
Application of Next Generation Semiconductor-Based Sequencing for the Identification of Apis mellifera Complementary Sex Determiner (csd) Alleles from Honey DNA

 ,
, 
 , , and
, , and
Abstract
:Simple Summary
Abstract

1. Introduction
2. Materials and Methods
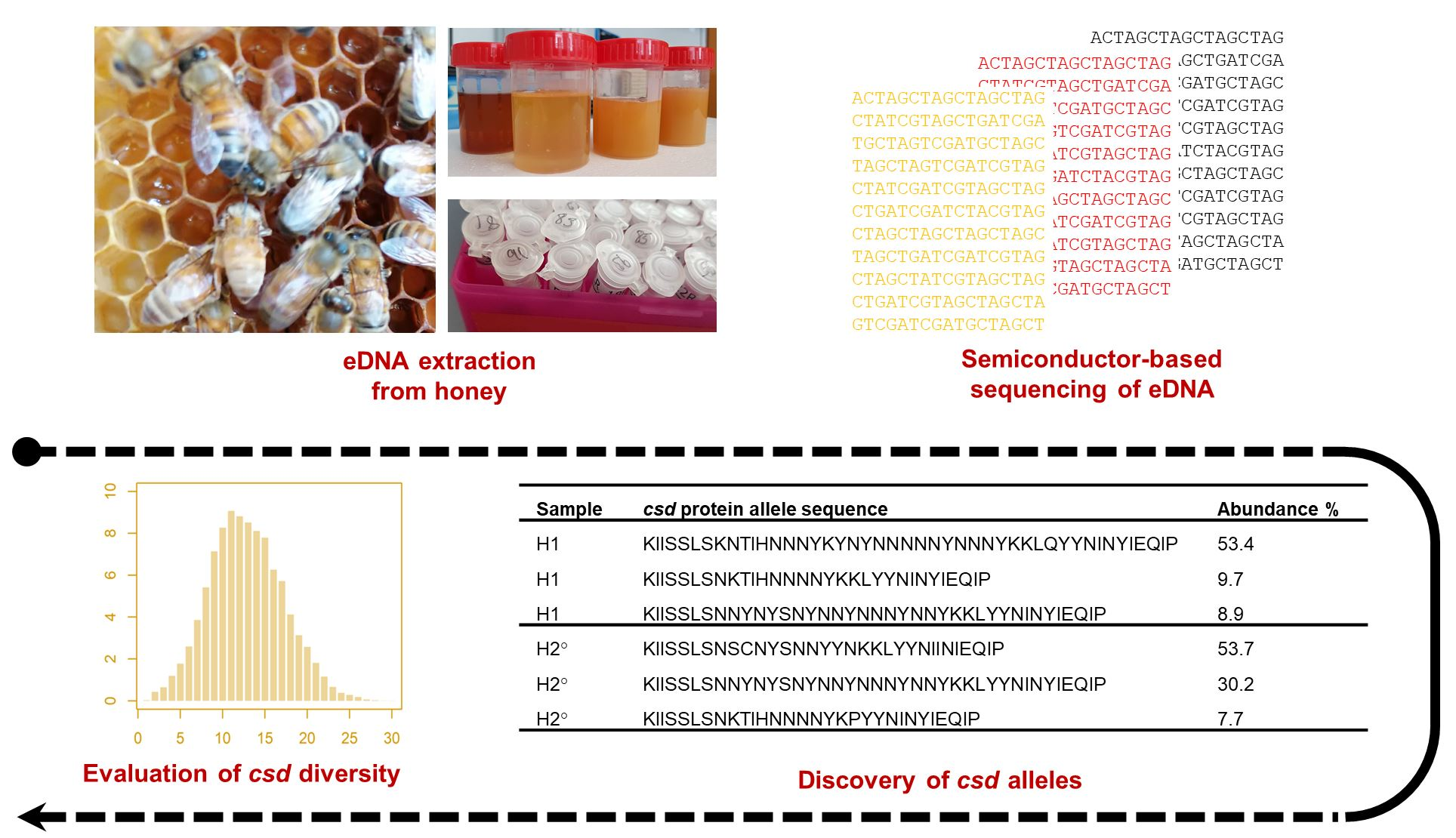
2.1. Honey Samples and DNA Extraction
2.2. PCR Amplification of the csd Region
2.3. Next Generation Sequencing
2.4. Bioinformatic and Data Analyses
2.4.1. Read Filtering and Identification of csd Sequences
2.4.2. Quality Control of csd Alleles
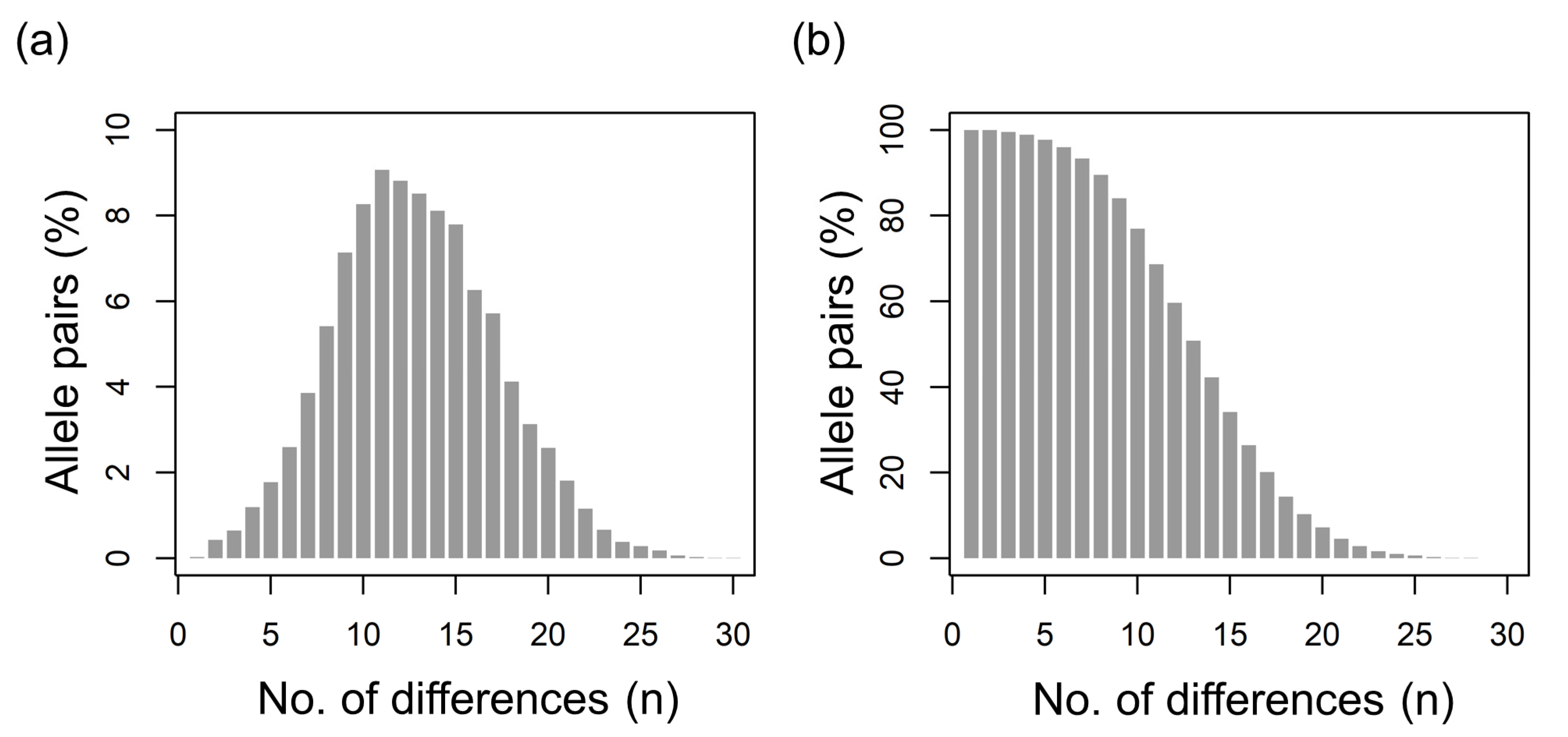
2.4.3. Evaluation of csd Polymorphims
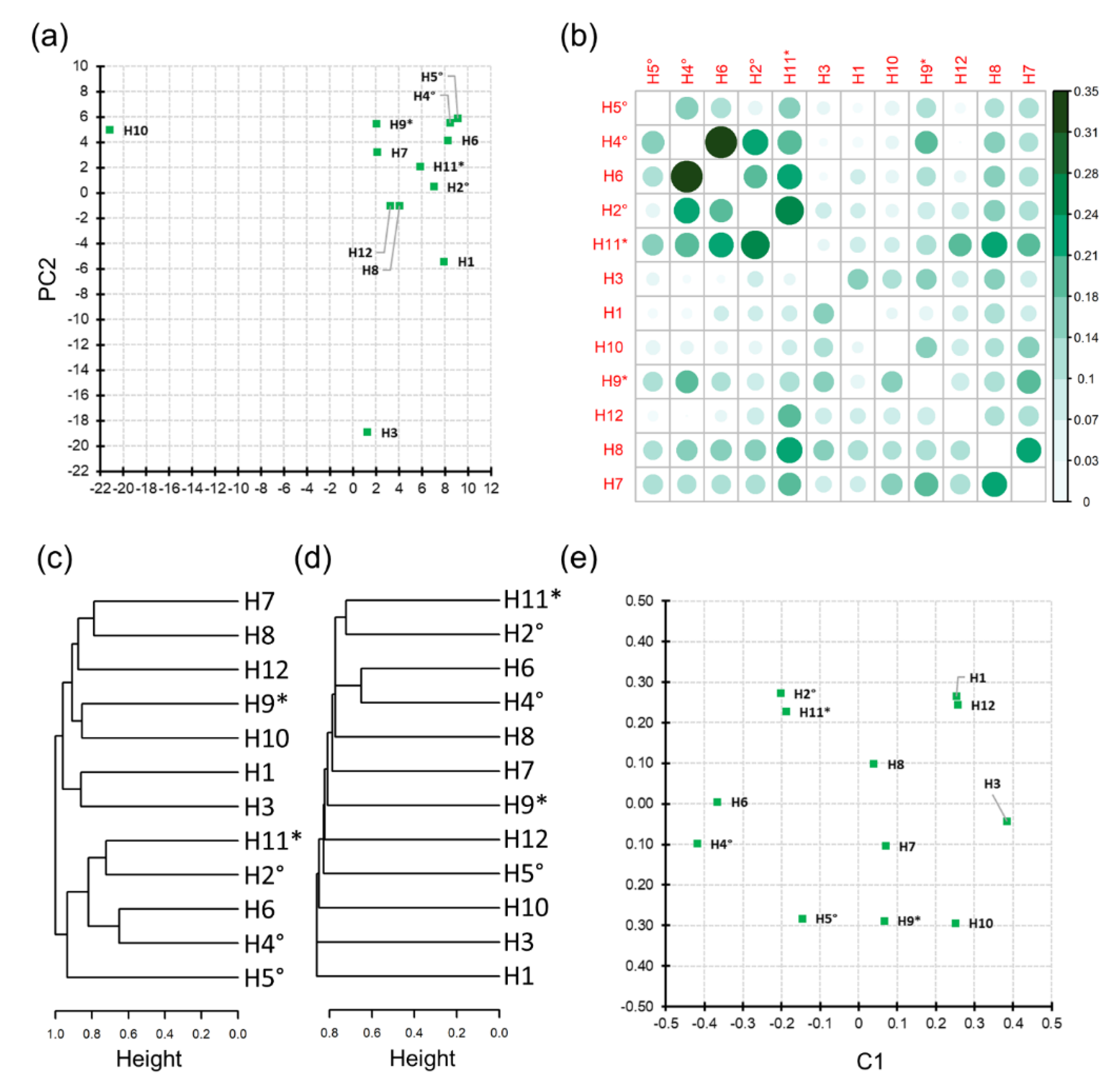
2.4.4. Evaluation of Honey Sample Similarity
3. Results
3.1. Sequenced Reads and Identified csd Alleles
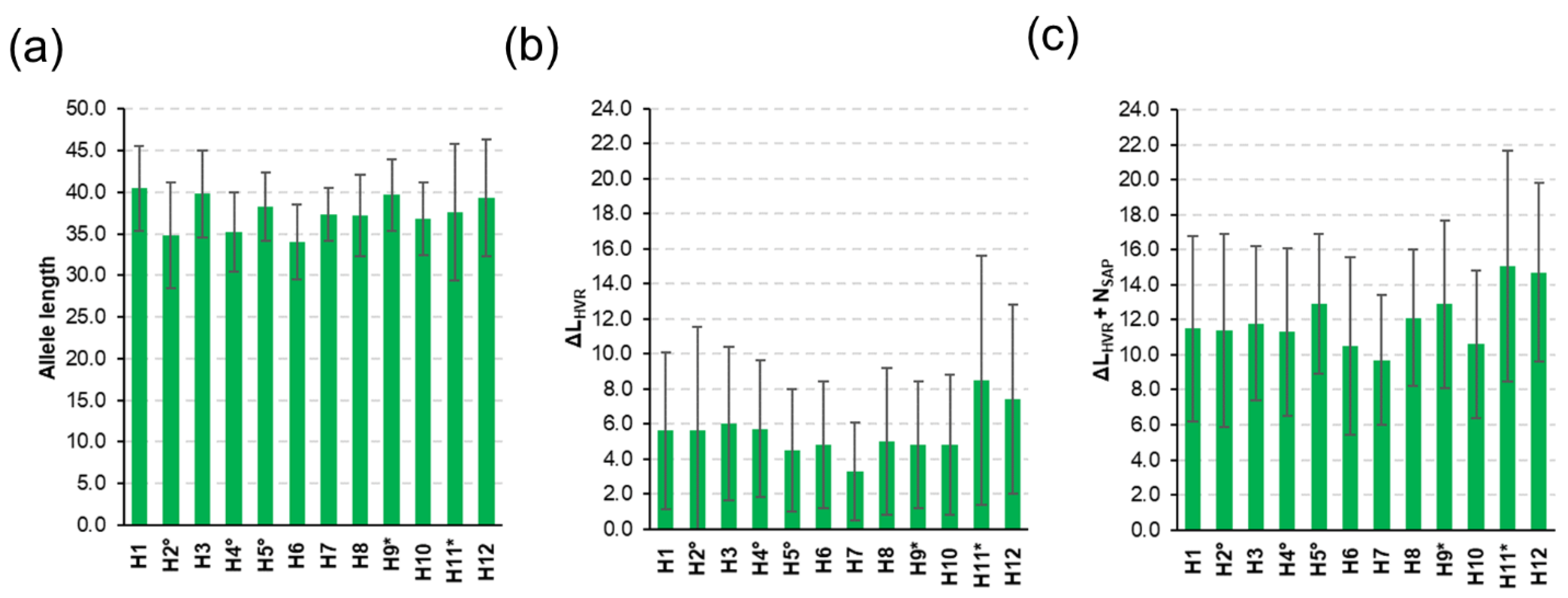
3.2. Diversity of csd Protein Alleles
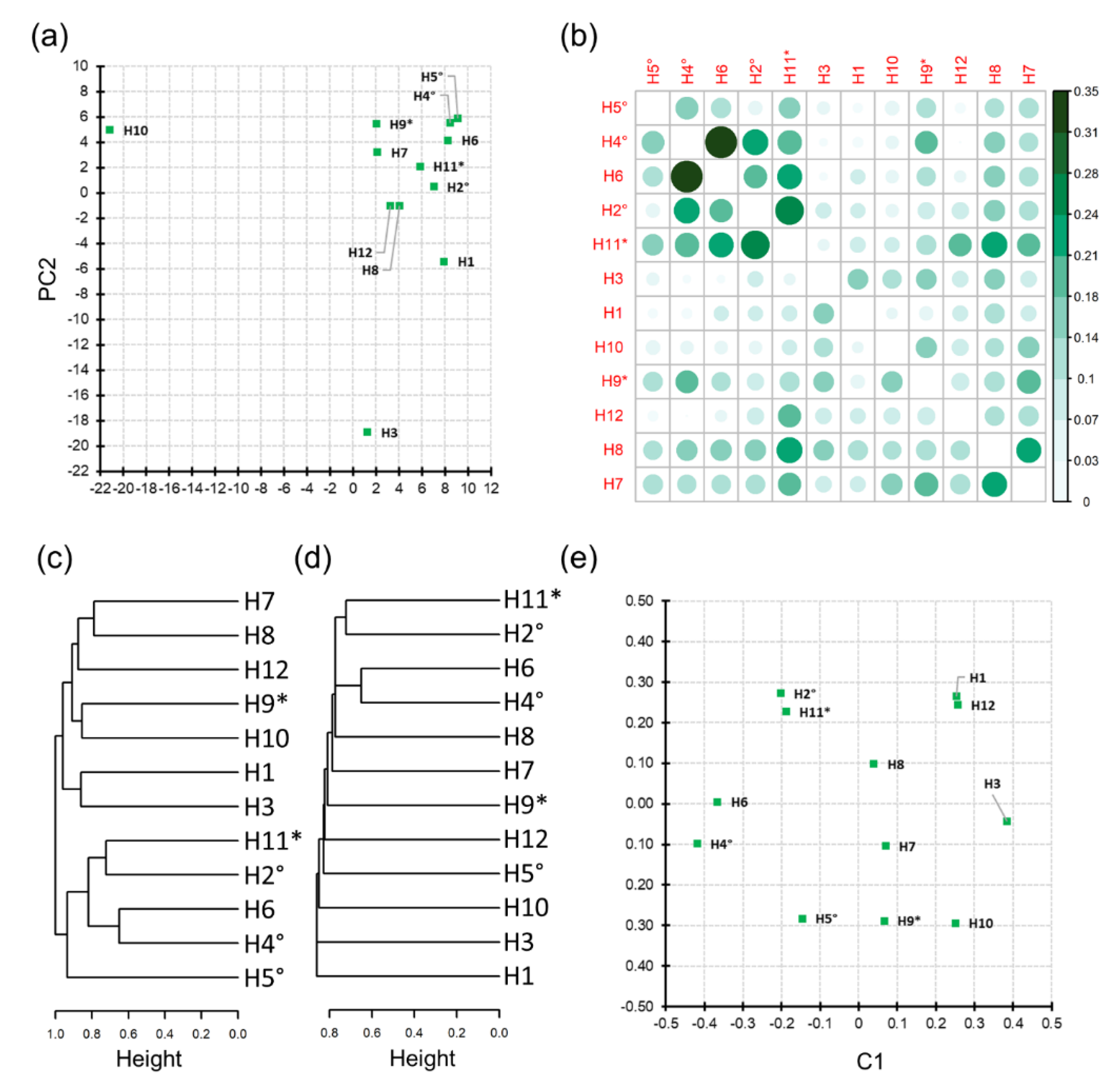
3.3. Honey Sample Similarity
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Heimpel, G.E.; de Boer, J.G. Sex determination in the Hymenoptera. Annu. Rev. Entomol. 2008, 53, 209–230. [Google Scholar] [CrossRef] [PubMed]
- Dzierzon, J. Gutachten über die von Herrn Direktor Stöhr im ersten und zweiten Kapitel des General-Gutachtens aufgestellten Fragen. Eichstädter Bienenztg. 1845, 1, 119–121. [Google Scholar]
- Beye, M.; Hasselmann, M.; Fondrk, M.K.; Page, R.E., Jr.; Omholt, S.W. The gene csd is the primary signal for sexual development in the honeybee and encodes an SR-type protein. Cell 2003, 114, 419–429. [Google Scholar] [CrossRef] [Green Version]
- Whiting, P.; Whiting, A.R. Diploid males from fertilized eggs in Hymenoptera. Science 1925, 62, 437. [Google Scholar] [CrossRef] [PubMed]
- Woyke, J.; Knytel, A. The chromosome number as proof that drones can arise from fertilizer eggs of the honeybee. J. Apic. Res. 1966, 5, 149–154. [Google Scholar] [CrossRef]
- Mackensen, O. Viability and sex determination in the honey bee (Apis mellifera L.). Genetics 1951, 36, 500–509. [Google Scholar] [CrossRef]
- Woyke, J. Drone larvae from fertilised eggs of the honeybee. J. Apic. Res. 1963, 2, 19–24. [Google Scholar] [CrossRef]
- Woyke, J. Effect of sex allele homo-heterozygosity on honeybee colony population and on their honey production. 1. Favourable development conditions and unrestricted queens. J. Apic. Res. 1980, 19, 51–63. [Google Scholar] [CrossRef]
- Woyke, J. Effect of sex allele homo-heterozygosity on honeybee colony population and on their honey production. 2. Unfavourable development conditions and restricted queens. J. Apic. Res. 1981, 20, 148–155. [Google Scholar] [CrossRef]
- Tarpy, D.R.; Page, R.E. The curious promiscuity of queen honey bees (Apis mellifera): Evolutionary and behavioral mechanisms. Ann. Zool. Fenn. 2001, 38, 255–265. [Google Scholar]
- Page, R.E.J.; Marks, R.W. The population genetics of sex determination in honey bees: Random mating in closed populations. Heredity 1982, 48, 263–270. [Google Scholar] [CrossRef] [Green Version]
- Kalmus, H.; Smith, C.A.B. Production of pure lines in bees. J. Genet. 1949, 49, 153–158. [Google Scholar] [CrossRef] [PubMed]
- Mackensen, O. Further studies on a lethal series in the honey bee. J. Hered. 1955, 46, 72–74. [Google Scholar] [CrossRef]
- Hachinohe, Y.; Jimbu, M. Occurrence of the lethal eggs in the honeybee. Bull. Nat. Inst. Agric. Sci. Ser. G. 1958, 14, 123–130. [Google Scholar]
- Laidlaw, H.H., Jr.; Gomes, F.P.; Kerr, W.E. Estimation of the number of lethal alleles in a panmictic population of Apis mellifera L. Genetics 1956, 41, 179–188. [Google Scholar] [CrossRef] [PubMed]
- Adams, J.; Rothman, E.D.; Kerr, W.E.; Paulino, Z.L. Estimation of the number of sex alleles and queen matings from diploid male frequencies in a population of Apis mellifera. Genetics 1977, 86, 583–596. [Google Scholar] [CrossRef]
- Yokohama, S.; Nei, M. Population dynamics of sex-determining alleles in honey bees and self-incompatibility alleles in plants. Genetics 1979, 91, 609–626. [Google Scholar] [CrossRef]
- Hasselmann, M.; Beye, M. Signatures of selection among sex-determining alleles of the honey bee. Proc. Natl. Acad. Sci. USA 2004, 101, 4888–4893. [Google Scholar] [CrossRef] [Green Version]
- Hasselmann, M.; Beye, M. Pronounced differences of recombination activity at the sex determination locus of the honeybee, a locus under strong balancing selection. Genetics 2006, 174, 1469–1480. [Google Scholar] [CrossRef] [Green Version]
- Hasselmann, M.; Vekemans, X.; Pflugfelder, J.; Koeniger, N.; Koeniger, G.; Tingek, S.; Beye, M. Evidence for convergent nucleotide evolution and high allelic turnover rates at the complementary sex determiner gene of Western and Asian honeybees. Mol. Biol. Evol. 2008, 25, 696–708. [Google Scholar] [CrossRef] [Green Version]
- Lechner, S.; Ferretti, L.; Schöning, C.; Kinuthia, W.; Willemsen, D.; Hasselmann, M. Nucleotide variability at its limit? Insights into the number and evolutionary dynamics of the sex-determining specificities of the honey bee Apis mellifera. Mol. Biol. Evol. 2014, 31, 272–287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, S.; Huang, Z.Y.; Green, D.R.; Smith, D.R.; Zhang, J. Evolution of the complementary sex-determination gene of honey bees: Balancing selection and trans-species polymorphisms. Genome Res. 2006, 16, 1366–1375. [Google Scholar] [CrossRef] [Green Version]
- Zareba, J.; Blazej, P.; Laszkiewicz, A.; Sniezewski, L.; Majkowski, M.; Janik, S.; Cebrat, M. Uneven distribution of complementary sex determiner (csd) alleles in Apis mellifera population. Sci. Rep. 2017, 7, 2317. [Google Scholar] [CrossRef]
- Kaskinova, M.D.; Gataullin, A.R.; Saltykova, E.S.; Gaifullina, L.R.; Poskryakov, A.V.; Nikolenko, A.G. Polymorphism of the hypervariable region of the csd gene in the Apis mellifera L. population in Southern Urals. Russian J. Gen. 2019, 55, 267–270. [Google Scholar] [CrossRef]
- Wang, Z.; Liu, Z.; Wu, X.; Yan, W.; Zeng, Z. Polymorphism analysis of csd gene in six Apis mellifera subspecies. Mol. Biol. Rep. 2012, 39, 3067–3071. [Google Scholar] [CrossRef] [PubMed]
- Bilodeau, L.; Avalos, A.; Danka, R.G. Genetic diversity of the complementary sex-determiner (csd) gene in two closed breeding stocks of Varroa-resistant honey bees. Apidologie 2020, 51, 1125–1132. [Google Scholar] [CrossRef]
- Bilodeau, L.; Elsik, C. A scientific note defining allelic nomenclature standards for the highly diverse complementary sex-determiner (csd) locus in honey bees. Apidologie 2021, 52, 749–754. [Google Scholar] [CrossRef]
- Hyink, O.; Laas, F.; Dearden, P.K. Genetic tests for alleles of complementary-sex-determiner to support honeybee breeding programmes. Apidologie 2013, 44, 306–313. [Google Scholar] [CrossRef] [Green Version]
- Kolics, É.; Parrag, T.; Házi, F.; Szepesi, K.; Heltai, B.; Mátyás, K.; Kutasy, B.; Virág, E.; Taller, J.; Orbán, L.; et al. An alternative, high throughput method to identify csd alleles of the honey bee. Insects 2020, 11, 483. [Google Scholar] [CrossRef] [PubMed]
- Utzeri, V.J.; Schiavo, G.; Ribani, A.; Tinarelli, S.; Bertolini, F.; Bovo, S.; Fontanesi, L. Entomological signatures in honey: An environmental DNA metabarcoding approach can disclose information on plant-sucking insects in agricultural and forest landscapes. Sci. Rep. 2018, 8, 9996. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bovo, S.; Ribani, A.; Utzeri, V.J.; Schiavo, G.; Bertolini, F.; Fontanesi, L. Shotgun metagenomics of honey DNA: Evaluation of a methodological approach to describe a multi-kingdom honey bee derived environmental DNA signature. PLoS ONE 2018, 13, e0205575. [Google Scholar] [CrossRef] [PubMed]
- Bovo, S.; Utzeri, V.J.; Ribani, A.; Cabbri, R.; Fontanesi, L. Shotgun sequencing of honey DNA can describe honey bee derived environmental signatures and the honey bee hologenome complexity. Sci. Rep. 2020, 10, 9279. [Google Scholar] [CrossRef]
- Utzeri, V.J.; Ribani, A.; Fontanesi, L. Authentication of honey based on a DNA method to differentiate Apis mellifera subspecies: Application to Sicilian honey bee (A. m. siciliana) and Iberian honey bee (A. m. iberiensis) honeys. Food Control 2018, 91, 294–301. [Google Scholar] [CrossRef]
- Utzeri, V.J.; Ribani, A.; Taurisano, V.; Banqué, C.H.i.; Fontanesi, L. Distribution of the main Apis mellifera mitochondrial DNA lineages in Italy assessed using an environmental DNA approach. Insects 2021, 12, 620. [Google Scholar] [CrossRef] [PubMed]
- Utzeri, V.J.; Ribani, A.; Schiavo, G.; Bertolini, F.; Bovo, S.; Fontanesi, L. Application of next generation semiconductor based sequencing to detect the botanical composition of monofloral, polyfloral and honeydew honey. Food Control 2018, 86, 342–349. [Google Scholar] [CrossRef]
- Utzeri, V.J.; Schiavo, G.; Ribani, A.; Bertolini, F.; Bovo, S.; Fontanesi, L. A next generation sequencing approach for targeted Varroa destructor (Acari: Varroidae) mitochondrial DNA analysis based on honey derived environmental DNA. J. Invertebr. Pathol. 2019, 161, 47–53. [Google Scholar] [CrossRef] [PubMed]
- Ribani, A.; Utzeri, V.J.; Taurisano, V.; Fontanesi, L. Honey as a source of environmental DNA for the detection and monitoring of honey bee pathogens and parasites. Vet. Sci. 2020, 7, 113. [Google Scholar] [CrossRef]
- Ribani, A.; Utzeri, V.J.; Taurisano, V.; Galuppi, R.; Fontanesi, L. Analysis of honey environmental DNA indicates that the honey bee (Apis mellifera L.) trypanosome parasite Lotmaria passim is widespread in the apiaries of the North of Italy. J. Invertebr. Pathol. 2021, 184, 107628. [Google Scholar] [CrossRef] [PubMed]
- Ribani, A.; Schiavo, G.; Utzeri, V.J.; Bertolini, F.; Geraci, C.; Bovo, S.; Fontanesi, L. Application of next generation semiconductor based sequencing for species identification in dairy products. Food Chem. 2018, 246, 90–98. [Google Scholar] [CrossRef]
- Ribani, A.; Schiavo, G.; Utzeri, V.J.; Bertolini, F.; Geraci, C.; Bovo, S.; Fontanesi, L. Application of next generation semiconductor based sequencing for species identification and analysis of within-species mitotypes useful for authentication of meat derived products. Food Control 2018, 91, 58–67. [Google Scholar] [CrossRef]
- Bertolini, F.; Ghionda, M.C.; D’Alessandro, E.; Geraci, C.; Chiofalo, V.; Fontanesi, L. A next generation semiconductor based sequencing approach for the identification of meat species in DNA mixtures. PLoS ONE 2015, 10, e0121701. [Google Scholar] [CrossRef] [PubMed]
- UniProt Consortium. UniProt: The universal protein knowledgebase in 2021. Nucleic Acids Res. 2021, 49, D480–D489. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Rozewicki, J.; Yamada, K.D. MAFFT online service: Multiple sequence alignment, interactive sequence choice and visualization. Brief Bioinform. 2019, 20, 1160–1166. [Google Scholar] [CrossRef] [Green Version]
- Waterhouse, A.M.; Procter, J.B.; Martin, D.M.A.; Clamp, M.; Barton, G.J. Jalview Version 2—A multiple sequence alignment editor and analysis workbench. Bioinformatics 2009, 25, 1189–1191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2019; Available online: https://www.R-project.org/ (accessed on 2 August 2021).
- Landgraf, A.J.; Lee, Y. Dimensionality reduction for binary data through the projection of natural parameters. J. Mult. Anal. 2020, 180, 104668. [Google Scholar] [CrossRef]
- Porter, T.M.; Hajibabaei, M. Scaling up: A guide to high throughput genomic approaches for biodiversity analysis. Mol. Ecol. 2018, 27, 313–338. [Google Scholar] [CrossRef] [Green Version]
- Neumann, P.; Moritz, R.F.; van Praagh, J. Queen mating frequency in different types of honey bee mating apiaries. J. Apic. Res. 1999, 38, 11–18. [Google Scholar] [CrossRef]
- Estoup, A.; Solignac, M.; Cornuet, J.M. Precise assessment of the number of patrilines and of genetic relatedness in honeybee colonies. Proc. R. Soc. Lond. B 1994, 258, 1–7. [Google Scholar] [CrossRef]
- Oleksa, A.; Chybicki, I.; Tofilski, A.; Burczyk, J. Nuclear and mitochondrial patterns of introgression into native dark bees (Apis mellifera mellifera) in Poland. J. Apic. Res. 2011, 50, 116–129. [Google Scholar] [CrossRef] [Green Version]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample ID 1 | Province | No. of Reads | No. of Retained Reads (%) 2 | No. of csd Protein Alleles 3 | No. of Private csd Alleles (%) 4 | No. of New csd Alleles (Private) 5 |
|---|---|---|---|---|---|---|
| H1 | Bologna | 173,064 | 149,678 (86.4) | 17 | 8 (47.1) | 9 (7) |
| H2 ° | Reggio Emilia | 75,454 | 72,027 (95.4) | 13 | 4 (30.8) | 5 (3) |
| H3 | Piacenza | 414,002 | 269,785 (64.8) | 40 | 16 (40.0) | 13 (11) |
| H4 ° | Reggio Emilia | 84,307 | 82,056 (97.3) | 14 | 2 (14.3) | 5 (0) |
| H5 ° | Reggio Emilia | 397,333 | 296,982 (74.7) | 20 | 7 (35.0) | 5 (3) |
| H6 | Ferrara | 34,350 | 33,503 (97.5) | 13 | 3 (23.1) | 7 (3) |
| H7 | Piacenza | 99,979 | 48,369 (48.3) | 23 | 11 (47.8) | 12 (11) |
| H8 | Bologna | 41,741 | 37,431 (89.7) | 17 | 4 (23.5) | 3 (1) |
| H9 * | Piacenza | 97,735 | 73,994 (75.7) | 33 | 13 (39.4) | 16 (11) |
| H10 | Rimini | 70,307 | 53,728 (75.1) | 61 | 41 (67.2) | 40 (37) |
| H11 * | Piacenza | 35,183 | 29,865 (84.9) | 10 | 2 (20.0) | 3 (1) |
| H12 | Piacenza | 1341 | 1220 (91.0) | 10 | 3 (30.0) | 0 (0) |
| Sample ID 1 | csd Protein Allele Sequence 2 | Abundance % | Novel 3 |
|---|---|---|---|
| H1 | KIISSLSKNTIHNNNYKYNYNNNNNYNNNYKKLQYYNINYIEQIP | 53.4 | |
| H1 | KIISSLSNKTIHNNNNYKKLYYNINYIEQIP | 9.7 | |
| H1 | KIISSLSNNYNYSNYNNYNNNYNNYKKLYYNINYIEQIP ‡ | 8.9 | |
| H2 ° | KIISSLSNSCNYSNNYYNKKLYYNIINIEQIP † | 53.7 | |
| H2 ° | KIISSLSNNYNYSNYNNYNNNYNNYKKLYYNINYIEQIP ‡ | 30.2 | |
| H2 ° | KIISSLSNKTIHNNNNYKPYYNINYIEQIP ** | 7.7 | |
| H3 | KIISSLSNNYKYSNYNNYNNYNNKKLYYNIINIEQIP | 40.0 | Yes |
| H3 | KIISSLSNKTIHNNNNYNNYKKLYYNIINIEQIP | 12.4 | Yes |
| H3 | KIISSLSNKTIHNNNYKYNYNNNNNYKKLQYYNIINIEQIP | 6.3 | |
| H4 ° | KIISSLSNKTIHNNNNYKPYYNINYIEQIP ** | 83.6 | |
| H5 ° | KIISSLSSNYNSNNYNNYNNYKQLCYNINYIEQIP @ | 39.6 | |
| H5 ° | KIISSLSNNYKYSNYNNYNNYNKKLYYKNYIINIEQIP | 12.0 | |
| H5 ° | KIISSLSNNYNYNNKYNYNNNYNKKLYYNIINIEQIP § | 8.5 | |
| H6 | KIISSLSNKTIHNNNNYKPYYNINYIEQIP ** | 93.1 | |
| H7 | KIISSLSNNYNYNNKYNYNNNYNKKLYYNIINIEQIP § | 38.2 | |
| H7 | KIISSLSNKTIHNNNKYNYNNNYNNNCKKLYYNINYIEQIP | 8.9 | Yes |
| H8 | KIISSLSNKTIHNNNNYKPYYNINYIEQIP ** | 33.3 | |
| H8 | KIISSLSSNYNSNNYNNYNNYKQLCYNINYIEQIP @ | 25.9 | |
| H8 | KITSSLSNNYNSNNYNKYNYNNSKKLYYNINYIEQIP | 13.2 | |
| H8 | KIISSLSNKTIHNNNNYKYNYNNNNYKNYNNYKKLYYNINYIEQIP | 5.8 | |
| H9 * | KIISSLSNKTIHNNNNYKYNYNNNNYKPYYNINYIEQIP | 45.0 | |
| H9 * | KIISSLSNKTIHNNNNYKYNYNNNYNNNNYSKKLYYNINYIEQIP | 10.3 | Yes |
| H9 * | KIISSLSNNYISNISNYNNNNNSKKLYYNINYIEQIP | 5.2 | |
| H10 | KIISSLSNSCNYSNNYYNKKLYYNIINIEQIP † | 17.6 | |
| H11 * | KIISSLSSNYNSNNYNNYNNYKQLCYNINYIEQIP @ | 43.8 | |
| H11 * | KIISSLSNKTIHNNNNYKPYYNINYIEQIP ** | 32.5 | |
| H11 * | KIISSLSNNYNYNNKYNYNNNYNKKLYYNIINIEQIP § | 6.7 | |
| H12 | KITSSLSNNYNSNSYNNYNNNYKKLQYYNIINIEQIP | 37.5 | |
| H12 | KIISSLSNNYNYSNYNNYNNYNNNYNNYNNNYNNYKKLYYNINYIEQIP | 31.5 | |
| H12 | KIISSLSNKTIHNNNNYKYNYNNNNYNNNNYNNNYNNNCKKLYYNINYIEQIP | 5.4 | |
| H12 | KIISSLSNNYKYSNYNNYNNYNNNSKKLYKNYIINIEQIP | 5.3 | |
| H12 | KIISSLSNSCNYSNNYYNKKLYYNIINIEQIP † | 5.1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bovo, S.; Ribani, A.; Utzeri, V.J.; Taurisano, V.; Schiavo, G.; Bolner, M.; Fontanesi, L. Application of Next Generation Semiconductor-Based Sequencing for the Identification of Apis mellifera Complementary Sex Determiner (csd) Alleles from Honey DNA. Insects 2021, 12, 868. https://doi.org/10.3390/insects12100868
Bovo S, Ribani A, Utzeri VJ, Taurisano V, Schiavo G, Bolner M, Fontanesi L. Application of Next Generation Semiconductor-Based Sequencing for the Identification of Apis mellifera Complementary Sex Determiner (csd) Alleles from Honey DNA. Insects. 2021; 12(10):868. https://doi.org/10.3390/insects12100868
Chicago/Turabian StyleBovo, Samuele, Anisa Ribani, Valerio Joe Utzeri, Valeria Taurisano, Giuseppina Schiavo, Matteo Bolner, and Luca Fontanesi. 2021. "Application of Next Generation Semiconductor-Based Sequencing for the Identification of Apis mellifera Complementary Sex Determiner (csd) Alleles from Honey DNA" Insects 12, no. 10: 868. https://doi.org/10.3390/insects12100868
APA StyleBovo, S., Ribani, A., Utzeri, V. J., Taurisano, V., Schiavo, G., Bolner, M., & Fontanesi, L. (2021). Application of Next Generation Semiconductor-Based Sequencing for the Identification of Apis mellifera Complementary Sex Determiner (csd) Alleles from Honey DNA. Insects, 12(10), 868. https://doi.org/10.3390/insects12100868