
Combined Transcriptomic and Proteomic Analysis of Myzus persicae, the Green Peach Aphid, Infected with Cucumber Mosaic Virus


Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Plant and CMV Inoculation
2.2. Aphids and Sample Collection
2.3. RNA Extraction, Nucleotide Sequencing, and Raw Data Processing
2.4. Identification of Differentially Expressed Genes (DEGs)
2.5. Protein Extraction, Digestion, and iTRAQ Labeling
2.6. Reverse Phase Separation and Nano-Liquid Chromatography-Coupled MS/MS
2.7. Protein Quantification and Correlation Analysis
2.8. Functional and Pathway Enrichment Analysis
2.9. Quantitative Real-Time PCR (qRT-PCR)
3. Results
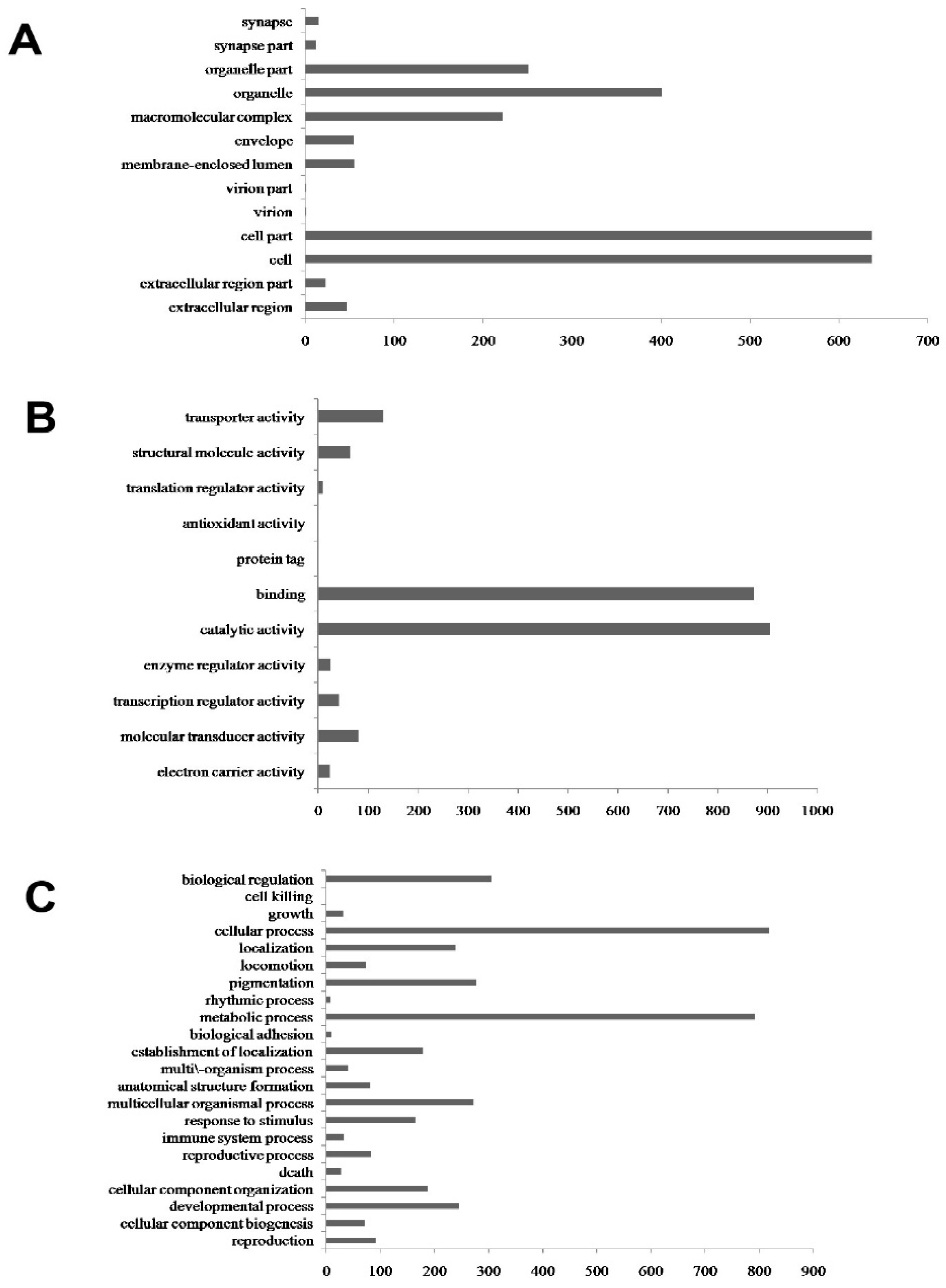
3.1. Transcriptome Difference Analysis
3.2. Validation of the DEGs
3.3. Proteomics Analysis
3.4. Analysis of the Correlation between the Transcriptome and Proteome
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Blackman, R.L.; Eastop, V.F. Aphids on the World’s Crops: An Identification and Information Guide, 2nd ed.; John Wiley and Sons: New York, NY, USA, 2000. [Google Scholar]
- Moran, P.J.; Cheng, Y.F.; Cassell, J.L.; Thompson, G.A. Gene expression profiling of Arabidopsis thaliana in compatible plant-aphid interactions. Arch. Insect Biochem. Physiol. 2002, 51, 182–203. [Google Scholar] [CrossRef] [PubMed]
- Van Emden, H.F.; Harrington, R. Aphids as Crop Pests; CABI Press: Wallingford, UK, 2017. [Google Scholar]
- Peñaflor, M.F.G.V.; Mauck, K.E.; Alves, K.J.; DeMoraes, C.M.; Mescher, M.C.; Bell, J.R. Effects of single and mixed infections of Bean pod mottle virus and Soybean mosaic virus on host-plant chemistry and host–vector interactions. Funct. Ecol. 2016, 30, 1648–1659. [Google Scholar] [CrossRef]
- Carmo-Sousa, M.; Moreno, A.; Garzo, E.; Fereres, A. A non-persistently transmitted-virus induces a pull-push strategy in its aphid vector to optimize transmission and spread. Virus Res. 2014, 186, 38–46. [Google Scholar] [CrossRef]
- Bouhida, M.; Lockhart, B.E. Increased importance of cucumber mosaic virus infection in greenhouse-grown bananas in Morocco. Phytopathology 1990, 80, 981. [Google Scholar]
- Edwardson, J.R.; Christie, R.G. Cucumoviruses. In CRC Handbook of Viruses Infecting Legumes; Edwardson, J.R., Christie, R.G., Eds.; CRC Press Inc.: Boca Raton, FL, USA, 1991; pp. 293–319. [Google Scholar]
- Chen, B.; Francki, R.I.B. Cucumovirus transmission by the aphid Myzus persicae is determined solely by the viral coat protein. J. Gen. Virol. 1990, 71, 939–944. [Google Scholar] [CrossRef]
- Suzuki, M.; Kuwata, S.; Kataoaka, J.; Masuta, C.; Nitta, N.; Takanami, Y. Functional analysis of deletion mutants of Cucumber mosaic virus RNA3 using an in vitro transcription system. Virology 1991, 183, 106–113. [Google Scholar] [CrossRef]
- Ziebell, H.; Murphy, A.M.; Groen, S.C.; Tungadi, T.; Westwood, J.H.; Lewsey, M.G.; Moulin, M.; Kleczkowski, A.; Smith, A.G.; Stevens, M.; et al. Cucumber mosaic virus and its 2b RNA silencing suppressor modify plant-aphid interactions in tobacco. Sci. Rep. 2011, 1, 187. [Google Scholar] [CrossRef] [PubMed]
- Tungadi, T.; Groen, S.C.; Murphy, A.M.; Pate, A.E.; Iqbal, J.; Bruce, J.A.; Cunniffe, N.J.; Carr, J.P. Cucumber mosaic virus and its 2b protein alter emission of host volatile organic compounds but not aphid vector settling in tobacco. Virol. J. 2017, 14, 91. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Gu, L.; Liu, F.; Chen, F.; Ge, F.; Sun, Y. Aphid-borne viral spread is enhanced by virus-induced accumulation of plant reactive oxygen species. Plant Physiol. 2018, 179, 143–155. [Google Scholar] [CrossRef] [PubMed]
- Megahed, E.S.; Pirone, T.P. Comparative transmission of cucumber mosaic virus acquired by aphids from plants or through a membrane. Phytopathology 1966, 56, 1420–1421. [Google Scholar]
- Wamonje, F.O.; Donnelly, R.; Tungadi, T.D.; Murphy, A.M.; Pate, A.E.; Woodcock, C. Different plant viruses induce changes in feeding behavior of specialist and generalist aphids on common bean that are likely to enhance virus transmission. Front. Plant Sci. 2019, 10, 1811. [Google Scholar] [CrossRef] [PubMed]
- Worrall, E.A.; Wamonje, F.O.; Mukeshimana, G.; Harvey, J.J.; Carr, J.P.; Mitter, N. Bean Common Mosaic Virus and Bean Common Mosaic Necrosis Virus: Relationships, Biology, and Prospects for Control. Adv. Virus Res. 2015, 93, 1–46. [Google Scholar] [PubMed]
- Wang, H.; Wu, K.; Liu, Y.; Wu, Y.; Wang, X. Integrative proteomics to understand the transmission mechanism of barley yellow dwarf virus-gpv by its insect vector rhopalosiphum padi. Sci. Rep. 2015, 5, 10971. [Google Scholar] [CrossRef] [PubMed]
- Zhong, X.; Wang, Z.Q.; Xiao, R.; Wang, Y.; Xie, Y.; Zhou, X. Itraq analysis of the tobacco leaf proteome reveals that rna-directed dna methylation (rddm) has important roles in defense against geminivirus-betasatellite infection. J. Proteom. 2017, 152, 88–101. [Google Scholar] [CrossRef] [PubMed]
- Kumar, B.K.; Kanakala, P.; Malathi, S.; Gopal, V.G.; Usha, P.R. Transcriptomic and proteomic analysis of yellow mosaic diseased soybean. J. Plant Biochem. Biotechnol. 2017, 26, 224–234. [Google Scholar] [CrossRef]
- Ye, X.; Wang, H.; Chen, P.; Fu, B.; Zhang, M.; Li, J.D.; Zheng, X.B.; Tan, B.; Feng, J.C. Combination of itraq proteomics and RNA-seq transcriptomics reveals multiple levels of regulation in phytoplasma-infected Ziziphus jujuba Mill. Hortic. Res. 2017, 4, 17080. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.Z.; Li, Y.Y.; An, F.X.T.; Huang, F.X.; Wang, M.Q.; Liu, C.X.; Mao, J.J.; Zhang, L.S. Comparative transcriptome and iTRAQ proteome analyses reveal the mechanisms of diapause in Aphidius gifuensis Ashmead (Hymenoptera: Aphidiidae). Front. Physiol. 2018, 9, 1697. [Google Scholar] [CrossRef]
- Zhou, A.M.; Sun, H.W.; Dai, S.Y.; Feng, S.; Zhang, J.Z.; Gong, S.F.; Wang, J.J. Identification of Transcription Factors Involved in the Regulation of Flowering in Adonis amurensis through Combined RNA-seq Transcriptomics and iTRAQ Proteomics. Genes 2019, 10, 305. [Google Scholar] [CrossRef]
- Zhao, J.; Chi, Y.; Zhang, X.J.; Lei, T.; Wang, X.W.; Liu, S.S. Comparative proteomic analysis provides new insight into differential transmission of two begomoviruses by a whitefly. J. Virol. 2019, 16, 32. [Google Scholar] [CrossRef]
- Cilia, M.; Tamborindeguy, C.; Fish, T.; Howe, K.; Thannhauser, T.W.; Gray, S. Genetics coupled to quantitative intact proteomics links heritable aphid and endosymbiont protein expression to circulative polerovirus transmission. J. Virol. 2011, 85, 2148–2166. [Google Scholar] [CrossRef]
- Tamborindeguy, C.; Bereman, M.S.; DeBlasio, S.; Igwe, D.; Smith, D.M.; White, F.; MacCoss, M.J.; Gray, S.M.; Cilia, M. Genomic and proteomic analysis of Schizaphis graminum reveals cyclophilin proteins are involved in the transmission of cereal yellow dwarf virus. PLoS ONE 2013, 8, e71620. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Xu, Z.; Zhang, Y.; Chai, L.; Yi, H.; Deng, X. An integrative analysis of the transcriptome and proteome of the pulp of a spontaneous late-ripening sweet orange mutant and its wild type improves our understanding of fruit ripening in citrus. J. Exp. Bot. 2014, 65, 1651–1671. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Guo, W.; Feng, L.; Ye, X.; Xie, W.; Huang, X.; Liu, J. Transcriptome and proteome analysis of Eucalyptus infected with Calonectria pseudoreteaudii. J. Proteom. 2015, 115, 117–131. [Google Scholar] [CrossRef] [PubMed]
- Stare, T.; Stare, K.; Weckwerth, W.; Wienkoop, S.; Gruden, K. Comparison between proteome and transcriptome response in potato (Solanum tuberosum L.) leaves following potato virus y (PVY) infection. Proteomes 2017, 5, 14. [Google Scholar] [CrossRef] [PubMed]
- Ng, J.C.K.; Perry, K.L. Transmission of plant viruses by aphid vectors. Mol. Plant Pathol. 2004, 5, 505–511. [Google Scholar] [CrossRef]
- Mortazavi, A.; Williams, B.A.; McCue, K.; Schaeffer, L.; Wold, B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 2008, 5, 621–628. [Google Scholar] [CrossRef] [PubMed]
- Wei, D.H.; Zeng, Y.Y.; Xing, X.H.; Liu, H.Z.; Lin, M.J.; Liu, X.L.; Liu, J.F. The proteome differences between hepatitis b virus genotype b and genotype c induced hepatocellular carcinoma revealed by itraq based quantitative proteomics. J. Proteome Res. 2015, 15, 487–498. [Google Scholar] [CrossRef]
- Mok, L.; Wynne1, J.W.; Ford, K.; Shiell, B.; Bacic, A.; Michalski, W.P. Proteomic analysis of pteropus alectokidney cells in response to the viral mimic, poly i:c. J. Pest Sci. 2019, 13, 25. [Google Scholar]
- Yang, N.; Xie, W.; Yang, X.; Wang, S.; Wu, Q.; Li, R.M.; Pan, H.P.; Liu, B.M.; Shi, X.B.; Fang, Y.; et al. Transcriptomic and Proteomic Responses of Sweet potato Whitefly, Bemisia tabaci, to Thiamethoxam. PLoS ONE 2013, 8, e61820. [Google Scholar]
- Kanehisa, M.; Araki, M.; Goto, S.; Hattori, M.; Hirakawa, M.; Itoh, M.; Katayama, T.; Kawashima, S.; Okuda, S.; Tokimatsu, T.; et al. KEGG for linking genomes to life and the environment. Nucleic Acids Res. 2007, 36, 480–484. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Peng, X.; Zuo, Y.; Li, Y.; Chen, M. Molecular Cloning, Expression Pattern and Polymorphisms of NADPH-Cytochrome P450 Reductase in the Bird Cherry-Oat Aphid Rhopalosiphum padi (L.). PLoS ONE 2016, 11, e0154633. [Google Scholar] [CrossRef] [PubMed]
- Puinean, A.M.; Foster, S.P.; Oliphant, L.; Denholm, I.; Field, L.M.; Millar, N.S.; Williamson, M.S.; Bass, C. Amplification of a cytochrome p450 gene is associated with resistance to neonicotinoid insecticides in the aphid Myzus persicae. PLoS Genet. 2011, 6, e1000999. [Google Scholar] [CrossRef] [PubMed]
- Bass, C.; Puinean, A.M.; Andrews, M.; Cutler, P.; Daniels, M.; Elias, J.; Paul, V.L.; Crossthwaite, A.J.; Denholm, I.; Field, L.M.; et al. Mutation of a nicotinic acetylcholine receptor β subunit is associated with resistance to neonicotinoid insecticides in the aphid Myzus persicae. BMC Neurosci. 2011, 12, 51. [Google Scholar] [CrossRef] [PubMed]
- Lycett, G.J.; McLaughlin, L.A.; Ranson, H.; Hemingway, J.; Kafatos, F.C.; Loukeris, T.G.; Paine, M.J.I. Anopheles gambiae P450 reductase is highly expressed in oenocytes and in vivo knockdown increases permethrin susceptibility. Insect Mol. Biol. Rep. 2006, 15, 321–327. [Google Scholar] [CrossRef] [PubMed]
- Lian, L.Y.; Widdowson, P.; McLaughlin, L.A.; Paine, M.J. Biochemical comparison of Anopheles gambiae and human NADPH P 450 reductases reveals different 2′-5′-ADP and FMN binding traits. PLoS ONE 2011, 6, e20574. [Google Scholar] [CrossRef]
- Deshoux, M.; Monsion, B.; Uzest, M. Insect cuticular proteins and their role in transmission of phytoviruses. Curr. Opin. Virol. 2018, 33, 137–143. [Google Scholar] [CrossRef] [PubMed]
- Dombrovsky, A.; Sobolev, I.; Chejanovsky, N.; Raccah, B. Characterization of RR-1 and RR-2 cuticular proteins from Myzus persicae. Comp. Biochem. Physiol. B Biochem. Mol. Biol. 2007, 146, 256–264. [Google Scholar] [CrossRef] [PubMed]
- Uzest, M.; Gargani, D.; Dombrovsky, A.; Cazevieille, C.; Cot, D.; Blanc, S. The ‘acrostyle’: A newly described anatomical structure in aphid stylets. Arthropod Struct. Dev. 2010, 39, 221–229. [Google Scholar] [CrossRef]
- Liang, Y.; Gao, X.W. The cuticle protein gene MPCP4 of Myzus persicae (Homoptera: Aphididae) plays a critical role in cucumber mosaic virus acquisition. J. Econ. Entomol. 2017, 110, 848–853. [Google Scholar] [CrossRef] [PubMed]
- Webster, C.G.; Pichon, E.; Munster, M.; Monsion, B.; Deshoux, M.; Gargani, D.; Calevro, F.; Jimenez, J.; Moreno, A.; Krenz, B.; et al. Identification of plant virus receptor candidates in the stylets of their aphid vectors. J. Virol. 2018, 92, 14. [Google Scholar] [CrossRef] [PubMed]
- Rebers, J.E.; Riddiford, L.M. Structure and expression of a Manduca sexta larval cuticle gene homologous to Drosophila cuticle genes. J. Mol. Biol. 1988, 203, 411–423. [Google Scholar] [CrossRef]
- Andersen, S.O.; Højrup, P.; Roepstorff, P. Insect cuticular proteins. Insect Biochem. Mol. Biol. 1995, 25, 153–176. [Google Scholar] [CrossRef]
- Fernández-Calvino, L.; Goytia, E.; López-Abella, D.; Giner, A.; Urizarna, M.; Vilaplana, L.; Lopez-Moya, J.J. The helper-component protease transmission factor of tobacco etch potyvirus binds specifically to an aphid ribosomal protein homologous to the laminin receptor precursor. J. Gen. Virol. 2010, 91, 2862–2873. [Google Scholar] [CrossRef] [PubMed]
- Pesapane, A.; Giovanni, C.D.; Rossi, F.W.; Alfano, D.; Formisano, L.; Ragno, P.; Selleri, C.; Montuori, N.; Lavecchia, A. Discovery of new small molecules inhibiting 67 kDa laminin receptor interaction with laminin and cancer cell invasion. Oncotarget 2015, 6, 18116–18133. [Google Scholar] [CrossRef] [PubMed]
- Scott, J.G. Cytochromes P450 and insecticide resistanc. Insect Biochem. Mol. Biol. 1999, 29, 757–777. [Google Scholar] [CrossRef]
- Zhao, L.M.; Barry, A.; Shin, D.Y.; Yu, F.H. The effect of permethrin resistance on Aedes aegypti transcriptome following ingestion of zika virus infected blood. Viruses 2018, 10, 470. [Google Scholar] [CrossRef] [PubMed]
- Christian, Z.; Shakhawat, B. Archaea signal recognition particle shows the way. Archaea 2010. [Google Scholar] [CrossRef]
- Fathi, A.; Pakzad, M.; Taei, A.; Brink, T.C.; Pirhaji, L.; Ruiz, G.; Bordbar, M.S.T.; Gourabi, H.; Adjaye, J.; Baharvand, H.; et al. Comparative proteome and transcriptome analyses of embryonic stem cells during embryoid body-based differentiation. Proteomics 2009, 9, 4859–4870. [Google Scholar] [CrossRef] [PubMed]
- Lan, P.; Li, W.; Wen, T.N.; Shiau, J.Y.; Wu, Y.C.; Lin, W.; Schmidt, W. iTRAQ protein profile analysis of Arabidopsis roots reveals new aspects critical for iron homeostasis. Plant Physiol. 2011, 155, 821–834. [Google Scholar] [CrossRef]
- Galland, M.; Huguet, R.; Arc, E.; Cueff, G.; Job, D.; Rajjou, L. Dynamic proteomics emphasizes the importance of selective mRNA translation and Protein turnover during Arabidopsis seed germination. Mol. Cell. Proteom. 2014, 13, 252–268. [Google Scholar] [CrossRef] [PubMed]
- Waggoner, S.A.; Liebhaber, S.A. Regulation of alpha-globin mRNA stability. Exp. Biol. Med. 2003, 228, 387–395. [Google Scholar] [CrossRef] [PubMed]
- Beyer, A.; Hollunder, J.; Nasheuer, H.P.; Wilhelm, T. Posttranscriptional expression regulation in the yeast Saccharomyces cerevisiae on a genomic scale. Mol. Cell. Proteom. 2004, 3, 1083–1092. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pathway | Number of Genes | p-Value | Pathway ID | |
|---|---|---|---|---|
| DEGs † | Expressed ‡ | |||
| Up-Regulated | ||||
| Antigen processing and presentation | 25 | 156 | 6.68 × 10−5 | ko04612 |
| cAMP signaling pathway | 32 | 270 | 0.001033478 | ko04024 |
| Lysosome | 26 | 225 | 0.003930307 | ko04142 |
| Vasopressin-regulated water reabsorption | 12 | 73 | 0.004136451 | ko04962 |
| Steroid hormone biosynthesis | 22 | 183 | 0.005010783 | ko00140 |
| Metabolism of xenobiotics by cytochrome P450 | 23 | 196 | 0.005434029 | ko00980 |
| Bile secretion | 19 | 151 | 0.005667970 | ko04976 |
| Retinol metabolism | 21 | 175 | 0.006122107 | ko00830 |
| Drug metabolism—other enzymes | 24 | 211 | 0.006467250 | ko00983 |
| Chemical carcinogenesis | 23 | 200 | 0.006738707 | ko05204 |
| Legionellosis | 15 | 111 | 0.007453879 | ko05134 |
| Drug metabolism—cytochrome P450 | 22 | 192 | 0.008218267 | ko00982 |
| Starch and sucrose metabolism | 29 | 277 | 0.008386653 | ko00500 |
| Pentose and glucuronate interconversions | 24 | 218 | 0.009174987 | ko00040 |
| Ascorbate and aldarate metabolism | 21 | 184 | 0.010028922 | ko00053 |
| Porphyrin and chlorophyll metabolism | 22 | 202 | 0.013560873 | ko00860 |
| Hematopoietic cell lineage | 10 | 70 | 0.01909453 | ko04640 |
| Galactose metabolism | 13 | 103 | 0.01942347 | ko00052 |
| Measles | 15 | 131 | 0.0259525 | ko05162 |
| ABC transporters | 18 | 168 | 0.027132284 | ko02010 |
| Calcium signaling pathway | 17 | 156 | 0.027367424 | ko04020 |
| Ubiquinone and other terpenoid–quinone biosynthesis | 4 | 17 | 0.03233652 | ko00130 |
| Fatty acid biosynthesis | 9 | 70 | 0.042834788 | ko00061 |
| Renin–angiotensin system | 12 | 107 | 0.048633847 | ko04614 |
| Jak-STAT signaling pathway | 9 | 72 | 0.048990908 | ko04630 |
| Down-Regulated | ||||
| Renin–angiotensin system | 13 | 107 | 0.004147531 | ko04614 |
| Vitamin digestion and absorption | 13 | 112 | 0.00585761 | ko04977 |
| Oxidative phosphorylation | 34 | 431 | 0.00730206 | ko00190 |
| Nicotine addiction | 6 | 32 | 0.00796758 | ko05033 |
| Hypertrophic cardiomyopathy (HCM) | 11 | 91 | 0.00827137 | ko05410 |
| Cardiac muscle contraction | 17 | 192 | 0.01963673 | ko04260 |
| Cutin, suberine, and wax biosynthesis | 7 | 57 | 0.02967602 | ko00073 |
| Gene ID | log2 Ratio | Annotation |
|---|---|---|
| Cuticle Protein-Related Genes | ||
| c567 | 1.00 | RR1 cuticle protein 2 (Myzus persicae) |
| c31594 | 1.12 | RR1 cuticle protein 7 (Acyrthosiphon pisum) |
| c14422 | 1.32 | cuticle protein (Lipaphis erysimi) |
| c13658 | 1.79 | cuticular protein 70 (Acyrthosiphon pisum) |
| c16514 | 1.85 | cuticular protein 52 (Acyrthosiphon pisum) |
| c65979 | 1.99 | RR1 cuticle protein 11 (Acyrthosiphon pisum) |
| c46947 | 3.63 | Cuticle protein 6 (Blaberus craniifer) |
| c36838 | 6.08 | RR1 cuticle protein 6 (Acyrthosiphon pisum) |
| c53043 | 6.63 | structural constituent of cuticle |
| c66065 | −2.25 | cuticular protein 11 precursor (Acyrthosiphon pisum) |
| c78688 | −1.57 | cuticular protein 62 precursor (Acyrthosiphon pisum) |
| Ribosomal Protein-Related Genes | ||
| c81455 | 1.12 | large subunit ribosomal protein 6 (Plasmodium berghei) |
| c5335 | 1.20 | ribosomal protein L10Ae-like (Acyrthosiphon pisum) |
| c11420 | 1.26 | 28S ribosomal protein S18b (Acyrthosiphon pisum) |
| c69386 | 1.34 | 39S ribosomal protein L35 (Acyrthosiphon pisum) |
| c91354 | 1.74 | 40S ribosomal protein S21-like (Acyrthosiphon pisum) |
| c75408 | 1.83 | Ribosomal protein L15 |
| c14330 | 1.94 | ribosomal protein L27a (Acyrthosiphon pisum) |
| c27431 | 2.49 | 28S ribosomal protein S2 (Acyrthosiphon pisum) |
| c127488 | 4.8 | 28S ribosomal protein S29 (Acyrthosiphon pisum) |
| c31733 | −1.40 | ribosomal protein S27-1 (Acyrthosiphon pisum) |
| c116976 | −1.36 | ribosomal protein L41 (Drosophila melanogaster) |
| c51305 | −1.25 | 39S ribosomal protein L41 (Acyrthosiphon pisum) |
| c31252 | −1.09 | 40S ribosomal protein S7-like (Acyrthosiphon pisum) |
| r | Accession Number | Name | log2 Ratio |
|---|---|---|---|
| Up-regulated | E5LMN6 | Cytochrome b (fragment) | 0.75 |
| Q1ZZP7 | Cytochrome B5-like protein | 0.85 | |
| J9JMZ2 | NADPH--cytochrome P450 reductase | 1.47 | |
| V5SQ25 | Cytochrome P450 CYP6CY3 | 1.51 | |
| J9K284 | Ribosomal protein L15 | 1.54 | |
| Q9TFD9 | Cytochrome c oxidase subunit 2 (fragment) | 1.74 | |
| J9K6M4 | Calcium-transporting ATPase | 2.36 | |
| Down-regulated | Q45V96 | Tentative cuticle protein | −1.64 |
| J9JYX3 | ATP synthase subunit alpha | −1.25 | |
| J9KB74 | Glutamate dehydrogenase | −1.22 | |
| Q1W9N4 | Putative heat shock protein hslU | −0.81 | |
| Q8VUS1 | Chaperone protein DnaK (fragment) | −0.78 | |
| Q45V97 | RR2 cuticle protein 3 (fragment) | −0.76 | |
| B5LYP1 | Juvenile hormone binding protein | −0.74 | |
| Q45V95 | Cuticle protein 4 | −0.62 |
| Pathway | Number of Proteins | p-Value | ID | |
|---|---|---|---|---|
| DAPs † | Accumulated ‡ | |||
| Valine, leucine, and isoleucine degradation | 13 | 25 | 0.000235925 | api00280 |
| Fatty acid metabolism | 16 | 42 | 0.000751229 | api01212 |
| Alanine, aspartate, and glutamate metabolism | 9 | 15 | 0.001029258 | api00250 |
| Arginine and proline metabolism | 11 | 25 | 0.0020428 | api00330 |
| Fatty acid degradation | 10 | 21 | 0.002064943 | api00071 |
| Propanoate metabolism | 6 | 11 | 0.010188421 | api00640 |
| Fatty acid biosynthesis | 6 | 11 | 0.010188421 | api00061 |
| beta-Alanine metabolism | 6 | 13 | 0.018211309 | api00410 |
| Proteasome | 11 | 38 | 0.024134177 | api03050 |
| Spliceosome | 22 | 100 | 0.025505972 | api03040 |
| Carbon metabolism | 17 | 73 | 0.030987964 | api01200 |
| Biosynthesis of amino acids | 12 | 47 | 0.038879652 | api01230 |
| Pyruvate metabolism | 8 | 26 | 0.039544834 | api00620 |
| Nitrogen metabolism | 4 | 8 | 0.043309293 | api00910 |
| Gene ID | Protein ID | Log2 (Transcript Ratio) | Log2 (Protein Ratio) | E-Value | Annotation |
|---|---|---|---|---|---|
| c118109 | V5SQ25 | 1.769388711 | 1.506921778 | 2.00 × 10−69 | Cytochrome P450 CYP6CY3 (Myzus persicae) |
| c49464 | Q9TFD9 | 6.480679524 | 1.733540171 | 2.00 × 10−51 | Cytochrome c oxidase subunit 2 (fragment) |
| c52418 | J9K5U3 | 1.211651723 | 1.824611319 | 7.00 × 10−61 | Ubiquitin carboxyl-terminal hydrolase |
| c75408 | J9K284 | 1.832888752 | 1.5354622 | 3.00 × 10−28 | Ribosomal protein L15 |
| c45978 | J9K6M4 | 1.010053019 | 2.360933037 | 1.00 × 10−6 | Calcium-transporting ATPase |
| c38643 | J9K3N8 | 7.708471879 | 1.181065157 | 2.00 × 10−4 | Coatomer subunit alpha |
| c25651 | X1WJB6 | 9.50581 | 1.33764 | 3.00 × 10−52 | - |
| c35347 | Q64F38 | −1.36053284 | −1.74027779 | 1.00 × 10−4 | Tropomyosin (Myzus persicae) |
| c11315 | J9JYX3 | −1.21150443 | −1.23817987 | 6.00 × 10−6 | ATP synthase subunit alpha |
| c41875 | J9K071 | −3.07923 | −1.09732 | 3.00 × 10−7 | - |
| c34089 | Q45V96 | 0.4009223 | −1.6241371 | 1 × 10−10 | Tentative cuticle protein (Myzus persicae) |
| c30589 | X1WIC4 | 0.4215968 | 1.2683861 | 0 | Signal recognition particle subunit SRP72 |
| c49978 | J9KB74 | 0.1445756 | −1.2304454 | 0 | Glutamate dehydrogenase (Acyrthosiphon pisum) |
| c13794 | J9JM35 | −0.0390524 | 1.0775299 | 0 | Amino acid transporter |
| c41961 | J9JMR5 | 0.046467 | 1.1028535 | 1 × 10−120 | Protein BCCIP homolog |
| c51904 | J9JMZ2 | −0.0417784 | 1.4710292 | 0 | NADPH-cytochrome P450 reductase |
| c53037 | X1XJB2 | 0.253985 | −1.3529881 | 2 × 10−52 | Histone H2B (fragment) |
| c52517 | J9JWC3 | 0.0351009 | 2.2677165 | 0 | Eukaryotic translation initiation factor 3 subunit E (Acyrthosiphon pisum) |
| c28628 | J9KB65 | −0.0646973 | 2.1664679 | 0 | Lipase maturation factor |
| c49848 | Q8B4P7 | 1.6937694 | −1.2378697 | 4 × 10−89 | Putative structural protein |
| c36095 | J9K3Z0 | −1.7250354 | 1.8737054 | 0 | AP-3 complex subunit delta |
| c53987 | J9JRR5 | −1.1789701 | 1.6216423 | 0 | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liang, Y.; Ma, K.-S.; Liang, P.-Z.; Yang, L.-W.; Zhang, L.; Gao, X.-W. Combined Transcriptomic and Proteomic Analysis of Myzus persicae, the Green Peach Aphid, Infected with Cucumber Mosaic Virus. Insects 2021, 12, 372. https://doi.org/10.3390/insects12050372
Liang Y, Ma K-S, Liang P-Z, Yang L-W, Zhang L, Gao X-W. Combined Transcriptomic and Proteomic Analysis of Myzus persicae, the Green Peach Aphid, Infected with Cucumber Mosaic Virus. Insects. 2021; 12(5):372. https://doi.org/10.3390/insects12050372
Chicago/Turabian StyleLiang, Yan, Kang-Sheng Ma, Ping-Zhuo Liang, Li-Wen Yang, Lei Zhang, and Xi-Wu Gao. 2021. "Combined Transcriptomic and Proteomic Analysis of Myzus persicae, the Green Peach Aphid, Infected with Cucumber Mosaic Virus" Insects 12, no. 5: 372. https://doi.org/10.3390/insects12050372
APA StyleLiang, Y., Ma, K.-S., Liang, P.-Z., Yang, L.-W., Zhang, L., & Gao, X.-W. (2021). Combined Transcriptomic and Proteomic Analysis of Myzus persicae, the Green Peach Aphid, Infected with Cucumber Mosaic Virus. Insects, 12(5), 372. https://doi.org/10.3390/insects12050372