Characterization of Dendrolimus houi Lajonquiere (Lepidoptera: Lasiocampidae) Transcriptome across All Life Stages

 ,
,
Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Feeding
2.2. RNA Isolation and Illumina Sequencing
2.3. De Novo Transcriptome Assembly and Annotation
2.4. Sequencing and Analysis of Differential Gene Expression Profile
2.5. Microsatellite Identification
2.6. Single Nucleotide Polymorphism
3. Results
3.1. Sequencing and de Novo Transcriptome Assembly
3.2. Predictive Protein Annotation
3.3. Functional Classification and Metabolic Pathway Analysis of Unigenes
3.4. Comparison and Analysis of Transcriptome at Different Developmental Stages of D. Houi
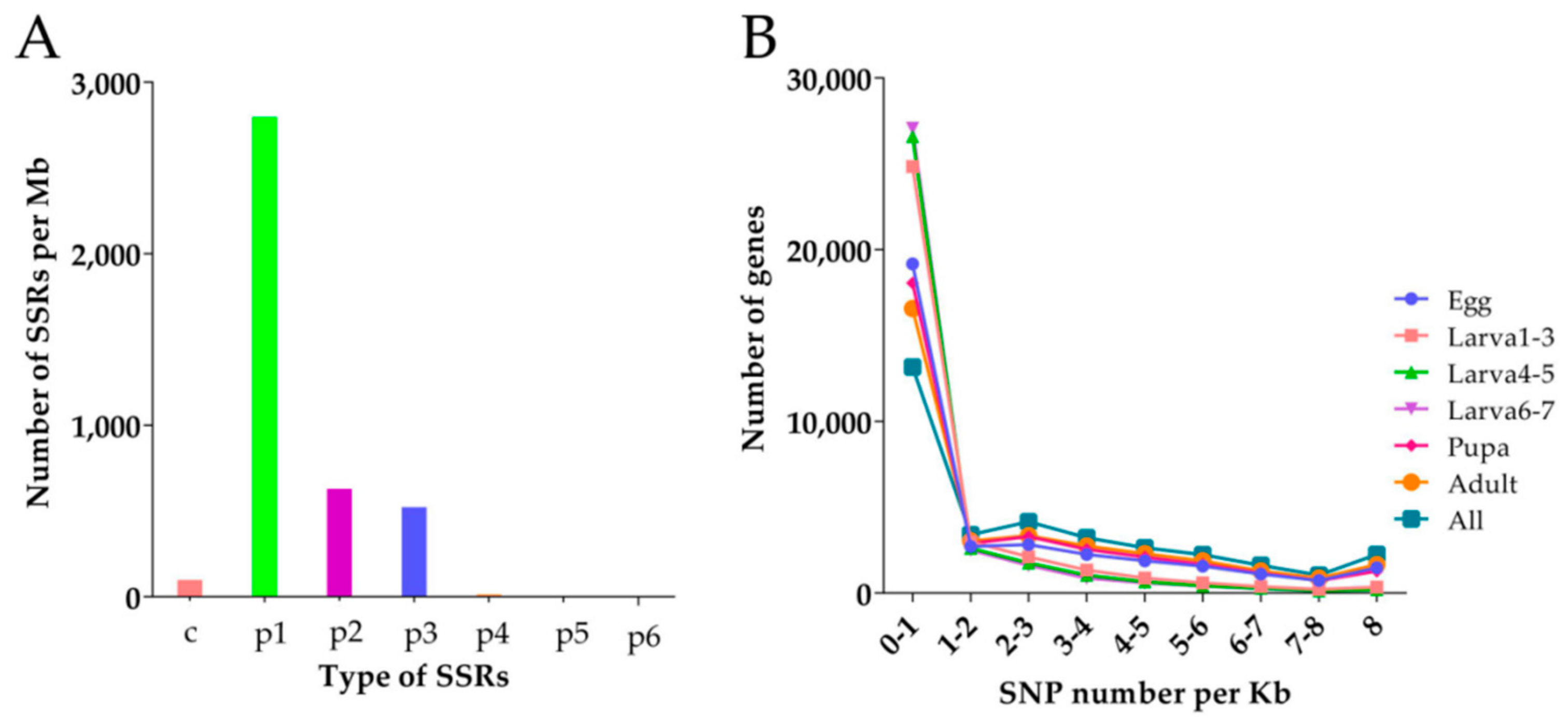
3.5. SSR and SNP Analysis of D. Houi
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Lin, H.Y.; Fu, L.Q.; Lin, J.H.; Hua, Y.; Han, X.H.; Zheng, J.X.; He, H.; Zhang, F.P.; Liang, G.H. Main species of parasitic natural enemy insects within Dendrolimus houi (Lajonquiere) in the forest of Cryptomeria fortunei (Hooibrenk). Chin. J. Biol. Control. 2017, 33, 842–848. [Google Scholar] [CrossRef]
- Liang, G.H.; Lin, H.Y.; Lu, C.D.; Han, H.H.; Hua, Y.; Huang, X.J.; Xie, Z.G.; Zhang, F.P.; Zhang, C.T. Morphology and biology of seven parasitic flies of Dendrolimus houi in China. Plant Prot. 2018, 44, 177–184. [Google Scholar] [CrossRef]
- Yang, H.; Shen, Y.; Deng, S.S.; Tang, Y.L.; Zhan, M.K.; Wang, J. Summary on IPM against Dendrolimus houi. Lajonquiere. Biol. Disaster Sci. 2017, 40, 231–234. [Google Scholar] [CrossRef]
- Zhou, Y.J.; Lin, H.Y.; Hua, Y.; Han, X.H.; Wei, Z.Y.; Zhang, F.P.; Liang, G.H. Occurrence regularity and biological characteristics of Dendrolimus houi in Fujian Province. Subtrop. Agric. Res. 2019, 15, 20–26. [Google Scholar] [CrossRef]
- Zhou, K.N. Reproductive Biology in the Pine Caterpilar, Dendrolimus Punctatus Walker (Lepidoptera: Lasiocampidae). Master’s Thesis, Jiangxi Agricultural University, Nanchang, China, 2013. [Google Scholar]
- Cao, X.C. Oviposition and feeding preference of Dendrolimus houi and Dendrolimus kikuchii to four species of pines. For. Pest. Dis. 2017, 36, 8–12. [Google Scholar]
- Chen, F.X.; Li, W.G.; Ma, X.L. Biological characteristics of Dendrolimus superans in Changbai Mountain Area. Prot. For. Sci. Technol. 2018, 40–42. [Google Scholar] [CrossRef]
- Li, H.S. Occurrence and Control of Dendrolimus Spectabilis. Agric. Sci. Technol. Equip. 2019, 14–15. [Google Scholar] [CrossRef]
- Shen, G.M.; Dou, W.; Niu, J.Z.; Jiang, H.B.; Yang, W.J.; Jia, F.X.; Hu, F.; Cong, L.; Wang, J.J. Transcriptome analysis of the oriental fruit fly (Bactrocera dorsalis). PLoS ONE 2011, 6, e29127. [Google Scholar] [CrossRef]
- Zhang, S.F.; Zhang, Z.; Wang, H.B.; Kong, X.B. Antennal transcriptome analysis and comparison of olfactory genes in two sympatric defoliators, Dendrolimus houi and Dendrolimus kikuchii (Lepidoptera: Lasiocampidae). Insect Biochem. Mol. Biol. 2014, 52, 69–81. [Google Scholar] [CrossRef]
- Chen, T.T.; Tan, L.R.; Hu, N.; Dong, Z.Q.; Hu, Z.G.; Qin, Q.; Long, J.Q.; Chen, P.; Xu, A.Y.; Pan, M.H.; et al. Specific genes related to nucleopolyhedrovirus in Bombyx mori susceptible and near-isogenic resistant strains through comparative transcriptome analysis. Insect Mol. Biol. 2019, 28, 473–484. [Google Scholar] [CrossRef]
- Neupane, S.; Mathew, F.M.; Varenhorst, A.J.; Nepal, M.P. Transcriptome profiling of interaction effects of soybean cyst nematodes and soybean aphids on soybean. Sci. Data 2019, 6, 13. [Google Scholar] [CrossRef] [PubMed]
- Dou, X.; Liu, S.; Soroker, V.; Harari, A.; Jurenka, R. Pheromone gland transcriptome of the pink bollworm moth, Pectinophora gossypiella: Comparison between a laboratory and field population. PLoS ONE 2019, 14, e0220187. [Google Scholar] [CrossRef] [PubMed]
- Moskalev, A.A.; Shaposhnikov, M.V.; Zemskaya, N.V.; Koval, L.А.; Schegoleva, E.V.; Guvatova, Z.G.; Kudryavtseva, A.V. Transcriptome analysis of Long-lived Drosophila melanogaster E(z) mutants sheds light on the molecular mechanisms of longevity. Sci. Rep. 2019, 9, 133. [Google Scholar] [CrossRef] [PubMed]
- Tian, C.H.; Tay, W.T.; Feng, H.; Wang, Y.; Hu, Y.; Li, G. Characterization of Adelphocoris suturalis (Hemiptera: Miridae) transcriptome from different developmental stages. Sci. Rep. 2015, 5, 11042. [Google Scholar] [CrossRef]
- Yang, C.H.; Yang, P.C.; Zhang, A.B. Transcriptome characterization of Dendrolimus punctatus and expression profiles at different developmental stages. PLoS ONE 2016, 11, e0161667. [Google Scholar] [CrossRef]
- Noriega, D.D.; Arias, P.L.; Barbosa, H.R.; Arraes, F.B.M.; Ossa, G.A.; Villegas, B.; Valencia-Jiménez, A. Transcriptome and gene expression analysis of three developmental stages of the coffee berry borer, Hypothenemus hampei. Sci. Rep. 2019, 9, 12804. [Google Scholar] [CrossRef]
- Mccarthy, C.B.; Cabrera, N.A.; Virla, E.G. Metatranscriptomic Analysis of larval guts from field-collected and laboratory-reared, Spodoptera frugiperda from the South American subtropical region. Genome Announc. 2015, 3, e00777–15. [Google Scholar] [CrossRef]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, I.A.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.D.; Chen, Z.H.; et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef]
- Pertea, G.; Huang, X.Q.; Liang, F.; Antonescu, V.; Sultana, R.; Karamycheva, S.; Lee, Y.D.; White, J.; Cheung, F.; Parvizi, B. TIGR gene indices clustering tools (TGICL): A software system for fast clustering of large EST datasets. Bioinformatics 2003, 19, 651–652. [Google Scholar] [CrossRef]
- Li, L.T.; Zhu, Y.B.; Ma, J.F.; Li, Z.Y.; Dong, Z.P. An Analysis of the Athetis lepigone transcriptome from four developmental stages. PLoS ONE 2013, 8, e73911. [Google Scholar] [CrossRef]
- Götz, S.; García-Gómez, J.M.; Terol, J.; Williams, T.D.; Nagaraj, S.H.; Nueda, M.J.; Robles, M.; Talón, M.; Dopazo, J.; Conesa, A. High-throughput functional annotation and data mining with the Blast2GO suite. Nucleic Acids Res. 2008, 36, 3420–3435. [Google Scholar] [CrossRef]
- Benjamini, Y.; Yekutieli, D. The control of the false discovery rate inmultiple testing under dependency. Ann. Stat. 2001, 29, 1165–1188. [Google Scholar] [CrossRef]
- Lin, T.; Wu, H.; Luo, L. The gene expression profile of Monochamus alternatus in response to deltamethrin exposure. J. Asia-Pac. Entomol. 2014, 17, 893–899. [Google Scholar] [CrossRef]
- Tang, P.A.; Wu, H.J.; Xue, H.; Ju, X.R.; Song, W.; Zhang, Q.L. Characterization of transcriptome in the indian meal moth Plodia interpunctella (Lepidoptera: Pyralidae) and gene expression analysis during developmental stages. Gene 2017, 622, 29–41. [Google Scholar] [CrossRef]
- Liu, Z.; Gong, P.; Heckel, D.G.; Wei, W.; Sun, J.; Li, D. Effects of larval host plants on over-wintering physiological dynamics and survival of the cotton bollworm, Helicoverpa armigera (Hübner) (Lepidoptera: Noctuidae). J. Insect Physiol. 2009, 55, 1–9. [Google Scholar] [CrossRef]
- Zhang, B.G.; Li, Y.P.; Ma, R.; Cheng, P.; Guo, X.X.; Wang, H.F.; Liu, L.J. Effects of low temperature on trehalose and trehalase contents in Culex Pipiens Pallens. Chin. J. Schistosomiasis Control 2019, 31, 510–512. [Google Scholar] [CrossRef]
- Liu, L.; Zhang, B.; Cheng, P.; Wang, H.; Guo, X.; Zhang, C.; Gong, M. Overwintering of Culex pipiens pallens (Diptera: Culicidae) in Shandong, China. J. Entomol. Sci. 2016, 51, 314–320. [Google Scholar] [CrossRef]
- Xie, J.; Xiong, W.; Hu, X.; Gu, S.; Zhang, S.; Gao, S.; Li, B. Characterization and functional analysis of hsp21.8b: An orthologous small heat shock protein gene in Tribolium castaneum. J. Appl. Entomol. 2018, 142, 654–666. [Google Scholar] [CrossRef]
- Wu, Y.K.; Zou, C.; Fu, D.M.; Zhang, W.N.; Xiao, H.J. Molecular characterization of three Hsp90 from Pieris and expression patterns in response to cold and thermal stress in summer and winter diapause of Pieris melete. Insect Sci. 2017, 25, 273–283. [Google Scholar] [CrossRef]
- Dudaniec, R.Y.; Chuan, J.Y.; Lancaster, L.T.; Svensson, E.I.; Hansson, B. Signatures of local adaptation along environmental gradients in a range-expanding damselfly (Ischnura elegans). Mol. Ecol. 2018, 27, 2576–2593. [Google Scholar] [CrossRef]
- Togawa, T.; Augustine, D.W.; Emmons, A.C. CPF and CPFL, two related gene families encoding cuticular. proteins of Anopheles gambiae and other insects. Insect Biochem. Mol. Biol. 2007, 37, 675–688. [Google Scholar] [CrossRef]
- Dubrovsky, E.B.; Bernardo, T.J. The juvenile hormone receptor and molecular mechanisms of juvenile hormone action. Adv. Insect Physiol. 2014, 46, 305–388. [Google Scholar] [CrossRef]
- Zhou, X.; Tarver, M.M. Hexamerin-based regulation of juvenile hormone-dependent gene expression. underlies phenotypic plasticity in a social insect. Development 2007, 134, 601–610. [Google Scholar] [CrossRef]
- Wang, Z.B.; Bai, J.H.; Liu, Y.J.; Li, H.; Zhan, S.; Xiao, Q. Transcriptomic analysis reveals insect hormone biosynthesis pathway involved in desynchronized development phenomenon in hybridized sibling species of tea geometrids (Ectropis grisescens and Ectropis obliqua). Insects 2019, 10, 381. [Google Scholar] [CrossRef]
- Han, X.H.; Lu, C.D.; Hua, Y.; Lin, H.Y.; Shi, Y.F.; Wu, S.Q.; Zhang, F.P.; Liang, G.H. Phylogenetic analysis of transcriptome and three detoxification enzyme families related genes in Anoplophora chinensis (Coleoptera: Cerambycidae). Sci. Silvae Sin. 2019, 55, 104–113. [Google Scholar] [CrossRef]
- Cheng, J.; Lu, Z.H.; Wang, C.Y.; Chen, J.X.; Lin, T. De novo analysis of Heortia vitessoides (Lepidoptera: Crambidae) transcriptome and identification of putative cytochrome P450 monooxygenase genes. J. Entomol. Sci. 2019, 54, 293–315. [Google Scholar] [CrossRef]
- Liu, J.; Lu, L.; Liu, H.; Li, Y.; Su, X.; Zhang, B.; Chen, X. De novo assembly of the transcriptome for Greenbug (Schizaphis graminum Rondani) and analysis on insecticide resistance-related genes. Entomol. Res. 2019, 49, 363–373. [Google Scholar] [CrossRef]
- Wang, X.G.; Ruan, Y.W.; Gong, C.W.; Xiang, X.; Xu, X.; Zhang, Y.M.; Shen, L.T. Transcriptome analysis of. Sogatella furcifera (Homoptera: Delphacidae) in response to Sulfoxaflor and functional verification of resistance-related P450 genes. Int. J. Mol. Sci. 2019, 20, 4573. [Google Scholar] [CrossRef]
- Pick, C.; Burmester, T. A putative hexamerin from a Campodea sp. suggests an independent origin of haemocyanin-related storage proteins in Hexapoda. Insect Mol. Biol. 2009, 18, 673–679. [Google Scholar] [CrossRef]
- Hahn, D.A.; James, L.N.; Milne, K.R.; Hatle, J.D. Life-history plasticity after attaining a dietary threshold for reproduction is associated with protein storage in flesh flies. Funct. Ecol. 2008, 22, 1081–1090. [Google Scholar] [CrossRef]
- Allen, M.L.; Rhoades, J.H.; Sparks, M.E.; Grodowitz, M.J. Differential gene expression in red imported fire ant (Solenopsis invicta) (Hymenoptera: Formicidae) larval and pupal stages. Insects 2018, 9, 185. [Google Scholar] [CrossRef] [PubMed]
- Xie, K.; Li, J.P.; Zhou, R.; Tian, Z.M.; Huang, Z.J.; Yang, W.Y.; Li, S.; Cao, Y. Biological significance of the. occurrence of autophagy and the source of autophagosomal membrane in insects. Acta Entomol. Sin. 2013, 56, 697–707. [Google Scholar] [CrossRef]
- Anderson, R.S.; Cook, M.L. Induction of lysozymelike activity in the hemolymph and hemocytes of an insect, Spodoptera eridania. J. Invertebr. Pathol. 1979, 33, 197–203. [Google Scholar] [CrossRef]
- Adamo, S.A. Estimating disease resistance in insects: Phenoloxidase and lysozyme-like activity and disease resistance in the cricket Gryllus texensis. J. Insect Physiol. 2004, 50, 209–216. [Google Scholar] [CrossRef] [PubMed]
- Sardiello, M.; Palmieri, M.; Di Ronza, A.; Medina, D.L.; Valenza, M.; Gennarino, V.A. A gene network regulating lysosomal biogenesis and function. Science 2009, 325, 473–477. [Google Scholar] [CrossRef] [PubMed]
- Jones, M.E.; Haire, M.F.; Kloetzel, P.M.; Mykles, D.L.; Schwartz, L.M. Changes in the structure and function of the multicatalytic proteinase (proteasome) during programmed cell death in the intersegmental muscles of the hawkmoth, Manduca sexta. Dev. Biol. 1995, 169, 436–447. [Google Scholar] [CrossRef]
- Xue, M.; Momiji, H.; Rabbani, N.; Barker, G.; Bretschneider, T.; Shmygol, A.; Rand, D.A.; Thornalley, P.J. Frequency modulated. translocational oscillations of nrf2 mediate the antioxidant response element cytoprotective transcriptional response. Antioxid. Redox Signal. 2014, 23, 613–629. [Google Scholar] [CrossRef]
- Xu, J.W.; Zhu, X.Y.; Chao, Q.J.; Zhang, Y.J.; Yang, Y.X.; Wang, R.R.; Zhang, Y.; Xie, M.Z.; Ge, Y.T.; Wu, X.L. Chemosensory gene families in the oligophagous pear pest Cacopsylla chinensis (Hemiptera: Psyllidae). Insects 2019, 10, 175. [Google Scholar] [CrossRef]
- Wang, X.T.; Zhang, Y.J.; Qiao, L.; Chen, B. Comparative analyses of simple sequence repeats (SSRs) in 23 mosquito species genomes: Identification, characterization, and distribution (Diptera: Culicidae). Insect Sci. 2018, 26, 607–619. [Google Scholar] [CrossRef]
- Li, M.N.; Long, R.C.; Feng, Z.R.; Liu, F.Q.; Sun, Y.; Zhang, K.; Kang, J.M.; Wang, Z.; Cao, S.H. Transcriptome analysis of salt-responsive genes and SSR marker exploration in Carex rigescens using RNA-seq. J. Integr. Agric. 2018, 17, 184–196. [Google Scholar] [CrossRef]
- Jin, Y.L.; Cong, B.; Wang, L.Y.; Dong, H. An analysis of the transcriptome of Epacromius coerulipes (Orthoptera: Acrididae). Acta Entomol. Sin. 2015, 58, 817–825. [Google Scholar] [CrossRef]
- Wei, D.D.; Shi, J.X.; Zhang, X.X.; Chen, S.C.; Wei, D.; Wang, J.J. Analysis of microsatellite loci from Bactrocera dorsalis based on transcriptome dataset. Chin. J. Appl. Ecol. 2014, 25, 1799–1805. [Google Scholar] [CrossRef]
- Qin, H.W.; Men, X.Y.; Yu, Y.; Lu, Z.B.; Sun, T.L.; Zhou, X.H.; Li, L.L. Effects of host plants on the cold hardiness of Athetis lepigone (Lepidoptera: Noctuidae) larvae. Acta Entomol. Sin. 2017, 60, 205–210. [Google Scholar] [CrossRef]
- Zhang, R.G.; Jian, Y.D.; Li, S.G. Damage of trees in Yunnan province to low temperature and analysis on cold resistance of these specie. J. West China For. Sci. 2009, 38, 1–8. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Clean Data | Raw Data | GC (%) | Q20 (%) |
|---|---|---|---|---|
| Adult | 22,239,714 | 6,659,218,426 | 42.01 | 95.8 |
| Egg | 25,298,693 | 7,572,136,450 | 38.5 | 95.94 |
| 1st–3rd instar larva | 20,748,014 | 6,211,582,592 | 51.25 | 96.21 |
| 4th–5th instar larva | 22,182,643 | 6,635,257,832 | 53.01 | 96.42 |
| 6th–7th instar larva | 26,275,877 | 7,872,265,062 | 50.66 | 96.04 |
| Pupa | 21,595,820 | 6,470,712,932 | 43.97 | 96.42 |
| Sequence Length (bp) | Transcript Number | Unigene Number |
|---|---|---|
| 300–500 | 15,369(23.59%) | 11,705(34.71%) |
| 500–1000 | 15,563(23.89%) | 8014(23.77%) |
| 1000–2000 | 16,025(24.60%) | 6552(19.43%) |
| 2000+ | 18,189(27.92%) | 7446(22.08%) |
| Total Number | 65,149 | 33,720 |
| Total Length (bp) | 108,319,868 | 47,972,049 |
| N50 Length (bp) | 2659 | 2534 |
| Mean Length (bp) | 1662.65 | 1422.66 |
| Anno_Database | Annotated_Number | 300≤ Length <1000 | Length ≥1000 |
|---|---|---|---|
| COG_Annotation | 8435 | 4094 | 4341 |
| GO_Annotation | 7639 | 2400 | 5239 |
| KEGG_Annotation | 6230 | 1653 | 4577 |
| KOG_Annotation | 9597 | 2569 | 7028 |
| Pfam_Annotation | 13,459 | 4880 | 8579 |
| Swiss-Prot_Annotation | 8618 | 2216 | 6402 |
| eggNOG_Annotation | 15,310 | 5631 | 9679 |
| nr_Annotation | 15,911 | 5693 | 10,218 |
| All_Annotated | 17,797 | 7495 | 10,302 |
| SSR Type | Repeat Motif | Number | Frequency |
|---|---|---|---|
| Mono-nucleotide | A/T/G/C | 2812 | 67.96% |
| Di-nucleotide | AT/GT/TA/TC Other types | 644 | 15.56% |
| Trinucleotide | GTG/GTC/GAA/GAT/AAT/CGG/TTC/TAT Other types | 536 | 12.95% |
| Tetra-nucleotide | ATTC/AGGA/GTAT/TCAC/TTAC Other types | 28 | 0.68% |
| Penta-nucleotide | ATATC/CTCTG/GTACA/AAATG | 4 | 0.097% |
| Hexa-nucleotide | TTCTCC | 1 | 0.023% |
| Compound SSR | (A)10(AT)7 Other types | 113 | 2.73% |
| Total | 4138 | 100% |
| Samples | HomoSNP | HeteSNP | AllSNP |
|---|---|---|---|
| Egg | 47,721 | 67,256 | 114,977 |
| 1st–3rd instar larva | 35,301 | 25,968 | 61,269 |
| 4th–5th instar larva | 30,498 | 17,964 | 48,462 |
| 6th–7th instar larva | 31,420 | 12,572 | 43,992 |
| Pupa | 60,348 | 61,627 | 121,975 |
| Adult | 74,160 | 57,616 | 131,776 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Han, X.; Lu, C.; Geib, S.M.; Zheng, J.; Wu, S.; Zhang, F.; Liang, G. Characterization of Dendrolimus houi Lajonquiere (Lepidoptera: Lasiocampidae) Transcriptome across All Life Stages. Insects 2019, 10, 442. https://doi.org/10.3390/insects10120442
Han X, Lu C, Geib SM, Zheng J, Wu S, Zhang F, Liang G. Characterization of Dendrolimus houi Lajonquiere (Lepidoptera: Lasiocampidae) Transcriptome across All Life Stages. Insects. 2019; 10(12):442. https://doi.org/10.3390/insects10120442
Chicago/Turabian StyleHan, Xiaohong, Ciding Lu, Scott M. Geib, Junxian Zheng, Songqing Wu, Feiping Zhang, and Guanghong Liang. 2019. "Characterization of Dendrolimus houi Lajonquiere (Lepidoptera: Lasiocampidae) Transcriptome across All Life Stages" Insects 10, no. 12: 442. https://doi.org/10.3390/insects10120442
APA StyleHan, X., Lu, C., Geib, S. M., Zheng, J., Wu, S., Zhang, F., & Liang, G. (2019). Characterization of Dendrolimus houi Lajonquiere (Lepidoptera: Lasiocampidae) Transcriptome across All Life Stages. Insects, 10(12), 442. https://doi.org/10.3390/insects10120442