High-Resolution Profiling of Gut Bacterial Communities in an Invasive Beetle using PacBio SMRT Sequencing System


Abstract
1. Introduction
2. Materials and Methods
2.1. Insects and Samples
2.2. DNA Extraction, PCR, PacBio Sequencing
2.3. Sequence Data Analysis
3. Results
3.1. Sequencing Information and Bacterial Diversity
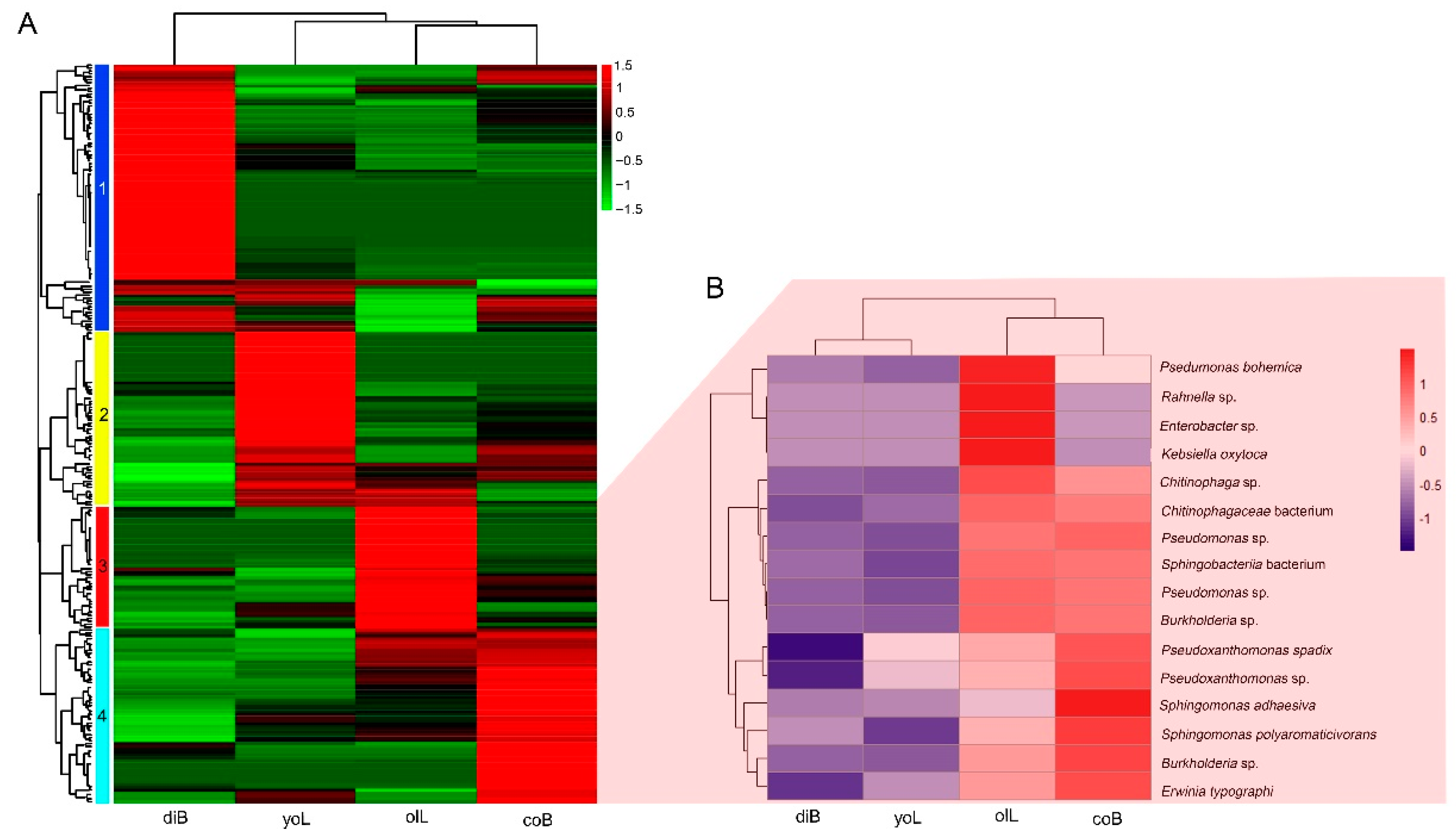
3.2. Bacterial Composition of Gut Samples at Different Life Stages
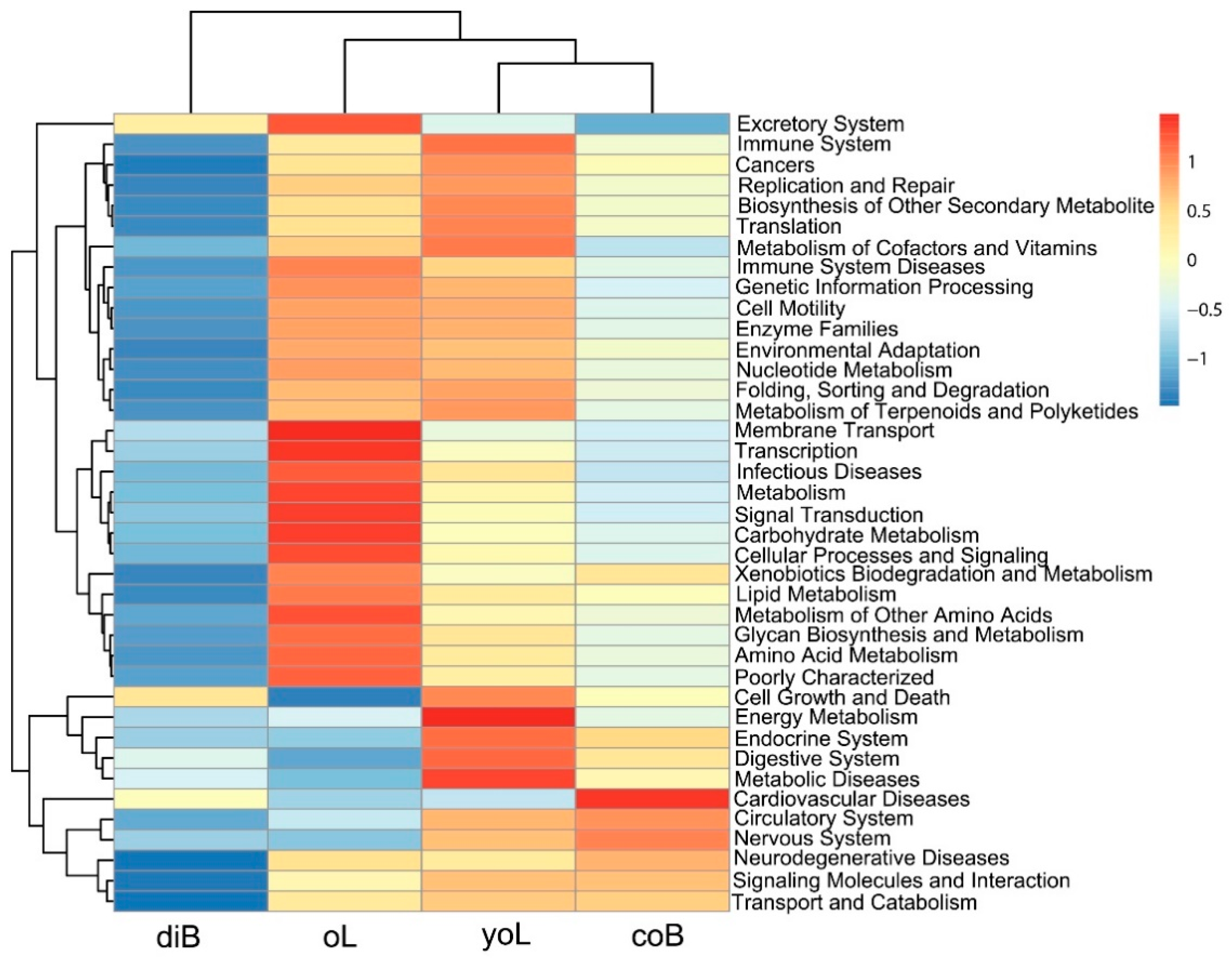
3.3. Prediction of the Functional Metagenome of D. valens Gut Bacterial Communities
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Yan, Z.; Sun, J.; Don, O.; Zhang, Z. The red turpentine beetle, Dendroctonus valens LeConte (Scolytidae): An exotic invasive pest of pine in China. Biodiver. Conserv. 2005, 14, 1735–1760. [Google Scholar] [CrossRef]
- Sun, J.; Lu, M.; Gillette, N.E.; Wingfield, M.J. Red turpentine beetle: Innocuous native becomes invasive tree killer in China. Annu. Rev. Entomol. 2013, 58, 293–311. [Google Scholar] [CrossRef] [PubMed]
- Lu, M.; Hulcr, J.; Sun, J. The Role of Symbiotic Microbes in Insect Invasions. Annu. Rev. Ecol. Evol. Syst. 2016, 47, 487–505. [Google Scholar] [CrossRef]
- Six, D.L.; Klepzig, K.D. Dendroctonus bark beetles as model systems for studies on symbiosis. Symbiosis 2004, 37, 207–232. [Google Scholar]
- Geib, S.M.; Filley, T.R.; Hatcher, P.G.; Hoover, K.; Carlson, J.E.; Jimenez-Gasco, M.d.M.; Nakagawa-Izumi, A.; Sleighter, R.L.; Tien, M. Lignin degradation in wood-feeding insects. Proc. Natl. Acad. Sci. USA 2008, 105, 12932–12937. [Google Scholar] [CrossRef] [PubMed]
- Delalibera, I., Jr.; Handelsman, J.; Raffa, K.F. Contrasts in cellulolytic activities of gut microorganisms between the wood borer, Saperda vestita (Coleoptera: Cerambycidae), and the bark beetles, Ips pini and Dendroctonus frontalis (Coleoptera: Curculionidae). Environ. Entomol. 2005, 34, 541–547. [Google Scholar] [CrossRef]
- Xu, L.; Liu, Y.; Xu, S.; Lu, M. Gut commensal bacteria in biological invasions. Int. Zool. 2019. [Google Scholar] [CrossRef] [PubMed]
- Suh, S.O.; Marshall, C.J.; Mchugh, J.V.; Blackwell, M. Wood ingestion by passalid beetles in the presence of xylose-fermenting gut yeasts. Mol. Ecol. 2003, 12, 3137–3145. [Google Scholar] [CrossRef]
- Morales-Jimenez, J.; Zuniga, G.; Villa-Tanaca, L.; Hernandez-Rodriguez, C. Bacterial community and nitrogen fixation in the red turpentine beetle, Dendroctonus valens LeConte (Coleoptera: Curculionidae: Scolytinae). Microb. Ecol. 2009, 58, 879–891. [Google Scholar] [CrossRef]
- Xu, L.; Lu, M.; Sun, J. Invasive bark beetle-associated microbes degrade a host defensive monoterpene. Insect Sci. 2016, 23, 183–190. [Google Scholar] [CrossRef]
- Boone, C.K.; Keefover-Ring, K.; Mapes, A.C.; Adams, A.S.; Bohlmann, J.; Raffa, K.F. Bacteria associated with a tree-killing insect reduce concentrations of plant defense compounds. J. Chem. Ecol. 2013, 39, 1003–1006. [Google Scholar] [CrossRef] [PubMed]
- Howe, M.; Keefover-Ring, K.; Raffa, K.F.; Louis, J. Pine engravers carry bacterial communities whose members reduce concentrations of host monoterpenes with variable degrees of redundancy, specificity, and capability. Environ. Entomol. 2018, 47, 638–645. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Deng, J.; Zhou, F.; Cheng, C.; Zhang, L.; Zhang, J.; Lu, M. Gut microbiota in an invasive bark beetle infected by a pathogenic fungus accelerates beetle mortality. J. Pest Sci. 2019, 92, 343–351. [Google Scholar] [CrossRef]
- McKenna, D.D.; Farrell, B.D. Beetles (Coleoptera). In The Timetree of Life; Hedges, S.B., Kumar, S., Eds.; Oxford University Press: Oxford, UK, 2009. [Google Scholar]
- Engel, P.; Moran, N.A. The gut microbiota of insects—Diversity in structure and function. FEMS Microbiol. Rev. 2013, 37, 699–735. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Lou, Q.; Cheng, C.; Lu, M.; Sun, J. Gut-associated bacteria of Dendroctonus valens and their involvement in verbenone production. Microb. Ecol. 2015, 70, 1012–1023. [Google Scholar] [CrossRef] [PubMed]
- Schloss, P.D.; Delalibera, I.; Handelsman, J.; Raffa, K.F. Bacteria Associated with the Guts of Two Wood-Boring Beetles: Anoplophora glabripennis and Saperda vestita (Cerambycidae). Environ. Entomol. 2006, 35, 625–629. [Google Scholar] [CrossRef]
- Vasanthakumar, A.; Delalibera, I.; Handelsman, J.; Klepzig, K.D.; Schloss, P.D.; Raffa, K.F. Characterization of Gut-Associated Bacteria in Larvae and Adults of the Southern Pine Beetle, Dendroctonus frontalis Zimmermann. Environ. Entomol. 2006, 35, 1710–1717. [Google Scholar] [CrossRef]
- Morales-Jimenez, J.; Zuniga, G.; Ramirez-Saad, H.C.; Hernandez-Rodriguez, C. Gut-associated bacteria throughout the life cycle of the bark beetle Dendroctonus rhizophagus Thomas and Bright (Curculionidae: Scolytinae) and their cellulolytic activities. Microb. Ecol. 2012, 64, 268–278. [Google Scholar] [CrossRef]
- Adams, A.S.; Adams, S.M.; Currie, C.R.; Gillette, N.E.; Raffa, K.F. Geographic variation in bacterial communities associated with the red turpentine beetle (Coleoptera: Curculionidae). Environ. Entomol. 2010, 39, 406–414. [Google Scholar] [CrossRef]
- Delalibera, I.J.; Vasanthakumar, A.; Burwitz, B.J.; Schloss, P.D.; Klepzig, K.D.; Handelsman, J.; Raffa, K.F. Composition of the bacterial community in the gut of the pine engraver, Ips pini (Say) (Coloptera) colonizing red pine. Symbiosis 2007, 43, 97–104. [Google Scholar]
- Yilmax, H.; Sezen, K.; Kati, H.; Demirbağ, Z. The first study on the bacterial flora of the european spruce bark beetle, Dendroctonus micans (Coleoptera: Scolytidae). Biologia 2006, 61, 679–686. [Google Scholar] [CrossRef]
- Tedersoo, L.; Tooming-Klunderud, A.; Anslan, S. PacBio metabarcoding of Fungi and other eukaryotes: Errors, biases and perspectives. New Phytol. 2017, 217, 1370. [Google Scholar] [CrossRef] [PubMed]
- Fichot, E.B.; Norman, R.S. Microbial phylogenetic profiling with the Pacific Biosciences sequencing platform. Microbiome 2013, 1, 10. [Google Scholar] [CrossRef] [PubMed]
- Hernández-García, J.A.; Briones-Roblero, C.I.; Rivera-Orduña, F.N.; Zúñiga, G. Revealing the gut bacteriome of Dendroctonus bark beetles (Curculionidae: Scolytinae): Diversity, core members and co-evolutionary patterns. Sci. Rep. 2017, 7, 13864. [Google Scholar] [CrossRef] [PubMed]
- Briones-Roblero, C.I.; Hernández-García, J.A.; Gonzalez-Escobedo, R.; Soto-Robles, L.V.; Rivera-Orduña, F.N.; Zúñiga, G. Structure and dynamics of the gut bacterial microbiota of the bark beetle, Dendroctonus rhizophagus (Curculionidae: Scolytinae) across their life stages. PLoS ONE 2017, 12, e0175470. [Google Scholar] [CrossRef] [PubMed]
- Kohl, K.D.; Brun, A.; Bordenstein, S.R.; Caviedes-Vidal, E.; Karasov, W.H. Gut microbes limit growth in house sparrow nestlings (Passer domesticus) but not through limitations in digestive capacity. Int. Zool. 2018, 13, 139–151. [Google Scholar] [CrossRef] [PubMed]
- Roberts, R.J.; Carneiro, M.O.; Schatz, M.C. The advantages of SMRT sequencing. Genome Biol. 2013, 14, 405. [Google Scholar] [CrossRef] [PubMed]
- Land, M.; Hauser, L.; Jun, S.R.; Nookaew, I.; Leuze, M.R.; Ahn, T.-H.; Karpinets, T.; Lund, O.; Kora, G.; Wassenaar, T.; et al. Insights from 20 years of bacterial genome sequencing. Funct. Integr. Genomics 2015, 15, 141–161. [Google Scholar] [CrossRef]
- Xu, D.; Xu, L.; Zhou, F.; Wang, B.; Wang, S.; Lu, M.; Sun, J. Gut Bacterial Communities of Dendroctonus valens and Monoterpenes and Carbohydrates of Pinus tabuliformis at Different Attack Densities to Host Pines. Front. Microbiol. 2018, 9, 1251. [Google Scholar] [CrossRef]
- Birgersson, G.; Schlyter, F.; Lofqvist, J.; Bergstrom, G. Quantitative variation of pheromone components in the spruce bark beetle Ips typographus from different attack phases. J. Chem. Ecol. 1984, 10, 1029–1055. [Google Scholar] [CrossRef]
- Edgar, R.C. UPARSE: Highly accurate OTU sequences from microbial amplicon reads. Nat. Methods 2013, 10, 996. [Google Scholar] [CrossRef] [PubMed]
- Langille, M.G.; Zaneveld, J.; Caporaso, J.G.; McDonald, D.; Knights, D.; Reyes, J.A.; Clemente, J.C.; Burkepile, D.E.; Thurber, R.L.V.; Knight, R. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat. Biotechnol. 2013, 31, 814–821. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Lu, M.; Xu, D.; Chen, L.; Sun, J. Sexual variation of bacterial microbiota of Dendroctonus valens guts and frass in relation to verbenone production. J. Insect Physiol. 2016, 95, 110–117. [Google Scholar] [CrossRef] [PubMed]
- Cheng, C.; Wickham, J.D.; Chen, L.; Xu, D.; Lu, M.; Sun, J. Bacterial microbiota protect an invasive bark beetle from a pine defensive compound. Microbiome 2018, 6, 132. [Google Scholar] [CrossRef] [PubMed]
- Cao, Q.; Wickham, J.D.; Chen, L.; Ahmad, F.; Lu, M.; Sun, J. Effect of Oxygen on Verbenone Conversion From cis-Verbenol by Gut Facultative Anaerobes of Dendroctonus valens. Front. Microbiol. 2018, 9, 464. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Wang, C.; Chen, H.; Ma, J. Differences in the structure of the gut bacteria communities in development stages of the Chinese white pine beetle (Dendroctonus armandi). Int. J. Mol. Sci. 2013, 14, 21006–21020. [Google Scholar] [CrossRef] [PubMed]
- Tremblay, J.; Singh, K.; Fern, A.; Kirton, E.; He, S.; Woyke, T.; Lee, J.; Chen, F.; Dangl, J.; Tringe, S. Primer and platform effects on 16S rRNA tag sequencing. Front. Microbiol. 2015, 6, 771. [Google Scholar] [CrossRef]
- Hansen, M.C.; Tolker-Nielsen, T.; Givskov, M.; Molin, S. Biased 16S rDNA PCR amplification caused by interference from DNA flanking the template region. FEMS Microbiol. Ecol. 1998, 26, 141–149. [Google Scholar] [CrossRef]
- Willner, D.; Daly, J.; Whiley, D.M.; Grimwood, K.; Wainwright, C.E.; Hugenholtz, P. Comparison of DNA Extraction Methods for Microbial Community Profiling with an Application to Pediatric Bronchoalveolar Lavage Samples. PLoS ONE 2012, 7, e34605. [Google Scholar] [CrossRef]
- Kennedy, N.A.; Walker, A.W.; Berry, S.H.; Duncan, S.H.; Farquarson, F.M.; Louis, P.; Thomson, J.M. The Impact of Different DNA Extraction Kits and Laboratories upon the Assessment of Human Gut Microbiota Composition by 16S rRNA Gene Sequencing. PLoS ONE 2014, 9, e88982. [Google Scholar] [CrossRef]
- Mao, D.P.; Zhou, Q.; Chen, C.Y.; Quan, Z.X. Coverage evaluation of universal bacterial primers using the metagenomic datasets. BMC Microbiol. 2012, 12, 66. [Google Scholar] [CrossRef]
- Rubin, B.E.R.; Sanders, J.G.; Hampton-Marcell, J.; Owens, S.M.; Gilbert, J.A.; Moreau, C.S. DNA extraction protocols cause differences in 16S rRNA amplicon sequencing efficiency but not in community profile composition or structure. MicrobiologyOpen 2014, 3, 910–921. [Google Scholar] [CrossRef]
- Lisdiyanti, P.; Yamada, Y.; Uchimura, T.; Komagata, K. Identification of Frateuria aurantia strains isolated from Indonesian sources. Microbiol. Cult. Coll. 2003, 19, 81–90. [Google Scholar]
- Zhou, F.; Lou, Q.; Wang, B.; Xu, L.; Cheng, C.; Lu, M.; Sun, J. Altered Carbohydrates Allocation by Associated Bacteria-fungi Interactions in a Bark Beetle-microbe Symbiosis. Sci. Rep. 2016, 6, 20135. [Google Scholar] [CrossRef]
- Colman, D.; Toolson, E.; Takacs-Vesbach, C. Do diet and taxonomy influence insect gut bacterial communities? Mol. Ecol. 2012, 21, 5124–5137. [Google Scholar] [CrossRef]
- Leufvén, A.; Birgersson, G. Quantitative variation of different monoterpenes around galleries of Ips typographus (Colleoptera: Scolytidae) attacking Norway spruce. Can. J. Bot. 1987, 65, 1038–1044. [Google Scholar] [CrossRef]
- Raffa, K.; Smalley, E. Interaction of pre-attack and induced monoterpene concentrations in host conifer defense against bark beetle-fungal complexes. Oecologia 1995, 102, 285–295. [Google Scholar] [CrossRef]
- Xu, B.B.; Liu, Z.D.; Sun, J.H. The effects of α-pinene on the feeding performance and pheromone production of Dendroctonus valens. Entomol. Exp. Appl. 2014, 150, 269–278. [Google Scholar] [CrossRef]
- Xu, L.; Shi, Z.; Wang, B.; Lu, M.; Sun, J. Pine Defensive Monoterpene alpha-Pinene Influences the Feeding Behavior of Dendroctonus valens and Its Gut Bacterial Community Structure. Int. J. Mol. Sci. 2016, 17, 1734. [Google Scholar] [CrossRef]
- Cheng, C.; Zhou, F.; Lu, M.; Sun, J. Inducible pine rosin defense mediates interactions between an invasive insect–fungal complex and newly acquired sympatric fungal associates. Int. Zool. 2015, 10, 453–464. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Species Richness Indices | Species Diversity Indices | ||
|---|---|---|---|---|
| OTUs Observed | Phylogenetic Diversity | Shannon Diversity | Simpson Diversity | |
| Young larvae (n = 9) | 45.89 ± 3.08 | 3.01 ± 0.21 | 2.40 ± 0.21 | 0.63 ± 0.06 |
| Old larvae (n = 7) | 53.14 ± 3.36 | 2.82 ± 0.15 | 2.71 ± 0.22 | 0.66 ± 0.05 |
| Dispersal adults (n = 7) | 45.29 ± 7.93 | 2.74 ± 0.40 | 2.56 ± 0.65 | 0.58 ± 0.14 |
| Colonized adults (n = 9) | 65.33 ± 4.30 | 3.58 ± 0.21 | 3.24 ± 0.29 | 0.73 ± 0.05 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xu, L.; Sun, L.; Zhang, S.; Wang, S.; Lu, M. High-Resolution Profiling of Gut Bacterial Communities in an Invasive Beetle using PacBio SMRT Sequencing System. Insects 2019, 10, 248. https://doi.org/10.3390/insects10080248
Xu L, Sun L, Zhang S, Wang S, Lu M. High-Resolution Profiling of Gut Bacterial Communities in an Invasive Beetle using PacBio SMRT Sequencing System. Insects. 2019; 10(8):248. https://doi.org/10.3390/insects10080248
Chicago/Turabian StyleXu, Letian, Liuwei Sun, Shihan Zhang, Shanshan Wang, and Min Lu. 2019. "High-Resolution Profiling of Gut Bacterial Communities in an Invasive Beetle using PacBio SMRT Sequencing System" Insects 10, no. 8: 248. https://doi.org/10.3390/insects10080248
APA StyleXu, L., Sun, L., Zhang, S., Wang, S., & Lu, M. (2019). High-Resolution Profiling of Gut Bacterial Communities in an Invasive Beetle using PacBio SMRT Sequencing System. Insects, 10(8), 248. https://doi.org/10.3390/insects10080248