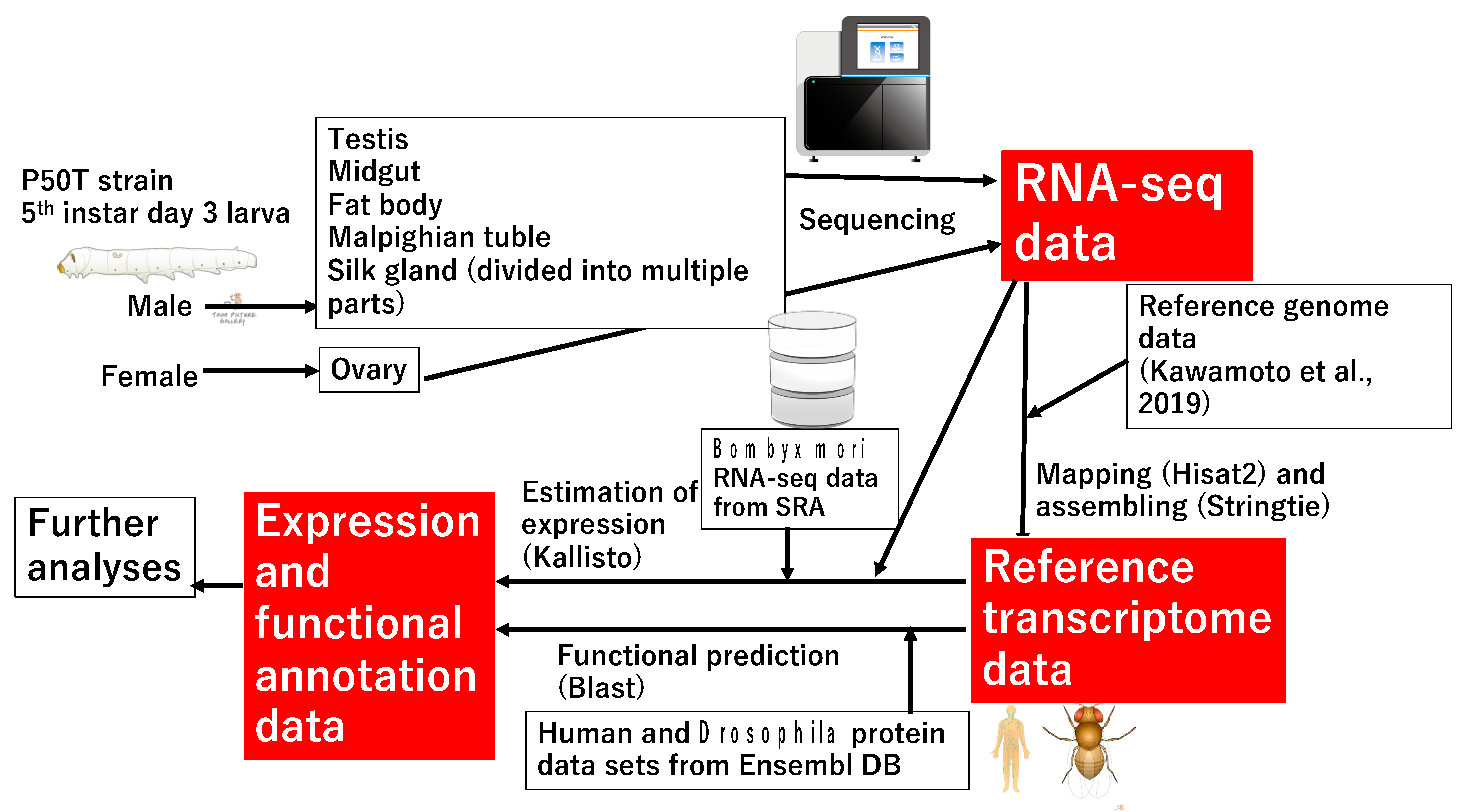
2.1. Silkworm Rearing, RNA Extraction, and Sequencing
2.2. Construction of RTD and Estimation of the Expression of Each Transcript
2.3. Annotation for the Reference Transcriptome and Functional Enrichment Analyses
2.4. Comparison of Gene Structures among Different Models

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | SRA Run ID | Strain | Sex | Reference |
|---|---|---|---|---|
| ASG-1,2,3 | DRR186474,DRR186475,DRR186476 | p50T | Male | This study |
| MSG_A-1,2,3 | DRR186477,DRR186478,DRR186479 | p50T | Male | This study |
| MSG_M-1,2,3 | DRR186480,DRR186481,DRR186482 | p50T | Male | This study |
| MSG_P-1,2,3 | DRR186483,DRR186484,DRR186485 | p50T | Male | This study |
| PSG-1,2,3 | DRR186486,DRR186487,DRR186488 | p50T | Male | This study |
| FB-1,2,3 | DRR186489,DRR186490,DRR186491 | p50T | Male | This study |
| MG-1,2,3 | DRR186492,DRR186493,DRR186494 | p50T | Male | This study |
| MT-1,2,3 | DRR186495,DRR186496,DRR186497 | p50T | Male | This study |
| TT-1,2,3 | DRR186498,DRR186499,DRR186500 | p50T | Male | This study |
| OV-1,2,3 | DRR186501,DRR186502,DRR186503 | p50T | Female | This study |
| BN_TT-1,2,3 | DRR068893,DRR068894,DRR068895 | o751 | Male | [25] |
| BN_FB-1,2,3 | DRR095105,DRR095106,DRR095107 | o751 | Male | [27] |
| BN_MG-1,2,3 | DRR095108,DRR095109,DRR095110 | o751 | Male | [26] |
| BN_MT-1,2,3 | DRR095111,DRR095112,DRR095113 | o751 | Male | [27] |
| BN_SG-1,2,3 | DRR095114,DRR095115,DRR095116 | o751 | Male | [27] |
2.5. RT-PCR
3. Results
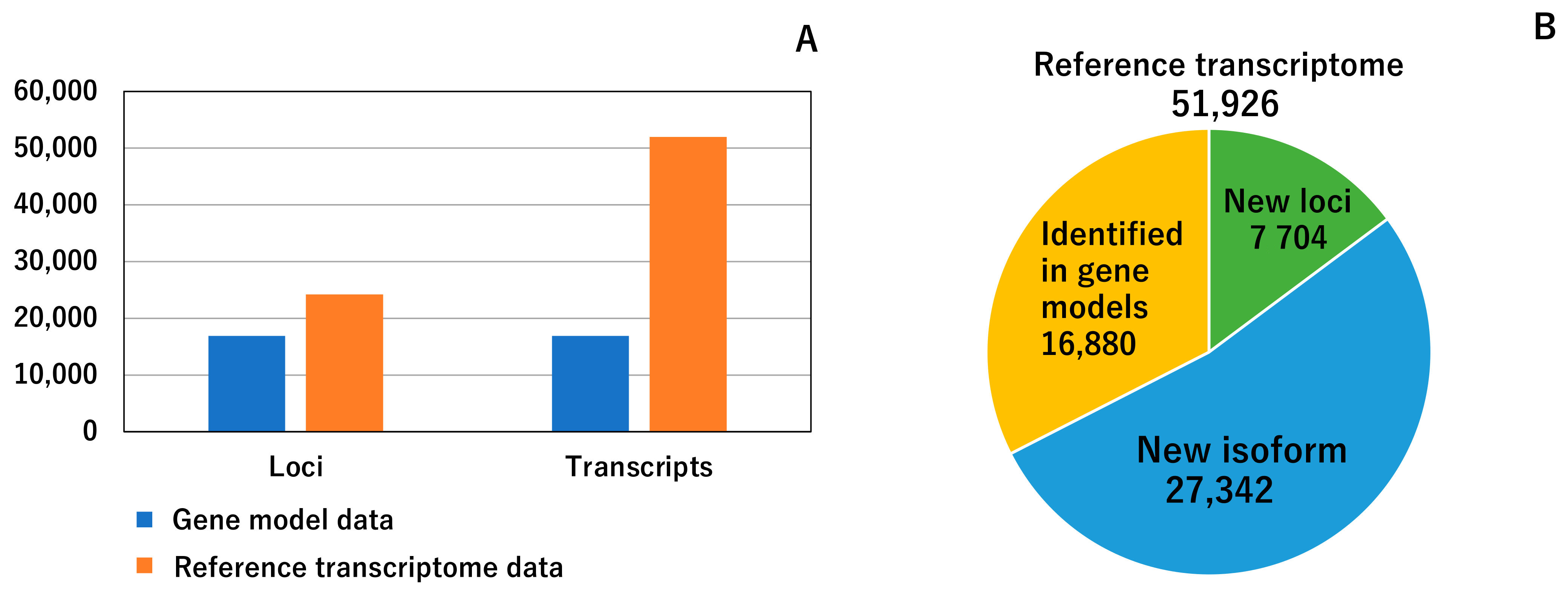
3.1. Reference Transcriptome Data
3.2. Comparison between Constructed Reference Transcriptome Data and Previous Gene Model Data
3.3. Elaborated Structures of Silk Genes
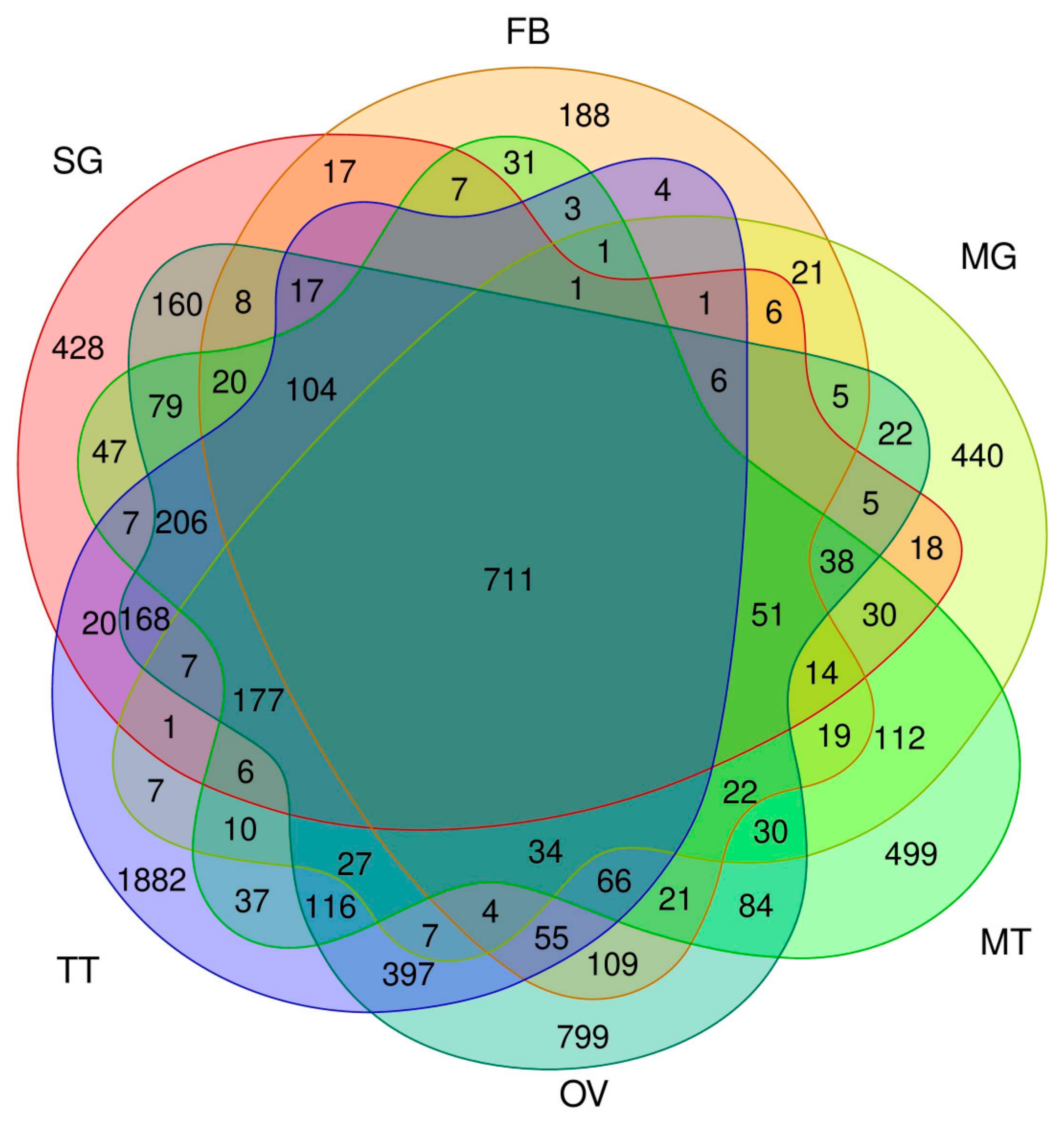
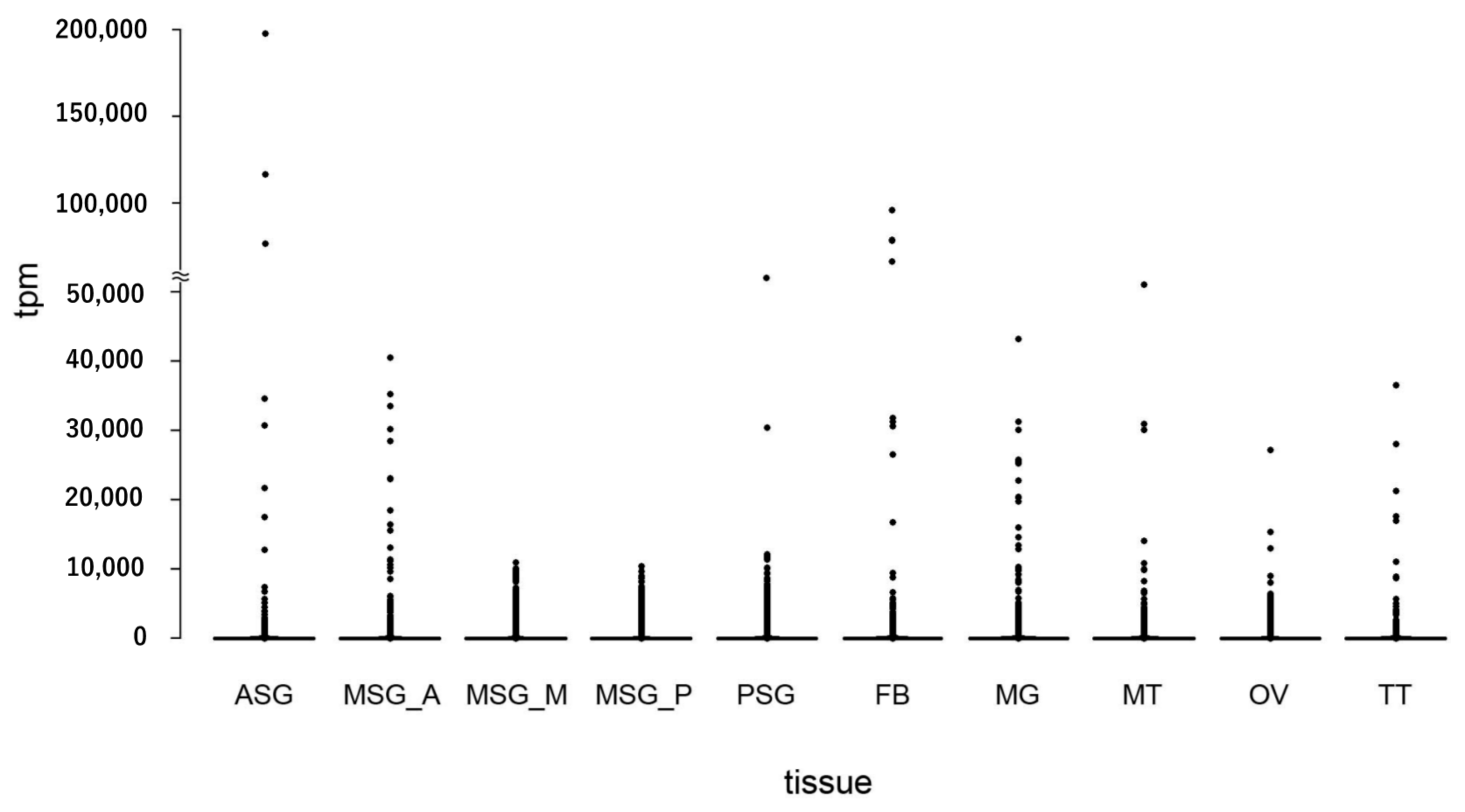
3.4. Estimating the Abundance of the Reference Transcriptome in Multiple Tissues
3.5. Transcript Abundance in Each Tissue and in the Silk Gland
3.6. Expression Analysis of Transcriptional Factor Genes in the Silkworm
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tan, A.; Tanaka, H.; Tamura, T.; Shiotsuki, T. Precocious metamorphosis in transgenic silkworms overexpressing juvenile hormone esterase. Proc. Natl. Acad. Sci. USA 2005, 102, 11751–11756. [Google Scholar] [CrossRef] [PubMed]
- Ito, K.; Kidokoro, K.; Sezutsu, H.; Nohata, J.; Yamamoto, K.; Kobayashi, I.; Uchino, K.; Kalyebi, A.; Eguchi, R.; Hara, W. De-letion of a Gene Encoding an Amino Acid Transporter in the Midgut Membrane Causes Resistance to a Bombyx Parvo-like Virus. Proc. Natl. Acad. Sci. USA 2008, 105, 7523–7527. [Google Scholar] [CrossRef]
- Sakurai, T.; Mitsuno, H.; Haupt, S.S.; Uchino, K.; Yokohari, F.; Nishioka, T.; Kobayashi, I.; Sezutsu, H.; Tamura, T.; Kanzaki, R. A Single Sex Pheromone Receptor Determines Chemical Response Specificity of Sexual Behavior in the Silkmoth Bombyx mori. PLoS Genet. 2011, 7, e1002115. [Google Scholar] [CrossRef] [PubMed]
- Daimon, T.; Uchibori, M.; Nakao, H.; Sezutsu, H.; Shinoda, T. Knockout silkworms reveal a dispensable role for juvenile hormones in holometabolous life cycle. Proc. Natl. Acad. Sci. USA 2015, 112, E4226–E4235. [Google Scholar] [CrossRef]
- KonDo, Y.; Yoda, S.; Mizoguchi, T.; Ando, T.; Yamaguchi, J.; Yamamoto, K.; Banno, Y.; Fujiwara, H. Toll ligand Spätzle3 controls melanization in the stripe pattern formation in caterpillars. Proc. Natl. Acad. Sci. USA 2017, 114, 8336–8341. [Google Scholar] [CrossRef]
- Yukuhiro, K.; Sezutsu, H.; Tsubota, T.; Takasu, Y.; Kameda, T.; Yonemura, N. Insect Silks and Cocoons: Structural and Mo-Lecular Aspects. In Extracellular Composite Matrices in Arthropods; Springer: Berlin, Germany, 2016; pp. 515–555. [Google Scholar]
- Tamura, T.; Thibert, C.; Royer, C.; Kanda, T.; Eappen, A.; Kamba, M.; Kômoto, N.; Thomas, J.-L.; Mauchamp, B.; Chavancy, G.; et al. Germline transformation of the silkworm Bombyx mori L. using a piggyBac transposon-derived vector. Nat. Biotechnol. 2000, 18, 81–84. [Google Scholar] [CrossRef] [PubMed]
- Tatematsu, K.-I.; Kobayashi, I.; Uchino, K.; Sezutsu, H.; Iizuka, T.; Yonemura, N.; Tamura, T. Construction of a binary transgenic gene expression system for recombinant protein production in the middle silk gland of the silkworm Bombyx mori. Transgenic Res. 2009, 19, 473–487. [Google Scholar] [CrossRef] [PubMed]
- Mita, K.; Kasahara, M.; Sasaki, S.; Nagayasu, Y.; Yamada, T.; Kanamori, H.; Namiki, N.; Kitagawa, M.; Yamashita, H.; Yasukochi, Y.; et al. The Genome Sequence of Silkworm, Bombyx mori. DNA Res. 2004, 11, 27–35. [Google Scholar] [CrossRef]
- Biology Analysis Group; Xia, Q.; Zhou, Z.; Lu, C.; Cheng, D.; Dai, F.-Y.; Liu, B.; Zhao, P.; Zha, X.; Cheng, T.; et al. A Draft Sequence for the Genome of the Domesticated Silkworm (Bombyx mori). Science 2004, 306, 1937–1940. [Google Scholar] [CrossRef]
- Consortium, I.S.G. The Genome of a Lepidopteran Model Insect, the Silkworm Bombyx mori. Insect Biochem. Mol. Biol. 2008, 38, 1036–1045. [Google Scholar]
- Xia, Q.; Cheng, D.; Duan, J.; Wang, G.; Cheng, T.; Zha, X.; Liu, C.; Zhao, P.; Dai, F.; Zhang, Z. Microarray-Based Gene Ex-pression Profiles in Multiple Tissues of the Domesticated Silkworm, Bombyx mori. Genome Biol. 2007, 8, 1–13. [Google Scholar] [CrossRef]
- Yamamoto, K.; Nohata, J.; Kadono-Okuda, K.; Narukawa, J.; Sasanuma, M.; Sasanuma, S.-I.; Minami, H.; Shimomura, M.; Suetsugu, Y.; Banno, Y.; et al. A BAC-based integrated linkage map of the silkworm Bombyx mori. Genome Biol. 2008, 9, R21. [Google Scholar] [CrossRef]
- Suetsugu, Y.; Futahashi, R.; Kanamori, H.; Kadono-Okuda, K.; Sasanuma, S.-I.; Narukawa, J.; Ajimura, M.; Jouraku, A.; Namiki, N.; Shimomura, M.; et al. Large Scale Full-Length cDNA Sequencing Reveals a Unique Genomic Landscape in a Lepidopteran Model Insect, Bombyx mori. G3 Genes Genomes Genet. 2013, 3, 1481–1492. [Google Scholar] [CrossRef] [PubMed]
- Kawamoto, M.; Jouraku, A.; Toyoda, A.; Yokoi, K.; Minakuchi, Y.; Katsuma, S.; Fujiyama, A.; Kiuchi, T.; Yamamoto, K.; Shimada, T. High-quality genome assembly of the silkworm, Bombyx mori. Insect Biochem. Mol. Biol. 2019, 107, 53–62. [Google Scholar] [CrossRef] [PubMed]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Kim, D.; Paggi, J.M.; Park, C.; Bennett, C.; Salzberg, S.L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 2019, 37, 907–915. [Google Scholar] [CrossRef] [PubMed]
- Pertea, M.; Kim, D.; Pertea, G.M.; Leek, J.T.; Salzberg, S.L. Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown. Nat. Protoc. 2016, 11, 1650–1667. [Google Scholar] [CrossRef]
- Bray, N.L.; Pimentel, H.; Melsted, P.; Pachter, L. Near-optimal probabilistic RNA-seq quantification. Nat. Biotechnol. 2016, 34, 525–527. [Google Scholar] [CrossRef] [PubMed]
- Ichino, F.; Bono, H.; Nakazato, T.; Toyoda, A.; Fujiyama, A.; Iwabuchi, K.; Sato, R.; Tabunoki, H. Construction of a simple evaluation system for the intestinal absorption of an orally administered medicine using Bombyx mori larvae. Drug Discov. Ther. 2018, 12, 7–15. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, Y.; Nojima, Y.; Sakamoto, T.; Iwabuchi, K.; Nakazato, T.; Bono, H.; Toyoda, A.; Fujiyama, A.; Kanost, M.; Tabunoki, H. Comparative analysis of seven types of superoxide dismutases for their ability to respond to oxidative stress in Bombyx mori. Sci. Rep. 2019, 9, 2170. [Google Scholar] [CrossRef]
- Kikuchi, A.; Nakazato, T.; Ito, K.; Nojima, Y.; Yokoyama, T.; Iwabuchi, K.; Bono, H.; Toyoda, A.; Fujiyama, A.; Sato, R. Iden-tification of Functional Enolase Genes of the Silkworm Bombyx mori from Public Databases with a Combination of Dry and Wet Bench Processes. BMC Genom. 2017, 18, 1–12. [Google Scholar] [CrossRef]
- Altschul, S.F.; Madden, T.L.; Schäffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef]
- Tabunoki, H.; Ono, H.; Ode, H.; Ishikawa, K.; Kawana, N.; Banno, Y.; Shimada, T.; Nakamura, Y.; Yamamoto, K.; Satoh, J.-I.; et al. Identification of Key Uric Acid Synthesis Pathway in a Unique Mutant Silkworm Bombyx mori Model of Parkinson’s Disease. PLoS ONE 2013, 8, e69130. [Google Scholar] [CrossRef]
- Kinsella, R.J.; Kahari, A.; Haider, S.; Zamora, J.; Proctor, G.; Spudich, G.; Almeida-King, J.; Staines, D.; Derwent, P.; Kerhornou, A.; et al. Ensembl BioMarts: A hub for data retrieval across taxonomic space. Database 2011, 2011, bar030. [Google Scholar] [CrossRef]
- Zhou, Y.; Zhou, B.; Pache, L.; Chang, M.; Khodabakhshi, A.H.; Tanaseichuk, O.; Benner, C.; Chanda, S.K. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat. Commun. 2019, 10, 1–10. [Google Scholar] [CrossRef]
- Yang, C.-C.; Yokoi, K.; Yamamoto, K.; Jouraku, A. An update of KAIKObase, the silkworm genome database. Database 2021, 2021. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.; Cheng, T.; Wu, Y.; Hu, W.; Long, R.; Liu, C.; Zhao, P.; Xia, Q. Transcriptomic Analysis of the Anterior Silk Gland in the Domestic Silkworm (Bombyx mori)—Insight into the Mechanism of Silk Formation and Spinning. PLoS ONE 2015, 10, e0139424. [Google Scholar] [CrossRef]
- Shi, R.; Ma, S.; He, T.; Peng, J.; Zhang, T.; Chen, X.; Wang, X.; Chang, J.; Xia, Q.; Zhao, P. Deep Insight into the Transcriptome of the Single Silk Gland of Bombyx mori. Int. J. Mol. Sci. 2019, 20, 2491. [Google Scholar] [CrossRef]
- Ishikawa, E.; Suzuki, Y. Tissue- and Stage-Specific Expression of Sericin Genes in the Middle Silk Gland of Bombyx mori. (sericin mRNA/northern blotting/gene regulation). Dev. Growth Differ. 1985, 27, 73–82. [Google Scholar] [CrossRef]
- Garel, A.; Deleage, G.; Prudhomme, J.C. Structure and organization of the Bombyx mori sericin 1 gene and of the sericins 1 deduced from the sequence of the Ser 1B cDNA. Insect Biochem. Mol. Biol. 1997, 27, 469–477. [Google Scholar] [CrossRef]
- Takasu, Y.; Yamada, H.; Tsubouchi, K. Isolation of Three Main Sericin Components from the Cocoon of the Silkworm, Bombyx mori. Biosci. Biotechnol. Biochem. 2002, 66, 2715–2718. [Google Scholar] [CrossRef]
- Zurovec, M.; Yonemura, N.; Kludkiewicz, B.; Sehnal, F.; Kodrík, D.; Vieira, L.C.; Kucerova, L.; Strnad, H.; Konik, P.; Sehadová, H. Sericin Composition in the Silk of Antheraea yamamai. Biomacromolecules 2016, 17, 1776–1787. [Google Scholar] [CrossRef]
- Takasu, Y.; Yamada, H.; Tsubouchi, K. The Silk Sericin Component with Low Crystallinity. Sanshi-Konchu Biotec 2006, 75, 133–139. [Google Scholar]
- Takasu, Y.; Yamada, H.; Tamura, T.; Sezutsu, H.; Mita, K.; Tsubouchi, K. Identification and characterization of a novel sericin gene expressed in the anterior middle silk gland of the silkworm Bombyx mori. Insect Biochem. Mol. Biol. 2007, 37, 1234–1240. [Google Scholar] [CrossRef]
- Dong, Z.; Guo, K.; Zhang, X.; Zhang, T.; Zhang, Y.; Ma, S.; Chang, H.; Tang, M.; An, L.; Xia, Q.; et al. Identification of Bombyx mori sericin 4 protein as a new biological adhesive. Int. J. Biol. Macromol. 2019, 132, 1121–1130. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Z.; Guan, Z.; Liu, G.; Wang, Y.; Zhang, Z. SGID: A comprehensive and interactive database of the silkworm. Database 2019, 2019. [Google Scholar] [CrossRef]
- Zhao, D.; Woolner, S.; Bownes, M. The Mirror transcription factor links signalling pathways in Drosophila oogenesis. Dev. Genes Evol. 2000, 210, 449–457. [Google Scholar] [CrossRef]
- Li, M.A.; Alls, J.D.; Avancini, R.M.; Koo, K.; Godt, D. The large Maf factor Traffic Jam controls gonad morphogenesis in Drosophila. Nat. Cell Biol. 2003, 5, 994–1000. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Mann, R.S. Requirement for two nearly identical TGIF-related homeobox genes in Drosophila spermatogenesis. Development 2003, 130, 2853–2865. [Google Scholar] [CrossRef]
- Nanda, S.; DeFalco, T.; Loh, S.H.Y.; Phochanukul, N.; Camara, N.; Van Doren, M.; Russell, S. Sox100B, a Drosophila Group E Sox-domain Gene, Is Required for Somatic Testis Differentiation. Sex. Dev. 2009, 3, 26–37. [Google Scholar] [CrossRef]
- Vaskivuo, T.E.; Anttonen, M.; Herva, R.; Billig, H.; Dorland, M.; Velde, E.R.T.; Stenbäck, F.; Heikinheimo, M.; Tapanainen, J.S. Survival of Human Ovarian Follicles from Fetal to Adult Life: Apoptosis, Apoptosis-Related Proteins, and Transcription Factor GATA-41. J. Clin. Endocrinol. Metab. 2001, 86, 3421–3429. [Google Scholar] [CrossRef] [PubMed]
- Mach, V.; Takiya, S.; Ohno, K.; Handa, H.; Imai, T.; Suzuki, Y. Silk gland factor-1 involved in the regulation of bombyx sericin-1 gene contains fork head motif. J. Biol. Chem. 1995, 270, 9340–9346. [Google Scholar] [CrossRef]
- Ohno, K.; Sawada, J.-I.; Takiya, S.; Kimoto, M.; Matsumoto, A.; Tsubota, T.; Uchino, K.; Hui, C.-C.; Sezutsu, H.; Handa, H.; et al. Silk Gland Factor-2, Involved in Fibroin Gene Transcription, Consists of LIM Homeodomain, LIM-interacting, and Single-stranded DNA-binding Proteins. J. Biol. Chem. 2013, 288, 31581–31591. [Google Scholar] [CrossRef]
- Kimoto, M.; Tsubota, T.; Uchino, K.; Sezutsu, H.; Takiya, S. Hox transcription factor Antp regulates sericin-1 gene expression in the terminal differentiated silk gland of Bombyx mori. Dev. Biol. 2014, 386, 64–71. [Google Scholar] [CrossRef]
- Kimoto, M.; Tsubota, T.; Uchino, K.; Sezutsu, H.; Takiya, S. LIM-homeodomain transcription factor Awh is a key component activating all three fibroin genes, fibH, fibL and fhx, in the silk gland of the silkworm, Bombyx mori. Insect Biochem. Mol. Biol. 2015, 56, 29–35. [Google Scholar] [CrossRef]
- Tsubota, T.; Tomita, S.; Uchino, K.; Kimoto, M.; Takiya, S.; Kajiwara, H.; Yamazaki, T.; Sezutsu, H. A Hox Gene, Antennapedia, Regulates Expression of Multiple Major Silk Protein Genes in the Silkworm Bombyx mori. J. Biol. Chem. 2016, 291, 7087–7096. [Google Scholar] [CrossRef] [PubMed]
- Baumgartner, S.; Bopp, D.; Burri, M.; Noll, M. Structure of two genes at the gooseberry locus related to the paired gene and their spatial expression during Drosophila embryogenesis. Genes Dev. 1987, 1, 1247–1267. [Google Scholar] [CrossRef]
- Grossniklaus, U.; Pearson, R.K.; Gehring, W.J. The Drosophila sloppy paired locus encodes two proteins involved in segmentation that show homology to mammalian transcription factors. Genes Dev. 1992, 6, 1030–1051. [Google Scholar] [CrossRef]
- Russell, S.; Sanchez-Soriano, N.; Wright, C.; Ashburner, M. The Dichaete gene of Drosophila melanogaster encodes a SOX-domain protein required for embryonic segmentation. Development 1996, 122, 3669–3676. [Google Scholar] [CrossRef]
- Couturier, L.; Mazouni, K.; Corson, F.; Schweisguth, F. Regulation of Notch output dynamics via specific E(spl)-HLH factors during bristle patterning in Drosophila. Nat. Commun. 2019, 10, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Yao, J.-G.; Weasner, B.M.; Wang, L.-H.; Jang, C.-C.; Weasner, B.; Tang, C.-Y.; Salzer, C.L.; Chen, C.-H.; Hay, B.; Sun, Y.H.; et al. Differential requirements for the Pax6(5a) genes eyegone and twin of eyegone during eye development in Drosophila. Dev. Biol. 2008, 315, 535–551. [Google Scholar] [CrossRef]
- Ferree, P.M.; Fang, C.; Mastrodimos, M.; Hay, B.A.; Amrhein, H.; Akbari, O.S. Identification of Genes Uniquely Expressed in the Germ-Line Tissues of the Jewel Wasp Nasonia vitripennis. G3 Genes Genomes Genet. 2015, 5, 2647–2653. [Google Scholar] [CrossRef] [PubMed]
- Kondo, S.; Vedanayagam, J.; Mohammed, J.; Eizadshenass, S.; Kan, L.; Pang, N.; Aradhya, R.; Siepel, A.; Steinhauer, J.; Lai, E.C. New genes often acquire male-specific functions but rarely become essential in Drosophila. Genes Dev. 2017, 31, 1841–1846. [Google Scholar] [CrossRef] [PubMed]
- Kaessmann, H. Origins, evolution, and phenotypic impact of new genes. Genome Res. 2010, 20, 1313–1326. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Cheng, T.; Hu, W.; Peng, Z.; Liu, C.; Xia, Q. Genome-wide identification and analysis of JHBP-domain family members in the silkworm Bombyx mori. Mol. Genet. Genom. 2016, 291, 2159–2171. [Google Scholar] [CrossRef]
- Hu, X.; Zhu, M.; Wang, S.; Zhu, L.; Xue, R.; Cao, G.; Gong, C. Proteomics analysis of digestive juice from silkworm during Bombyx mori nucleopolyhedrovirus infection. Proteomics 2015, 15, 2691–2700. [Google Scholar] [CrossRef] [PubMed]
- Horne, I.; Haritos, V.S. Multiple tandem gene duplications in a neutral lipase gene cluster in Drosophila. Gene 2008, 411, 27–37. [Google Scholar] [CrossRef] [PubMed]
- Julien, E.; Coulon-Bublex, M.; Garel, A.; Royer, C.; Chavancy, G.; Prudhomme, J.-C.; Couble, P. Silk Gland Development and Regulation of Silk Protein Genes. Compr. Mol. Insect Sci. 2005, 369–384. [Google Scholar] [CrossRef]
- Akai, H. Ultrastructural Morphology of Insects; University of Tokyo Press: Tokyo, Japan, 1976. [Google Scholar]
- Takasu, Y.; Hata, T.; Uchino, K.; Zhang, Q. Identification of Ser2 proteins as major sericin components in the non-cocoon silk of Bombyx mori. Insect Biochem. Mol. Biol. 2010, 40, 339–344. [Google Scholar] [CrossRef]
- Bello, B.; Horard, B.; Couble, P. The Selective Expression of Silk-Protein-Encoding Genes in Bombyx mori Silk Gland. Bull. de l’Institut Pasteur 1994, 92, 81–100. [Google Scholar]
- Akai, H. Porous Cocoon Filaments-Their Characteristics and Formation. Int. J. Wild Silkmoth Silk 2005, 10, 57–74. [Google Scholar]











| Transcript ID | Family | Human_Gname | Fly_Gname |
|---|---|---|---|
| KWMTBOMO02291.mrna1 | HMG | SOX14 | D |
| KWMTBOMO02290.mrna1 | HMG | SOX21 | Sox21b |
| KWMTBOMO07638.mrna1 | ETS | SPDEF | Ets98B |
| KWMTBOMO08935.mrna1 | zf-LITAF-like | CG30273 | |
| KWMTBOMO08827.mrna1 | zf-C2H2 | GLIS2 | sug |
| KWMTBOMO10947.mrna1 | zf-CCCH | ZFP36L1 | Tis11 |
| KWMTBOMO09826.mrna1 | zf-C2H2 | ZBTB49 | Clamp |
| KWMTBOMO12968.mrna1 | Fork_head | FOXN1 | jumu |
| KWMTBOMO15603.mrna1 | zf-C2H2 | ZNF606 | CG9215 |
| KWMTBOMO01121.mrna1 | zf-C2H2 | NAF1 | CG10341 |
| KWMTBOMO00301.mrna1 | zf-LITAF-like | CG32280 | |
| KWMTBOMO07252.mrna1 | zf-C2H2 | PRDM10 | |
| KWMTBOMO03284.mrna1 | zf-BED | ||
| KWMTBOMO09501.mrna1 | TRAM_LAG1_CLN8 | CERS5 | schlank |
| KWMTBOMO00651.mrna1 | Homeobox | LHX8 | Awh |
| KWMTBOMO07825.mrna1 | MYB | SMARCA1 | Iswi |
| KWMTBOMO08651.mrna1 | Homeobox | PBX1 | exd |
| KWMTBOMO11294.mrna1 | zf-C2H2 | ZNF891 | CG17328 |
| KWMTBOMO07831.mrna1 | PAX | PAX2 | toe |
| KWMTBOMO01266.mrna1 | PAX | PAX3 | gsb-n |
| KWMTBOMO02915.mrna1 | Fork_head | FOXB1 | fd96Ca |
| KWMTBOMO07945.mrna1 | zf-C2H2 | OVOL1 | ovo |
| KWMTBOMO01265.mrna1 | PAX | PAX3 | gsb-n |
| KWMTBOMO13459.mrna1 | Pou | POU3F4 | vvl |
| KWMTBOMO07731.mrna1 | zf-LITAF-like | LITAF | CG13510 |
| KWMTBOMO15317.mrna1 | TF_bZIP | FOSL1 | kay |
| KWMTBOMO07734.mrna1 | zf-LITAF-like | CG13510 | |
| KWMTBOMO10990.mrna1 | TF_bZIP | ATF3 | Atf3 |
| KWMTBOMO07931.mrna1 | ETS | ETV6 | aop |
| KWMTBOMO01705.mrna1 | zf-C2H2 | KLF10 | cbt |
| KWMTBOMO16597.mrna1 | zf-C2H2 | KLF18 | |
| KWMTBOMO02077.mrna1 | zf-C2H2 | GFI1B | sens |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yokoi, K.; Tsubota, T.; Jouraku, A.; Sezutsu, H.; Bono, H. Reference Transcriptome Data in Silkworm Bombyx mori. Insects 2021, 12, 519. https://doi.org/10.3390/insects12060519
Yokoi K, Tsubota T, Jouraku A, Sezutsu H, Bono H. Reference Transcriptome Data in Silkworm Bombyx mori. Insects. 2021; 12(6):519. https://doi.org/10.3390/insects12060519
Chicago/Turabian StyleYokoi, Kakeru, Takuya Tsubota, Akiya Jouraku, Hideki Sezutsu, and Hidemasa Bono. 2021. "Reference Transcriptome Data in Silkworm Bombyx mori" Insects 12, no. 6: 519. https://doi.org/10.3390/insects12060519
APA StyleYokoi, K., Tsubota, T., Jouraku, A., Sezutsu, H., & Bono, H. (2021). Reference Transcriptome Data in Silkworm Bombyx mori. Insects, 12(6), 519. https://doi.org/10.3390/insects12060519