Multiple Exon Skipping in the Duchenne Muscular Dystrophy Hot Spots: Prospects and Challenges




Abstract
1. Introduction
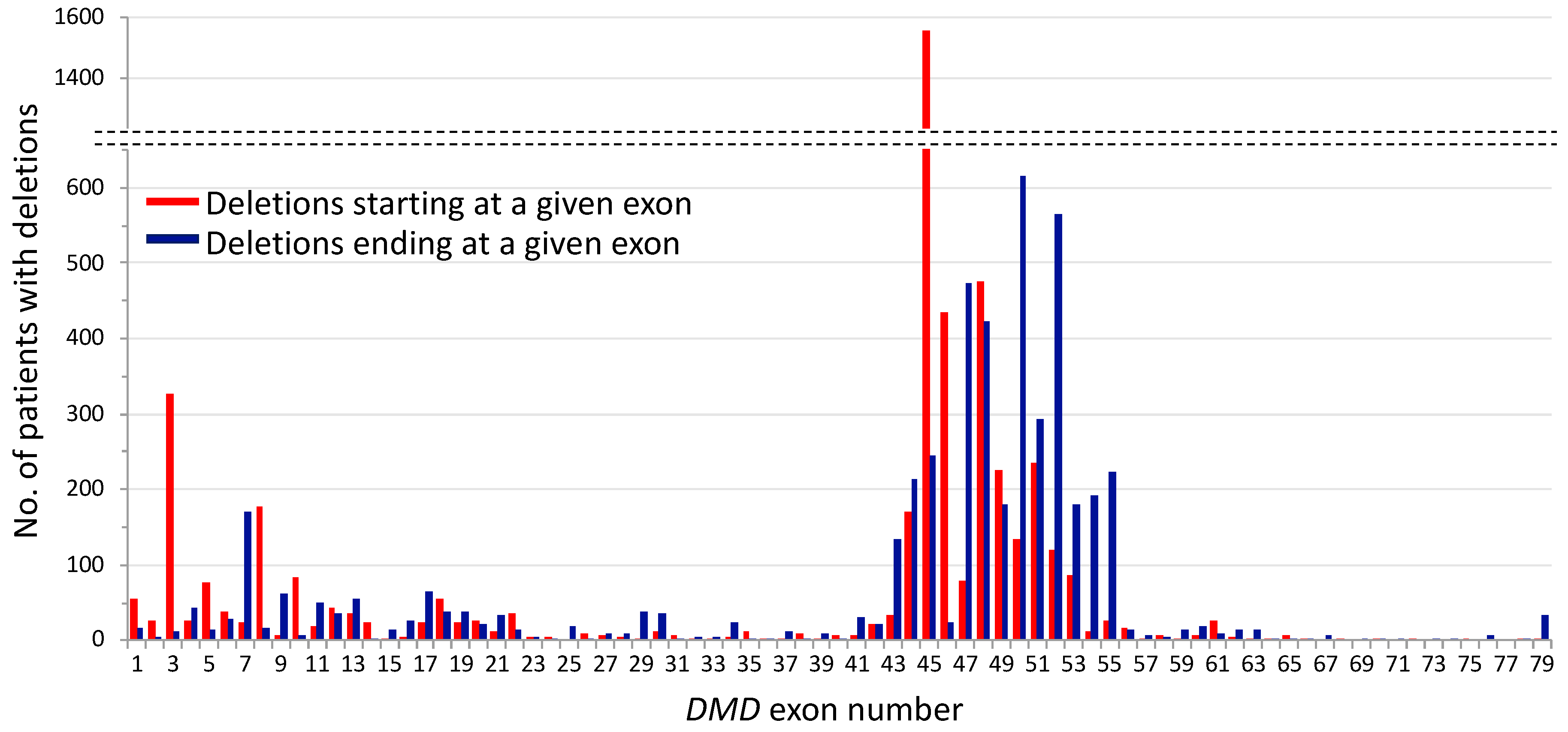
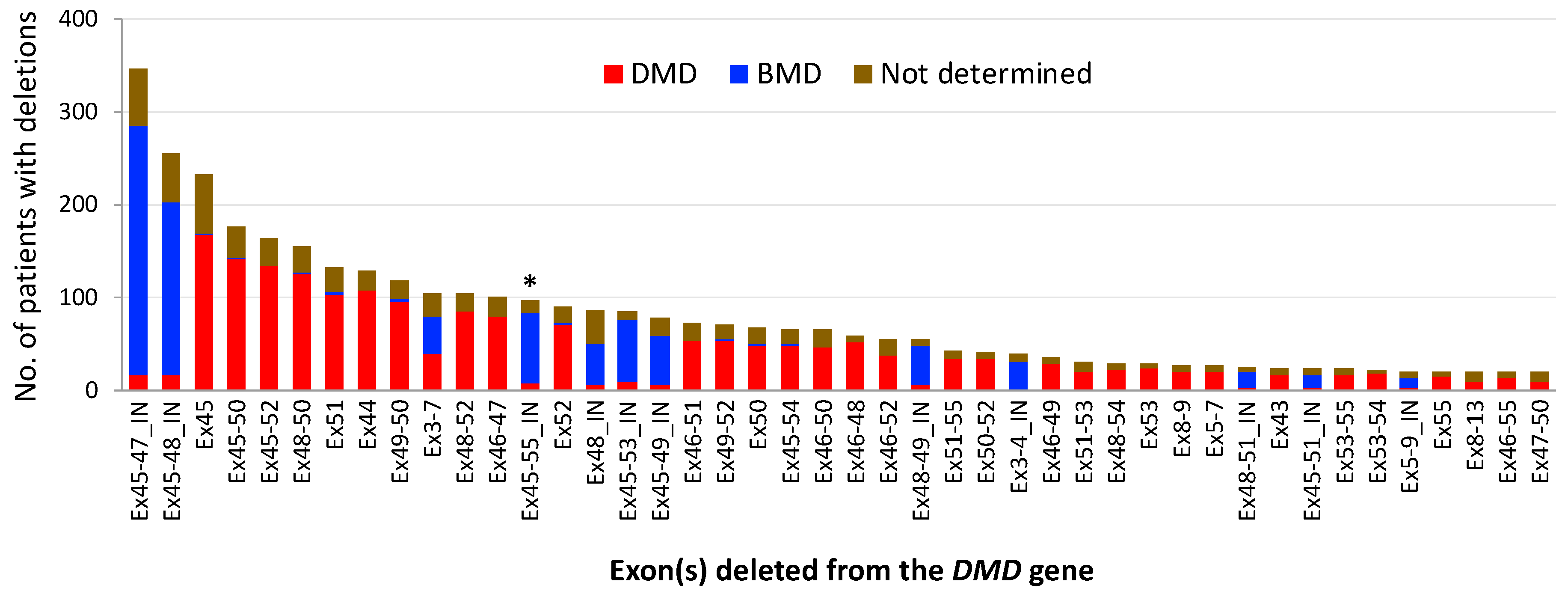
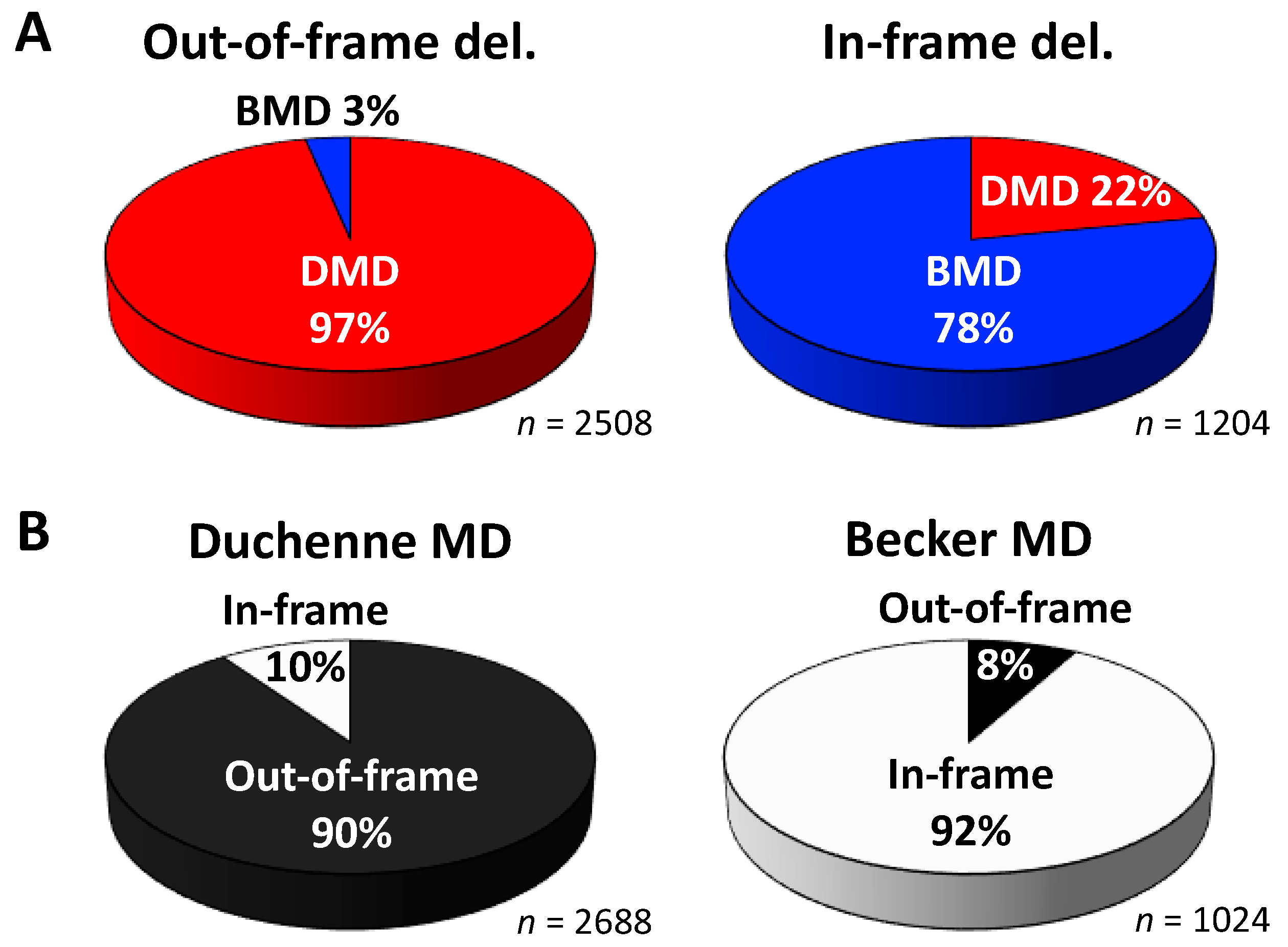
2. Mutational Hot Spots and Genotype-Phenotype Associations in the DMD Gene
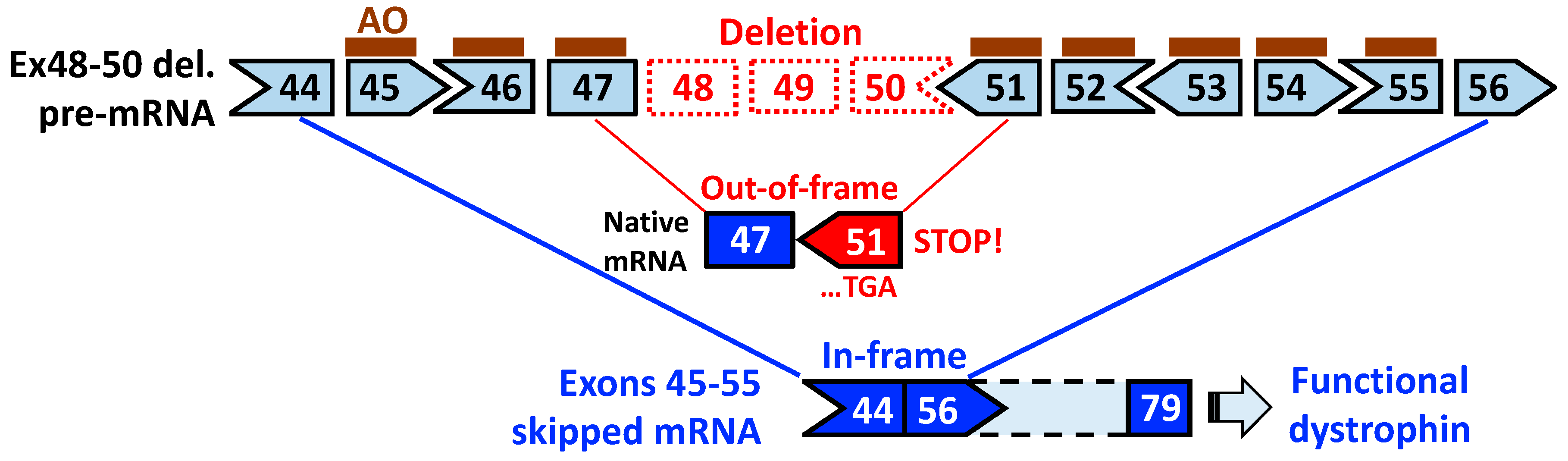
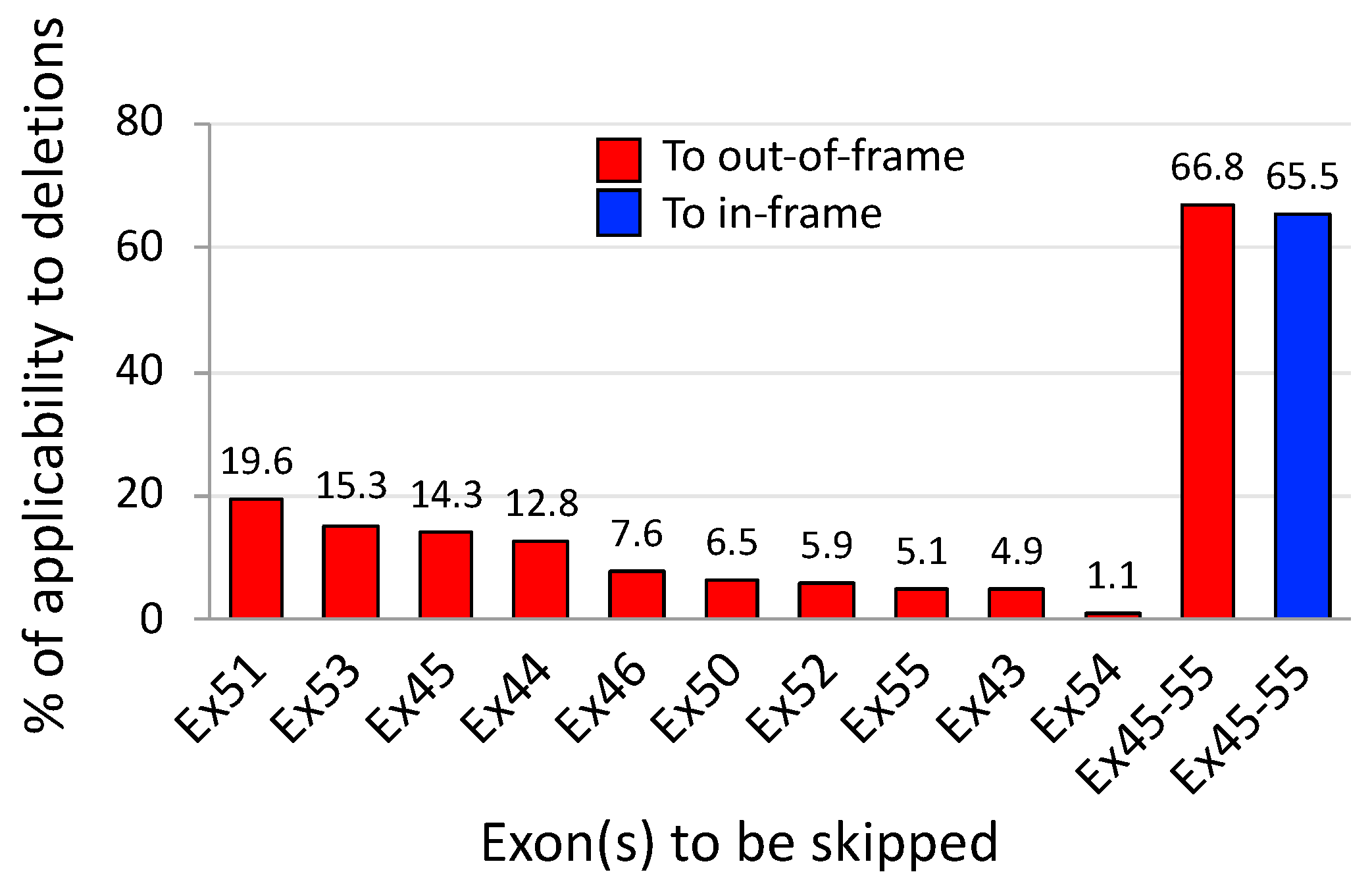
3. Exons 45–55 Skipping: Rationale and Pre-Clinical Tests
4. Expression Levels and Function of Exons 45–55-Deleted Dystrophin
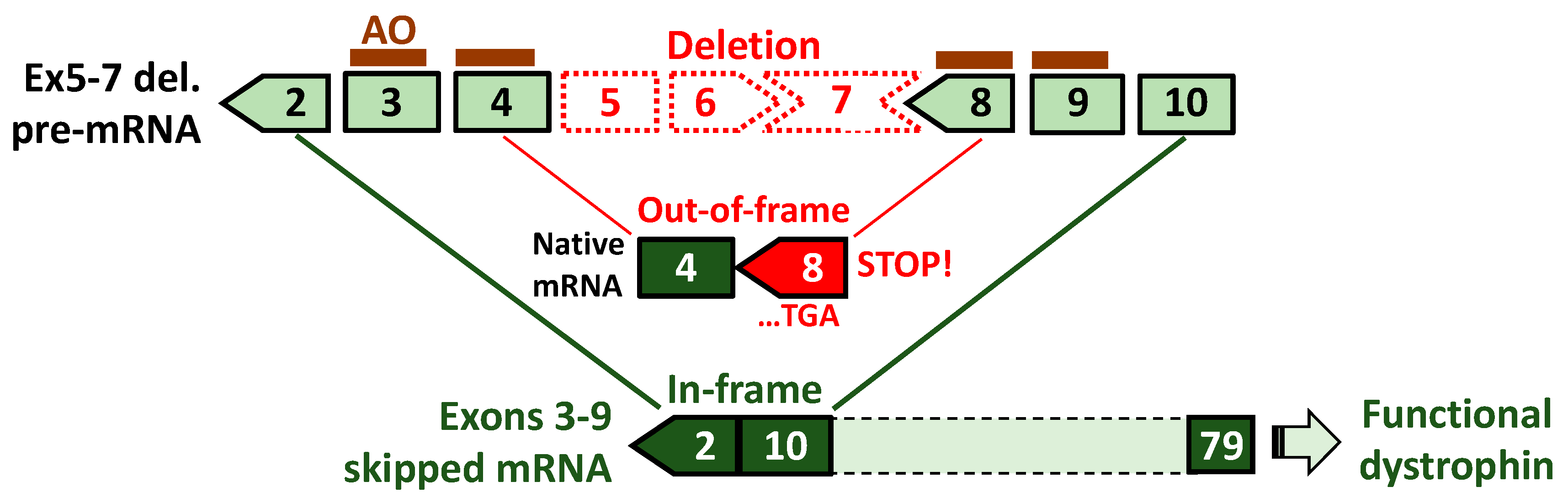
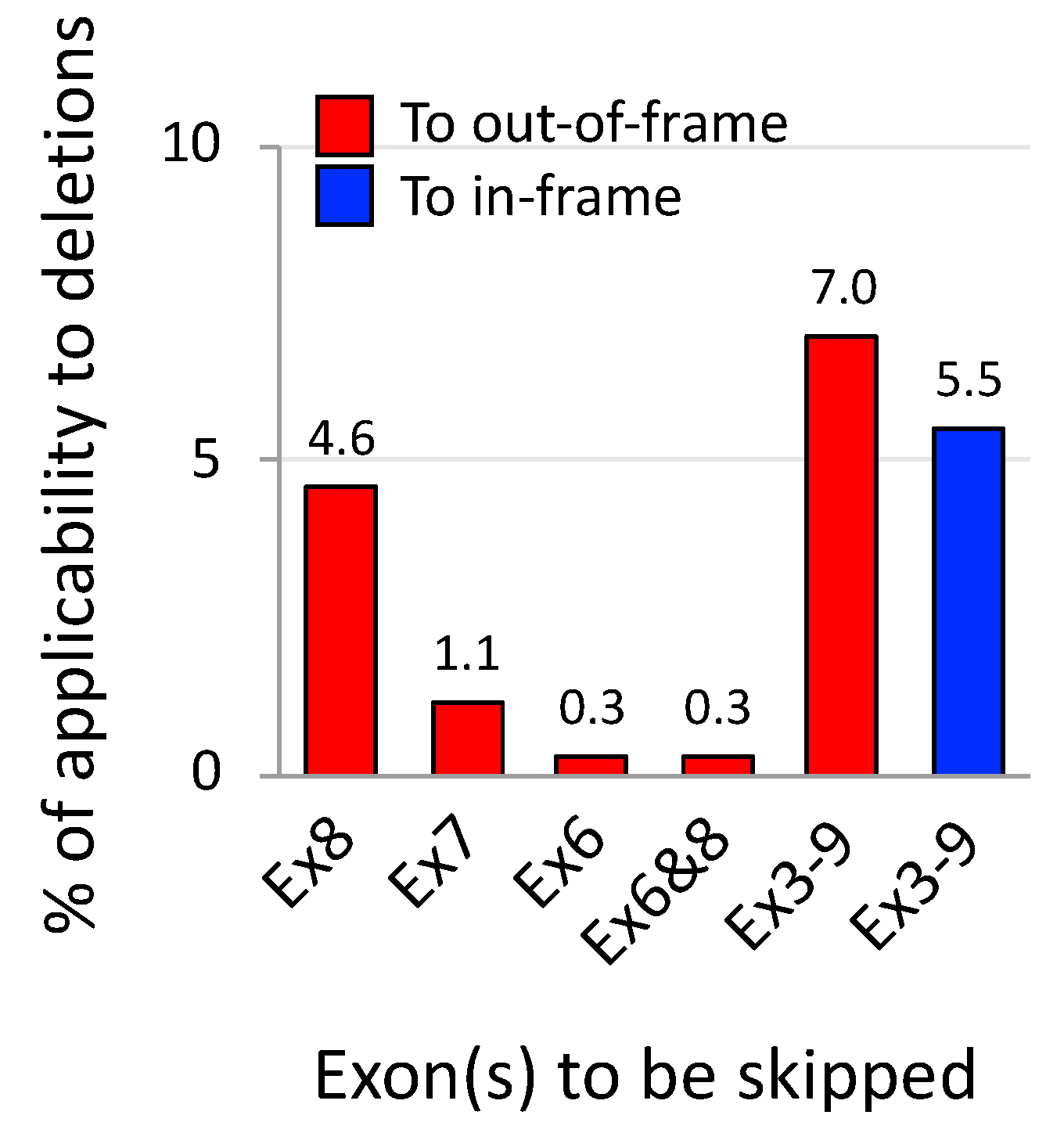
5. Exons 3–9 Skipping: Rationale and Challenges
6. Chemical Modification of AOs for Enhancing the Ability to Skip Multiple Exons
6.1. Vivo-PMOs
6.2. Cell-Penetrating Peptide-Conjugated PMOs
7. Design of Antisense Sequences and Cocktails for Multiple Exon Skipping
7.1. Screening and Optimization of Individual Antisense Oligonucleotide Sequences
7.2. Antisense Oligonucleotide Cocktails Tailored to Patient Mutations
8. Patient-Derived Cells and Humanized Animal Models for Testing Multiple Exon Skipping
8.1. Patient-Derived Muscle Cell Lines
8.2. Humanized Animal Models to Test Antisense Oligonucleotides Designed for Patients
9. Skipping of Exon Blocks Using Fewer Antisense Oligonucleotides
10. Conclusions
Funding
Conflicts of Interest
References
- Koenig, M.; Hoffman, E.P.; Bertelson, C.J.; Monaco, A.P.; Feener, C.; Kunkel, L.M. Complete cloning of the Duchenne muscular dystrophy (DMD) cDNA and preliminary genomic organization of the DMD gene in normal and affected individuals. Cell 1987, 50, 509–517. [Google Scholar] [CrossRef]
- Hoffman, E.P.; Brown, R.H., Jr.; Kunkel, L.M. Dystrophin: The protein product of the Duchenne muscular dystrophy locus. Cell 1987, 51, 919–928. [Google Scholar] [CrossRef]
- Mah, J.K.; Korngut, L.; Dykeman, J.; Day, L.; Pringsheim, T.; Jette, N. A systematic review and meta-analysis on the epidemiology of Duchenne and Becker muscular dystrophy. Neuromuscul. Disord. 2014, 24, 482–491. [Google Scholar] [CrossRef] [PubMed]
- Flanigan, K.M. Duchenne and Becker Muscular Dystrophies. Neurol. Clin. 2014, 32, 671–688. [Google Scholar] [CrossRef] [PubMed]
- Bello, L.; Kesari, A.; Gordish-Dressman, H.; Cnaan, A.; Morgenroth, L.P.; Punetha, J.; Duong, T.; Henricson, E.K.; Pegoraro, E.; McDonald, C.M.; et al. Genetic modifiers of ambulation in the CINRG duchenne natural history study. Ann. Neurol. 2015. [Google Scholar] [CrossRef]
- Nishizawa, H.; Shiba, N.; Nakamura, A. Importance of long-term motor function evaluation after prednisolone treatment for Duchenne muscular dystrophy. J. Phys. Ther. Sci. 2018, 30, 1211–1214. [Google Scholar] [CrossRef]
- Mostacciuolo, M.L.; Miorin, M.; Pegoraro, E.; Fanin, M.; Schiavon, F.; Vitiello, L.; Saad, F.A.; Angelini, C.; Danieli, G.A. Reappraisal of the incidence rate of Duchenne and Becker muscular dystrophies on the basis of molecular diagnosis. Neuroepidemiology 1993, 12, 326–330. [Google Scholar] [CrossRef]
- Ferreiro, V.; Giliberto, F.; Muniz, G.M.; Francipane, L.; Marzese, D.M.; Mampel, A.; Roque, M.; Frechtel, G.D.; Szijan, I. Asymptomatic Becker muscular dystrophy in a family with a multiexon deletion. Muscle Nerve 2009, 39, 239–243. [Google Scholar] [CrossRef]
- Anthony, K.; Cirak, S.; Torelli, S.; Tasca, G.; Feng, L.; Arechavala-Gomeza, V.; Armaroli, A.; Guglieri, M.; Straathof, C.S.; Verschuuren, J.J.; et al. Dystrophin quantification and clinical correlations in Becker muscular dystrophy: Implications for clinical trials. Brain 2011, 134, 3547–3559. [Google Scholar] [CrossRef]
- Taglia, A.; Petillo, R.; D’Ambrosio, P.; Picillo, E.; Torella, A.; Orsini, C.; Ergoli, M.; Scutifero, M.; Passamano, L.; Palladino, A.; et al. Clinical features of patients with dystrophinopathy sharing the 45-55 exon deletion of DMD gene. Acta Myol. 2015, 34, 9–13. [Google Scholar]
- Nakamura, A.; Shiba, N.; Miyazaki, D.; Nishizawa, H.; Inaba, Y.; Fueki, N.; Maruyama, R.; Echigoya, Y.; Yokota, T. Comparison of the phenotypes of patients harboring in-frame deletions starting at exon 45 in the Duchenne muscular dystrophy gene indicates potential for the development of exon skipping therapy. J. Hum. Genet. 2016, 62, 459. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, A. X-Linked Dilated Cardiomyopathy: A Cardiospecific Phenotype of Dystrophinopathy. Pharmaceuticals (Basel) 2015, 8, 303–320. [Google Scholar] [CrossRef] [PubMed]
- Beggs, A.H.; Hoffman, E.P.; Snyder, J.R.; Arahata, K.; Specht, L.; Shapiro, F.; Angelini, C.; Sugita, H.; Kunkel, L.M. Exploring the molecular basis for variability among patients with Becker muscular dystrophy: Dystrophin gene and protein studies. Am. J. Hum. Genet. 1991, 49, 54–67. [Google Scholar] [PubMed]
- Bushby, K.M.; Gardner-Medwin, D. The clinical, genetic and dystrophin characteristics of Becker muscular dystrophy. I. Natural history. J. Neurol. 1993, 240, 98–104. [Google Scholar] [CrossRef] [PubMed]
- Anthony, K.; Arechavala-Gomeza, V.; Ricotti, V.; Torelli, S.; Feng, L.; Janghra, N.; Tasca, G.; Guglieri, M.; Barresi, R.; Armaroli, A.; et al. Biochemical characterization of patients with in-frame or out-of-frame DMD deletions pertinent to exon 44 or 45 skipping. JAMA Neurol. 2014, 71, 32–40. [Google Scholar] [CrossRef] [PubMed]
- Koenig, M.; Beggs, A.H.; Moyer, M.; Scherpf, S.; Heindrich, K.; Bettecken, T.; Meng, G.; Muller, C.R.; Lindlof, M.; Kaariainen, H.; et al. The molecular basis for Duchenne versus Becker muscular dystrophy: Correlation of severity with type of deletion. Am. J. Hum. Genet. 1989, 45, 498–506. [Google Scholar] [PubMed]
- van den Bergen, J.C.; Wokke, B.H.; Janson, A.A.; van Duinen, S.G.; Hulsker, M.A.; Ginjaar, H.B.; van Deutekom, J.C.; Aartsma-Rus, A.; Kan, H.E.; Verschuuren, J.J. Dystrophin levels and clinical severity in Becker muscular dystrophy patients. J. Neurol. Neurosurg. Psychiatry 2013, 85, 747–753. [Google Scholar] [CrossRef] [PubMed]
- Nicolas, A.; Raguenes-Nicol, C.; Ben Yaou, R.; Ameziane-Le Hir, S.; Cheron, A.; Vie, V.; Claustres, M.; Leturcq, F.; Delalande, O.; Hubert, J.F.; et al. Becker muscular dystrophy severity is linked to the structure of dystrophin. Hum. Mol. Genet. 2014, 24, 1267–1279. [Google Scholar] [CrossRef]
- Monaco, A.P.; Bertelson, C.J.; Liechti-Gallati, S.; Moser, H.; Kunkel, L.M. An explanation for the phenotypic differences between patients bearing partial deletions of the DMD locus. Genomics 1988, 2, 90–95. [Google Scholar] [CrossRef]
- Aartsma-Rus, A.; Van Deutekom, J.C.; Fokkema, I.F.; Van Ommen, G.J.; Den Dunnen, J.T. Entries in the Leiden Duchenne muscular dystrophy mutation database: An overview of mutation types and paradoxical cases that confirm the reading-frame rule. Muscle Nerve 2006, 34, 135–144. [Google Scholar] [CrossRef]
- Tuffery-Giraud, S.; Beroud, C.; Leturcq, F.; Yaou, R.B.; Hamroun, D.; Michel-Calemard, L.; Moizard, M.P.; Bernard, R.; Cossee, M.; Boisseau, P.; et al. Genotype-phenotype analysis in 2405 patients with a dystrophinopathy using the UMD-DMD database: A model of nationwide knowledgebase. Hum. Mutat. 2009, 30, 934–945. [Google Scholar] [CrossRef] [PubMed]
- Bladen, C.L.; Salgado, D.; Monges, S.; Foncuberta, M.E.; Kekou, K.; Kosma, K.; Dawkins, H.; Lamont, L.; Roy, A.J.; Chamova, T.; et al. The TREAT-NMD DMD Global Database: Analysis of more than 7000 Duchenne muscular dystrophy mutations. Hum. Mutat. 2015, 36, 395–402. [Google Scholar] [CrossRef] [PubMed]
- Takeshima, Y.; Yagi, M.; Okizuka, Y.; Awano, H.; Zhang, Z.; Yamauchi, Y.; Nishio, H.; Matsuo, M. Mutation spectrum of the dystrophin gene in 442 Duchenne/Becker muscular dystrophy cases from one Japanese referral center. J. Hum. Genet. 2010, 55, 379–388. [Google Scholar] [CrossRef] [PubMed]
- Chelly, J.; Gilgenkrantz, H.; Lambert, M.; Hamard, G.; Chafey, P.; Recan, D.; Katz, P.; de la Chapelle, A.; Koenig, M.; Ginjaar, I.B.; et al. Effect of dystrophin gene deletions on mRNA levels and processing in Duchenne and Becker muscular dystrophies. Cell 1990, 63, 1239–1248. [Google Scholar] [CrossRef]
- Gangopadhyay, S.B.; Sherratt, T.G.; Heckmatt, J.Z.; Dubowitz, V.; Miller, G.; Shokeir, M.; Ray, P.N.; Strong, P.N.; Worton, R.G. Dystrophin in frameshift deletion patients with Becker muscular dystrophy. Am. J. Hum. Genet. 1992, 51, 562–570. [Google Scholar] [PubMed]
- Winnard, A.V.; Klein, C.J.; Coovert, D.D.; Prior, T.; Papp, A.; Snyder, P.; Bulman, D.E.; Ray, P.N.; McAndrew, P.; King, W.; et al. Characterization of translational frame exception patients in Duchenne/Becker muscular dystrophy. Hum. Mol. Genet. 1993, 2, 737–744. [Google Scholar] [CrossRef] [PubMed]
- Deburgrave, N.; Daoud, F.; Llense, S.; Barbot, J.C.; Recan, D.; Peccate, C.; Burghes, A.H.; Beroud, C.; Garcia, L.; Kaplan, J.C.; et al. Protein- and mRNA-based phenotype-genotype correlations in DMD/BMD with point mutations and molecular basis for BMD with nonsense and frameshift mutations in the DMD gene. Hum. Mutat. 2007, 28, 183–195. [Google Scholar] [CrossRef]
- Pramono, Z.A.; Takeshima, Y.; Alimsardjono, H.; Ishii, A.; Takeda, S.; Matsuo, M. Induction of exon skipping of the dystrophin transcript in lymphoblastoid cells by transfecting an antisense oligodeoxynucleotide complementary to an exon recognition sequence. Biochem. Biophys. Res. Commun. 1996, 226, 445–449. [Google Scholar] [CrossRef]
- Lim, K.R.; Maruyama, R.; Yokota, T. Eteplirsen in the treatment of Duchenne muscular dystrophy. Drug Des. Dev. Ther. 2017, 11, 533–545. [Google Scholar] [CrossRef]
- Rodrigues, M.; Yokota, T. An Overview of Recent Advances and Clinical Applications of Exon Skipping and Splice Modulation for Muscular Dystrophy and Various Genetic Diseases. Methods Mol. Biol. 2018, 1828, 31–55. [Google Scholar] [CrossRef]
- Komaki, H.; Nagata, T.; Saito, T.; Masuda, S.; Takeshita, E.; Sasaki, M.; Tachimori, H.; Nakamura, H.; Aoki, Y.; Takeda, S. Systemic administration of the antisense oligonucleotide NS-065/NCNP-01 for skipping of exon 53 in patients with Duchenne muscular dystrophy. Sci. Transl. Med. 2018, 10. [Google Scholar] [CrossRef] [PubMed]
- Aartsma-Rus, A.; Fokkema, I.; Verschuuren, J.; Ginjaar, I.; van Deutekom, J.; van Ommen, G.J.; den Dunnen, J.T. Theoretic applicability of antisense-mediated exon skipping for Duchenne muscular dystrophy mutations. Hum. Mutat. 2009, 30, 293–299. [Google Scholar] [CrossRef] [PubMed]
- Yokota, T.; Duddy, W.; Partridge, T. Optimizing exon skipping therapies for DMD. Acta Myol. 2007, 26, 179–184. [Google Scholar] [PubMed]
- Okubo, M.; Goto, K.; Komaki, H.; Nakamura, H.; Mori-Yoshimura, M.; Hayashi, Y.K.; Mitsuhashi, S.; Noguchi, S.; Kimura, E.; Nishino, I. Comprehensive analysis for genetic diagnosis of Dystrophinopathies in Japan. Orphanet J. Rare Dis. 2017, 12, 149. [Google Scholar] [CrossRef] [PubMed]
- Ma, P.; Zhang, S.; Zhang, H.; Fang, S.; Dong, Y.; Zhang, Y.; Hao, W.; Wu, S.; Zhao, Y. Comprehensive genetic characteristics of dystrophinopathies in China. Orphanet J. Rare Dis. 2018, 13, 109. [Google Scholar] [CrossRef] [PubMed]
- Mohammed, F.; Elshafey, A.; Al-Balool, H.; Alaboud, H.; Al Ben Ali, M.; Baqer, A.; Bastaki, L. Mutation spectrum analysis of Duchenne/Becker muscular dystrophy in 68 families in Kuwait: The era of personalized medicine. PLoS ONE 2018, 13, e0197205. [Google Scholar] [CrossRef] [PubMed]
- Beroud, C.; Tuffery-Giraud, S.; Matsuo, M.; Hamroun, D.; Humbertclaude, V.; Monnier, N.; Moizard, M.P.; Voelckel, M.A.; Calemard, L.M.; Boisseau, P.; et al. Multiexon skipping leading to an artificial DMD protein lacking amino acids from exons 45 through 55 could rescue up to 63% of patients with Duchenne muscular dystrophy. Hum. Mutat. 2007, 28, 196–202. [Google Scholar] [CrossRef] [PubMed]
- van den Bergen, J.C.; Schade van Westrum, S.M.; Dekker, L.; van der Kooi, A.J.; de Visser, M.; Wokke, B.H.; Straathof, C.S.; Hulsker, M.A.; Aartsma-Rus, A.; Verschuuren, J.J.; et al. Clinical characterisation of Becker muscular dystrophy patients predicts favourable outcome in exon-skipping therapy. J. Neurol. Neurosurg. Psychiatry 2014, 85, 92–98. [Google Scholar] [CrossRef]
- Mori-Yoshimura, M.; Mitsuhashi, S.; Nakamura, H.; Komaki, H.; Goto, K.; Yonemoto, N.; Takeuchi, F.; Hayashi, Y.K.; Murata, M.; Takahashi, Y.; et al. Characteristics of Japanese Patients with Becker Muscular Dystrophy and Intermediate Muscular Dystrophy in a Japanese National Registry of Muscular Dystrophy (Remudy): Heterogeneity and Clinical Variation. J. Neuromuscul. Dis. 2018, 5, 193–203. [Google Scholar] [CrossRef]
- Nakamura, A.; Fueki, N.; Shiba, N.; Motoki, H.; Miyazaki, D.; Nishizawa, H.; Echigoya, Y.; Yokota, T.; Aoki, Y.; Takeda, S. Deletion of exons 3–9 encompassing a mutational hot spot in the DMD gene presents an asymptomatic phenotype, indicating a target region for multiexon skipping therapy. J. Hum. Genet. 2016, 61, 663–667. [Google Scholar] [CrossRef]
- Heald, A.; Anderson, L.V.; Bushby, K.M.; Shaw, P.J. Becker muscular dystrophy with onset after 60 years. Neurology 1994, 44, 2388–2390. [Google Scholar] [CrossRef] [PubMed]
- Aoki, Y.; Yokota, T.; Nagata, T.; Nakamura, A.; Tanihata, J.; Saito, T.; Duguez, S.M.; Nagaraju, K.; Hoffman, E.P.; Partridge, T.; et al. Bodywide skipping of exons 45-55 in dystrophic mdx52 mice by systemic antisense delivery. Proc. Natl. Acad. Sci. USA 2012, 109, 13763–13768. [Google Scholar] [CrossRef] [PubMed]
- White, S.J.; den Dunnen, J.T. Copy number variation in the genome; the human DMD gene as an example. Cytogenet. Genome Res. 2006, 115, 240–246. [Google Scholar] [CrossRef] [PubMed]
- Bushby, K.; Finkel, R.; Birnkrant, D.J.; Case, L.E.; Clemens, P.R.; Cripe, L.; Kaul, A.; Kinnett, K.; McDonald, C.; Pandya, S.; et al. Diagnosis and management of Duchenne muscular dystrophy, part 1: Diagnosis, and pharmacological and psychosocial management. Lancet Neurol. 2010, 9, 77–93. [Google Scholar] [CrossRef]
- Nallamilli, B.R.; Ankala, A.; Hegde, M. Molecular diagnosis of duchenne muscular dystrophy. Curr. Protoc. Hum. Genet. 2014, 83, 9–25. [Google Scholar] [CrossRef]
- Aartsma-Rus, A.; Ginjaar, I.B.; Bushby, K. The importance of genetic diagnosis for Duchenne muscular dystrophy. J. Med. Genet. 2016, 53, 145–151. [Google Scholar] [CrossRef]
- Miyazaki, D.; Yoshida, K.; Fukushima, K.; Nakamura, A.; Suzuki, K.; Sato, T.; Takeda, S.; Ikeda, S. Characterization of deletion breakpoints in patients with dystrophinopathy carrying a deletion of exons 45-55 of the Duchenne muscular dystrophy (DMD) gene. J. Hum. Genet. 2009, 54, 127–130. [Google Scholar] [CrossRef]
- Oudet, C.; Hanauer, A.; Clemens, P.; Caskey, T.; Mandel, J.L. Two hot spots of recombination in the DMD gene correlate with the deletion prone regions. Hum. Mol. Genet. 1992, 1, 599–603. [Google Scholar] [CrossRef]
- Aoki, Y.; Yokota, T.; Wood, M.J. Development of multiexon skipping antisense oligonucleotide therapy for Duchenne muscular dystrophy. Biomed. Res. Int. 2013, 2013, 402369. [Google Scholar] [CrossRef]
- Findlay, A.R.; Wein, N.; Kaminoh, Y.; Taylor, L.E.; Dunn, D.M.; Mendell, J.R.; King, W.M.; Pestronk, A.; Florence, J.M.; Mathews, K.D.; et al. Clinical phenotypes as predictors of the outcome of skipping around DMD exon 45. Ann. Neurol. 2015, 77, 668–674. [Google Scholar] [CrossRef]
- Tsukamoto, H.; Inui, K.; Fukushima, H.; Nishigaki, T.; Taniike, M.; Tanaka, J.; Okada, S. Molecular study of Duchenne and Becker muscular dystrophies in Japanese. J. Inherit. Metab. Dis. 1991, 14, 819–824. [Google Scholar] [CrossRef] [PubMed]
- Yazaki, M.; Yoshida, K.; Nakamura, A.; Koyama, J.; Nanba, T.; Ohori, N.; Ikeda, S. Clinical characteristics of aged Becker muscular dystrophy patients with onset after 30 years. Eur. Neurol. 1999, 42, 145–149. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, A.; Yoshida, K.; Fukushima, K.; Ueda, H.; Urasawa, N.; Koyama, J.; Yazaki, Y.; Yazaki, M.; Sakai, T.; Haruta, S.; et al. Follow-up of three patients with a large in-frame deletion of exons 45–55 in the Duchenne muscular dystrophy (DMD) gene. J. Clin. Neurosci. 2008, 15, 757–763. [Google Scholar] [CrossRef] [PubMed]
- Kamdar, F.; Garry, D.J. Dystrophin-Deficient Cardiomyopathy. J. Am. Coll. Cardiol. 2016, 67, 2533–2546. [Google Scholar] [CrossRef] [PubMed]
- Colan, S.D. Evolving therapeutic strategies for dystrophinopathies: Potential for conflict between cardiac and skeletal needs. Circulation 2005, 112, 2756–2758. [Google Scholar] [CrossRef]
- van Vliet, L.; de Winter, C.L.; van Deutekom, J.C.; van Ommen, G.J.; Aartsma-Rus, A. Assessment of the feasibility of exon 45-55 multiexon skipping for Duchenne muscular dystrophy. BMC Med. Genet. 2008, 9, 105. [Google Scholar] [CrossRef] [PubMed]
- Echigoya, Y.; Aoki, Y.; Miskew, B.; Panesar, D.; Touznik, A.; Nagata, T.; Tanihata, J.; Nakamura, A.; Nagaraju, K.; Yokota, T. Long-term efficacy of systemic multiexon skipping targeting dystrophin exons 45–55 with a cocktail of vivo-morpholinos in mdx52 mice. Mol. Ther. Nucleic Acids 2015, 4, e225. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Echigoya, Y.; Duddy, W.; Saito, T.; Aoki, Y.; Takeda, S.; Yokota, T. Antisense PMO cocktails effectively skip dystrophin exons 45–55 in myotubes transdifferentiated from DMD patient fibroblasts. PLoS ONE 2018, 13, e0197084. [Google Scholar] [CrossRef] [PubMed]
- Ousterout, D.G.; Kabadi, A.M.; Thakore, P.I.; Majoros, W.H.; Reddy, T.E.; Gersbach, C.A. Multiplex CRISPR/Cas9-based genome editing for correction of dystrophin mutations that cause Duchenne muscular dystrophy. Nat. Commun. 2015, 6, 6244. [Google Scholar] [CrossRef]
- Young, C.S.; Mokhonova, E.; Quinonez, M.; Pyle, A.D.; Spencer, M.J. Creation of a Novel Humanized Dystrophic Mouse Model of Duchenne Muscular Dystrophy and Application of a CRISPR/Cas9 Gene Editing Therapy. J. Neuromuscul. Dis. 2017, 4, 139–145. [Google Scholar] [CrossRef]
- Young, C.S.; Hicks, M.R.; Ermolova, N.V.; Nakano, H.; Jan, M.; Younesi, S.; Karumbayaram, S.; Kumagai-Cresse, C.; Wang, D.; Zack, J.A.; et al. A Single CRISPR-Cas9 Deletion Strategy that Targets the Majority of DMD Patients Restores Dystrophin Function in hiPSC-Derived Muscle Cells. Cell Stem Cell 2016, 18, 533–540. [Google Scholar] [CrossRef] [PubMed]
- Center for Drug Evaluation and Research. Application Number 206488Orig1s000: Summary Review. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2016/206488_summary%20review_Redacted.pdf (accessed on 7 December 2018).
- Neri, M.; Torelli, S.; Brown, S.; Ugo, I.; Sabatelli, P.; Merlini, L.; Spitali, P.; Rimessi, P.; Gualandi, F.; Sewry, C.; et al. Dystrophin levels as low as 30% are sufficient to avoid muscular dystrophy in the human. Neuromuscul. Disord. 2007, 17, 913–918. [Google Scholar] [CrossRef] [PubMed]
- Gentil, C.; Leturcq, F.; Ben Yaou, R.; Kaplan, J.C.; Laforet, P.; Penisson-Besnier, I.; Espil-Taris, C.; Voit, T.; Garcia, L.; Pietri-Rouxel, F. Variable phenotype of del45-55 Becker patients correlated with nNOSmu mislocalization and RYR1 hypernitrosylation. Hum. Mol. Genet. 2012, 21, 3449–3460. [Google Scholar] [CrossRef] [PubMed]
- Sironi, M.; Bardoni, A.; Felisari, G.; Cagliani, R.; Robotti, M.; Comi, G.P.; Moggio, M.; Bresolin, N. Transcriptional activation of the non-muscle, full-length dystrophin isoforms in Duchenne muscular dystrophy skeletal muscle. J. Neurol. Sci. 2001, 186, 51–57. [Google Scholar] [CrossRef]
- Bello, L.; Campadello, P.; Barp, A.; Fanin, M.; Semplicini, C.; Soraru, G.; Caumo, L.; Calore, C.; Angelini, C.; Pegoraro, E. Functional changes in Becker muscular dystrophy: Implications for clinical trials in dystrophinopathies. Sci. Rep. 2016, 6, 32439. [Google Scholar] [CrossRef] [PubMed]
- Tanihata, J.; Nagata, T.; Ito, N.; Saito, T.; Nakamura, A.; Minamisawa, S.; Aoki, Y.; Ruegg, U.T.; Takeda, S.i. Truncated dystrophin ameliorates the dystrophic phenotype of mdx mice by reducing sarcolipin-mediated SERCA inhibition. Biochem. Biophys. Res. Commun. 2018, 505, 51–59. [Google Scholar] [CrossRef] [PubMed]
- Novakovic, I.; Bojic, D.; Todorovic, S.; Apostolski, S.; Lukovic, L.; Stefanovic, D.; Milasin, J. Proximal dystrophin gene deletions and protein alterations in becker muscular dystrophy. Ann. N. Y. Acad. Sci. 2005, 1048, 406–410. [Google Scholar] [CrossRef]
- Kaspar, R.W.; Allen, H.D.; Ray, W.C.; Alvarez, C.E.; Kissel, J.T.; Pestronk, A.; Weiss, R.B.; Flanigan, K.M.; Mendell, J.R.; Montanaro, F. Analysis of dystrophin deletion mutations predicts age of cardiomyopathy onset in becker muscular dystrophy. Circ. Cardiovasc. Genet. 2009, 2, 544–551. [Google Scholar] [CrossRef]
- Winnard, A.V.; Mendell, J.R.; Prior, T.W.; Florence, J.; Burghes, A.H. Frameshift deletions of exons 3–7 and revertant fibers in Duchenne muscular dystrophy: Mechanisms of dystrophin production. Am. J. Hum. Genet. 1995, 56, 158–166. [Google Scholar]
- Gurvich, O.L.; Maiti, B.; Weiss, R.B.; Aggarwal, G.; Howard, M.T.; Flanigan, K.M. DMD exon 1 truncating point mutations: Amelioration of phenotype by alternative translation initiation in exon 6. Hum. Mutat. 2009, 30, 633–640. [Google Scholar] [CrossRef]
- Hightower, R.M.; Alexander, M.S. Genetic modifiers of Duchenne and facioscapulohumeral muscular dystrophies. Muscle Nerve 2018, 57, 6–15. [Google Scholar] [CrossRef] [PubMed]
- Toh, Z.Y.; Thandar Aung-Htut, M.; Pinniger, G.; Adams, A.M.; Krishnaswarmy, S.; Wong, B.L.; Fletcher, S.; Wilton, S.D. Deletion of Dystrophin In-Frame Exon 5 Leads to a Severe Phenotype: Guidance for Exon Skipping Strategies. PLoS ONE 2016, 11, e0145620. [Google Scholar] [CrossRef] [PubMed]
- Henderson, D.M.; Lee, A.; Ervasti, J.M. Disease-causing missense mutations in actin binding domain 1 of dystrophin induce thermodynamic instability and protein aggregation. Proc. Natl. Acad. Sci. USA 2010, 107, 9632–9637. [Google Scholar] [CrossRef] [PubMed]
- Kyrychenko, V.; Kyrychenko, S.; Tiburcy, M.; Shelton, J.M.; Long, C.; Schneider, J.W.; Zimmermann, W.H.; Bassel-Duby, R.; Olson, E.N. Functional correction of dystrophin actin binding domain mutations by genome editing. JCI Insight 2017, 2. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Bao, B.; Echigoya, Y.; Yokota, T. Dystrophin-deficient large animal models: Translational research and exon skipping. Am. J. Transl. Res. 2015, 7, 1314–1331. [Google Scholar]
- Vieira, N.M.; Spinazzola, J.M.; Alexander, M.S.; Moreira, Y.B.; Kawahara, G.; Gibbs, D.E.; Mead, L.C.; Verjovski-Almeida, S.; Zatz, M.; Kunkel, L.M. Repression of phosphatidylinositol transfer protein alpha ameliorates the pathology of Duchenne muscular dystrophy. Proc. Natl. Acad. Sci. USA 2017, 114, 6080–6085. [Google Scholar] [CrossRef]
- Yokota, T.; Lu, Q.L.; Partridge, T.; Kobayashi, M.; Nakamura, A.; Takeda, S.; Hoffman, E. Efficacy of systemic morpholino exon-skipping in Duchenne dystrophy dogs. Ann. Neurol. 2009, 65, 667–676. [Google Scholar] [CrossRef]
- Echigoya, Y.; Nakamura, A.; Nagata, T.; Urasawa, N.; Lim, K.R.; Trieu, N.; Panesar, D.; Kuraoka, M.; Moulton, H.M.; Saito, T.; et al. Effects of systemic multiexon skipping with peptide-conjugated morpholinos in the heart of a dog model of Duchenne muscular dystrophy. Proc. Natl. Acad. Sci. USA 2017, 114, 4213–4218. [Google Scholar] [CrossRef]
- McClorey, G.; Moulton, H.M.; Iversen, P.L.; Fletcher, S.; Wilton, S.D. Antisense oligonucleotide-induced exon skipping restores dystrophin expression in vitro in a canine model of DMD. Gene Ther. 2006, 13, 1373–1381. [Google Scholar] [CrossRef]
- Saito, T.; Nakamura, A.; Aoki, Y.; Yokota, T.; Okada, T.; Osawa, M.; Takeda, S. Antisense PMO found in dystrophic dog model was effective in cells from exon 7-deleted DMD patient. PLoS ONE 2010, 5, e12239. [Google Scholar] [CrossRef]
- Bish, L.T.; Sleeper, M.M.; Forbes, S.C.; Wang, B.; Reynolds, C.; Singletary, G.E.; Trafny, D.; Morine, K.J.; Sanmiguel, J.; Cecchini, S.; et al. Long-term restoration of cardiac dystrophin expression in golden retriever muscular dystrophy following rAAV6-mediated exon skipping. Mol. Ther. 2012, 20, 580–589. [Google Scholar] [CrossRef]
- Barbash, I.M.; Cecchini, S.; Faranesh, A.Z.; Virag, T.; Li, L.; Yang, Y.; Hoyt, R.F.; Kornegay, J.N.; Bogan, J.R.; Garcia, L.; et al. MRI roadmap-guided transendocardial delivery of exon-skipping recombinant adeno-associated virus restores dystrophin expression in a canine model of Duchenne muscular dystrophy. Gene Ther. 2013, 20, 274–282. [Google Scholar] [CrossRef] [PubMed]
- Yokota, T.; Nakamura, A.; Nagata, T.; Saito, T.; Kobayashi, M.; Aoki, Y.; Echigoya, Y.; Partridge, T.; Hoffman, E.P.; Takeda, S. Extensive and prolonged restoration of dystrophin expression with vivo-morpholino-mediated multiple exon skipping in dystrophic dogs. Nucleic Acid Ther. 2012, 22, 306–315. [Google Scholar] [CrossRef] [PubMed]
- Vulin, A.; Barthelemy, I.; Goyenvalle, A.; Thibaud, J.L.; Beley, C.; Griffith, G.; Benchaouir, R.; le Hir, M.; Unterfinger, Y.; Lorain, S.; et al. Muscle function recovery in golden retriever muscular dystrophy after AAV1-U7 exon skipping. Mol. Ther. 2012, 20, 2120–2133. [Google Scholar] [CrossRef]
- Le Guiner, C.; Montus, M.; Servais, L.; Cherel, Y.; Francois, V.; Thibaud, J.L.; Wary, C.; Matot, B.; Larcher, T.; Guigand, L.; et al. Forelimb Treatment in a Large Cohort of Dystrophic Dogs Supports Delivery of a Recombinant AAV for Exon Skipping in Duchenne Patients. Mol. Ther. 2014, 22, 1923–1935. [Google Scholar] [CrossRef] [PubMed]
- Lim, K.R.Q.; Echigoya, Y.; Nagata, T.; Kuraoka, M.; Kobayashi, M.; Aoki, Y.; Partridge, T.; Maruyama, R.; Takeda, S.; Yokota, T. Efficacy of multi-exon skipping treatment in Duchenne muscular dystrophy dog model neonates. Mol. Ther. 2018. [Google Scholar] [CrossRef] [PubMed]
- Khvorova, A.; Watts, J.K. The chemical evolution of oligonucleotide therapies of clinical utility. Nat. Biotechnol. 2017, 35, 238–248. [Google Scholar] [CrossRef]
- Jirka, S.M.G.; Hoen, P.A.C.; Diaz Parillas, V.; Tanganyika-de Winter, C.L.; Verheul, R.C.; Aguilera, B.; de Visser, P.C.; Aartsma-Rus, A.M. Cyclic Peptides to Improve Delivery and Exon Skipping of Antisense Oligonucleotides in a Mouse Model for Duchenne Muscular Dystrophy. Mol. Ther. 2018, 26, 132–147. [Google Scholar] [CrossRef]
- Goemans, N.; Mercuri, E.; Belousova, E.; Komaki, H.; Dubrovsky, A.; McDonald, C.M.; Kraus, J.E.; Lourbakos, A.; Lin, Z.; Campion, G.; et al. A randomized placebo-controlled phase 3 trial of an antisense oligonucleotide, drisapersen, in Duchenne muscular dystrophy. Neuromuscul. Disord. 2018, 28, 4–15. [Google Scholar] [CrossRef]
- Echigoya, Y.; Lim, K.R.Q.; Trieu, N.; Bao, B.; Miskew Nichols, B.; Vila, M.C.; Novak, J.S.; Hara, Y.; Lee, J.; Touznik, A.; et al. Quantitative Antisense Screening and Optimization for Exon 51 Skipping in Duchenne Muscular Dystrophy. Mol. Ther. 2017, 25, 2561–2572. [Google Scholar] [CrossRef]
- McClorey, G.; Banerjee, S. Cell-Penetrating Peptides to Enhance Delivery of Oligonucleotide-Based Therapeutics. Biomedicines 2018, 6, 51. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Shen, X.; Dong, X.; Ran, N.; Han, G.; Cao, L.; Gu, B.; Yin, H. Peptide Nucleic Acid Promotes Systemic Dystrophin Expression and Functional Rescue in Dystrophin-deficient mdx Mice. Mol. Ther. Nucleic Acids 2015, 4, e255. [Google Scholar] [CrossRef] [PubMed]
- Shimo, T.; Maruyama, R.; Yokota, T. Designing Effective Antisense Oligonucleotides for Exon Skipping. Methods Mol. Biol. 2018, 1687, 143–155. [Google Scholar] [CrossRef] [PubMed]
- Relizani, K.; Goyenvalle, A. Use of Tricyclo-DNA Antisense Oligonucleotides for Exon Skipping. Methods Mol. Biol. 2018, 1828, 381–394. [Google Scholar] [CrossRef] [PubMed]
- Echigoya, Y.; Yokota, T. Skipping Multiple Exons of Dystrophin Transcripts Using Cocktail Antisense Oligonucleotides. Nucleic Acid Ther. 2014, 24, 57–68. [Google Scholar] [CrossRef] [PubMed]
- Aoki, Y.; Nakamura, A.; Yokota, T.; Saito, T.; Okazawa, H.; Nagata, T.; Takeda, S. In-frame dystrophin following exon 51-skipping improves muscle pathology and function in the exon 52-deficient mdx mouse. Mol. Ther. 2010, 18, 1995–2005. [Google Scholar] [CrossRef] [PubMed]
- Vila, M.C.; Klimek, M.B.; Novak, J.S.; Rayavarapu, S.; Uaesoontrachoon, K.; Boehler, J.F.; Fiorillo, A.A.; Hogarth, M.W.; Zhang, A.; Shaughnessy, C.; et al. Elusive sources of variability of dystrophin rescue by exon skipping. Skelet Muscle 2015, 5, 44. [Google Scholar] [CrossRef]
- Moulton, J.D.; Yan, Y.L. Using Morpholinos to control gene expression. Curr. Protoc. Mol. Biol. 2008, 83, 26–28. [Google Scholar] [CrossRef]
- Gong, P.; Wang, K.; Liu, Y.; Shepard, K.; Levicky, R. Molecular mechanisms in morpholino-DNA surface hybridization. J. Am. Chem. Soc. 2010, 132, 9663–9671. [Google Scholar] [CrossRef]
- Iwamoto, N.; Butler, D.C.D.; Svrzikapa, N.; Mohapatra, S.; Zlatev, I.; Sah, D.W.Y.; Meena; Standley, S.M.; Lu, G.; Apponi, L.H.; et al. Control of phosphorothioate stereochemistry substantially increases the efficacy of antisense oligonucleotides. Nat. Biotechnol. 2017, 35, 845–851. [Google Scholar] [CrossRef]
- Morcos, P.; Li, Y.; Jiang, S. Vivo-Morpholinos: A non-peptide transporter delivers Morpholinos into a wide array of mouse tissues. BioTechniques 2008, 45, 613–623. [Google Scholar] [CrossRef] [PubMed]
- Ferguson, D.P.; Dangott, L.J.; Lightfoot, J.T. Lessons learned from vivo-morpholinos: How to avoid vivo-morpholino toxicity. Biotechniques 2014, 56, 251–256. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, T.; Nakatani, M.; Narukawa, K.; Obika, S. Antisense drug discovery and development. Future Med. Chem. 2011, 3, 339–365. [Google Scholar] [CrossRef] [PubMed]
- Lehto, T.; Ezzat, K.; Wood, M.J.A.; El Andaloussi, S. Peptides for nucleic acid delivery. Adv. Drug Deliv. Rev. 2016, 106, 172–182. [Google Scholar] [CrossRef] [PubMed]
- Moulton, H.M.; Moulton, J.D. Morpholinos and their peptide conjugates: Therapeutic promise and challenge for Duchenne muscular dystrophy. Biochim. Biophys. Acta 2010, 1798, 2296–2303. [Google Scholar] [CrossRef] [PubMed]
- Moulton, J.D.; Jiang, S. Gene knockdowns in adult animals: PPMOs and vivo-morpholinos. Molecules 2009, 14, 1304–1323. [Google Scholar] [CrossRef] [PubMed]
- Amantana, A.; Moulton, H.M.; Cate, M.L.; Reddy, M.T.; Whitehead, T.; Hassinger, J.N.; Youngblood, D.S.; Iversen, P.L. Pharmacokinetics, biodistribution, stability and toxicity of a cell-penetrating peptide-morpholino oligomer conjugate. Bioconjug. Chem. 2007, 18, 1325–1331. [Google Scholar] [CrossRef]
- Burki, U.; Keane, J.; Blain, A.; O’Donovan, L.; Gait, M.J.; Laval, S.H.; Straub, V. Development and Application of an Ultrasensitive Hybridization-Based ELISA Method for the Determination of Peptide-Conjugated Phosphorodiamidate Morpholino Oligonucleotides. Nucleic Acid Ther. 2015, 25, 275–284. [Google Scholar] [CrossRef]
- Wu, B.; Lu, P.; Cloer, C.; Shaban, M.; Grewal, S.; Milazi, S.; Shah, S.N.; Moulton, H.M.; Lu, Q.L. Long-term rescue of dystrophin expression and improvement in muscle pathology and function in dystrophic mdx mice by peptide-conjugated morpholino. Am. J. Pathol. 2012, 181, 392–400. [Google Scholar] [CrossRef]
- Zincarelli, C.; Soltys, S.; Rengo, G.; Rabinowitz, J.E. Analysis of AAV serotypes 1–9 mediated gene expression and tropism in mice after systemic injection. Mol. Ther. 2008, 16, 1073–1080. [Google Scholar] [CrossRef]
- van der Bent, M.L.; Paulino da Silva Filho, O.; van Luijk, J.; Brock, R.; Wansink, D.G. Assisted delivery of antisense therapeutics in animal models of heritable neurodegenerative and neuromuscular disorders: A systematic review and meta-analysis. Sci. Rep. 2018, 8, 4181. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.; Wu, H.; McBride, J.L.; Jung, K.E.; Kim, M.H.; Davidson, B.L.; Lee, S.K.; Shankar, P.; Manjunath, N. Transvascular delivery of small interfering RNA to the central nervous system. Nature 2007, 448, 39–43. [Google Scholar] [CrossRef] [PubMed]
- Huey, R.; Hawthorne, S.; McCarron, P. The potential use of rabies virus glycoprotein-derived peptides to facilitate drug delivery into the central nervous system: A mini review. J. Drug Target. 2017, 25, 379–385. [Google Scholar] [CrossRef] [PubMed]
- Echigoya, Y.; Mouly, V.; Garcia, L.; Yokota, T.; Duddy, W. In silico screening based on predictive algorithms as a design tool for exon skipping oligonucleotides in duchenne muscular dystrophy. PLoS ONE 2015, 10, e0120058. [Google Scholar] [CrossRef] [PubMed]
- Shimo, T.; Tachibana, K.; Saito, K.; Yoshida, T.; Tomita, E.; Waki, R.; Yamamoto, T.; Doi, T.; Inoue, T.; Kawakami, J.; et al. Design and evaluation of locked nucleic acid-based splice-switching oligonucleotides in vitro. Nucleic Acids Res. 2014, 42, 8174–8187. [Google Scholar] [CrossRef] [PubMed]
- Touznik, A.; Maruyama, R.; Hosoki, K.; Echigoya, Y.; Yokota, T. LNA/DNA mixmer-based antisense oligonucleotides correct alternative splicing of the SMN2 gene and restore SMN protein expression in type 1 SMA fibroblasts. Sci. Rep. 2017, 7, 3672. [Google Scholar] [CrossRef] [PubMed]
- Aartsma-Rus, A.; De Winter, C.L.; Janson, A.A.; Kaman, W.E.; Van Ommen, G.J.; Den Dunnen, J.T.; Van Deutekom, J.C. Functional analysis of 114 exon-internal AONs for targeted DMD exon skipping: Indication for steric hindrance of SR protein binding sites. Oligonucleotides 2005, 15, 284–297. [Google Scholar] [CrossRef] [PubMed]
- Chow, G.; Morcos, P.A.; Moulton, H.M. Aggregation and Disaggregation of Morpholino Oligomers in Solution. Methods Mol. Biol. 2017, 1565, 31–38. [Google Scholar] [CrossRef] [PubMed]
- Reuter, J.S.; Mathews, D.H. RNAstructure: Software for RNA secondary structure prediction and analysis. BMC Bioinform. 2010, 11, 129. [Google Scholar] [CrossRef]
- Merlini, L.; Sabatelli, P. Improving clinical trial design for Duchenne muscular dystrophy. BMC Neurol. 2015, 15, 153. [Google Scholar] [CrossRef] [PubMed]
- Mamchaoui, K.; Trollet, C.; Bigot, A.; Negroni, E.; Chaouch, S.; Wolff, A.; Kandalla, P.K.; Marie, S.; Di Santo, J.; St Guily, J.L.; et al. Immortalized pathological human myoblasts: Towards a universal tool for the study of neuromuscular disorders. Skelet Muscle 2011, 1, 34. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, Q.; Yokota, T. Immortalized Muscle Cell Model to Test the Exon Skipping Efficacy for Duchenne Muscular Dystrophy. J. Pers. Med. 2017, 7, 13. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.J.A.; Saito, T.; Duddy, W.; Takeda, S.; Yokota, T. Direct Reprogramming of Human DMD Fibroblasts into Myotubes for In Vitro Evaluation of Antisense-Mediated Exon Skipping and Exons 45–55 Skipping Accompanied by Rescue of Dystrophin Expression. Methods Mol. Biol. 2018, 1828, 141–150. [Google Scholar] [CrossRef] [PubMed]
- Spaltro, G.; Vigorelli, V.; Casalnuovo, F.; Spinelli, P.; Castiglioni, E.; Rovina, D.; Paganini, S.; Di Segni, M.; Nigro, P.; Gervasini, C.; et al. Derivation of the Duchenne muscular dystrophy patient-derived induced pluripotent stem cell line lacking DMD exons 49 and 50 (CCMi001DMD-A-3, 49, 50). Stem Cell Res. 2017, 25, 128–131. [Google Scholar] [CrossRef] [PubMed]
- Eisen, B.; Ben Jehuda, R.; Cuttitta, A.J.; Mekies, L.N.; Reiter, I.; Ramchandren, S.; Arad, M.; Michele, D.E.; Binah, O. Generation of Duchenne muscular dystrophy patient-specific induced pluripotent stem cell line lacking exons 45–50 of the dystrophin gene (IITi001-A). Stem Cell Res. 2018, 29, 111–114. [Google Scholar] [CrossRef] [PubMed]
- Gowran, A.; Spaltro, G.; Casalnuovo, F.; Vigorelli, V.; Spinelli, P.; Castiglioni, E.; Rovina, D.; Paganini, S.; Di Segni, M.; Gervasini, C.; et al. Generation of induced pluripotent stem cells from a Becker muscular dystrophy patient carrying a deletion of exons 45–55 of the dystrophin gene (CCMi002BMD-A-9 45-55). Stem Cell Res. 2018, 28, 21–24. [Google Scholar] [CrossRef]
- Gee, P.; Xu, H.; Hotta, A. Cellular Reprogramming, Genome Editing, and Alternative CRISPR Cas9 Technologies for Precise Gene Therapy of Duchenne Muscular Dystrophy. Stem Cells Int. 2017, 2017, 8765154. [Google Scholar] [CrossRef]
- Shimo, T.; Hosoki, K.; Nakatsuji, Y.; Yokota, T.; Obika, S. A novel human muscle cell model of Duchenne muscular dystrophy created by CRISPR/Cas9 and evaluation of antisense-mediated exon skipping. J. Hum. Genet. 2018. [Google Scholar] [CrossRef]
- Shimo, T.; Tachibana, K.; Obika, S. Construction of a tri-chromatic reporter cell line for the rapid and simple screening of splice-switching oligonucleotides targeting DMD exon 51 using high content screening. PLoS ONE 2018, 13, e0197373. [Google Scholar] [CrossRef]
- Thorley, M.; Duguez, S.; Mazza, E.M.; Valsoni, S.; Bigot, A.; Mamchaoui, K.; Harmon, B.; Voit, T.; Mouly, V.; Duddy, W. Skeletal muscle characteristics are preserved in hTERT/cdk4 human myogenic cell lines. Skelet Muscle 2016, 6, 43. [Google Scholar] [CrossRef]
- Yuhuan, X.; Yingjun, X.; Yanting, X.; Yuchang, C.; Bing, S.; Shaoying, L.; Haoxian, L.; Yexing, X.; Shuming, O.; Zeyu, X.; et al. Generation of GZKHQi001-A and GZWWTi001-A, two induced pluripotent stem cell lines derived from peripheral blood mononuclear cells of Duchenne muscular dystrophy patients. Stem Cell Res. 2018, 28, 25–28. [Google Scholar] [CrossRef] [PubMed]
- Bremmer-Bout, M.; Aartsma-Rus, A.; de Meijer, E.J.; Kaman, W.E.; Janson, A.A.; Vossen, R.H.; van Ommen, G.J.; den Dunnen, J.T.; van Deutekom, J.C. Targeted exon skipping in transgenic hDMD mice: A model for direct preclinical screening of human-specific antisense oligonucleotides. Mol. Ther. 2004, 10, 232–240. [Google Scholar] [CrossRef] [PubMed]
- Veltrop, M.; van Vliet, L.; Hulsker, M.; Claassens, J.; Brouwers, C.; Breukel, C.; van der Kaa, J.; Linssen, M.M.; den Dunnen, J.T.; Verbeek, S.; et al. A dystrophic Duchenne mouse model for testing human antisense oligonucleotides. PLoS ONE 2018, 13, e0193289. [Google Scholar] [CrossRef] [PubMed]
- Tuffery-Giraud, S.; Miro, J.; Koenig, M.; Claustres, M. Normal and altered pre-mRNA processing in the DMD gene. Hum. Genet. 2017, 136, 1155–1172. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, H.; Aoki, Y.; Kameyama, T.; Saito, T.; Masuda, S.; Tanihata, J.; Nagata, T.; Mayeda, A.; Takeda, S.; Tsukahara, T. Endogenous Multiple Exon Skipping and Back-Splicing at the DMD Mutation Hotspot. Int. J. Mol. Sci. 2016, 17, 1722. [Google Scholar] [CrossRef] [PubMed]
- Surono, A.; Takeshima, Y.; Wibawa, T.; Pramono, Z.A.; Matsuo, M. Six novel transcripts that remove a huge intron ranging from 250 to 800 kb are produced by alternative splicing of the 5’ region of the dystrophin gene in human skeletal muscle. Biochem. Biophys. Res. Commun. 1997, 239, 895–899. [Google Scholar] [CrossRef] [PubMed]
- Surono, A.; Takeshima, Y.; Wibawa, T.; Ikezawa, M.; Nonaka, I.; Matsuo, M. Circular dystrophin RNAs consisting of exons that were skipped by alternative splicing. Hum. Mol. Genet. 1999, 8, 493–500. [Google Scholar] [CrossRef] [PubMed]
- Reiss, J.; Rininsland, F. An explanation for the constitutive exon 9 cassette splicing of the DMD gene. Hum. Mol. Genet. 1994, 3, 295–298. [Google Scholar] [CrossRef] [PubMed]
- Gazzoli, I.; Pulyakhina, I.; Verwey, N.E.; Ariyurek, Y.; Laros, J.F.; t Hoen, P.A.; Aartsma-Rus, A. Non-sequential and multi-step splicing of the dystrophin transcript. RNA Biol. 2016, 13, 290–305. [Google Scholar] [CrossRef] [PubMed]
- Aslesh, T.; Maruyama, R.; Yokota, T. Skipping Multiple Exons to Treat DMD-Promises and Challenges. Biomedicines 2018, 6, 1. [Google Scholar] [CrossRef]
- Aartsma-Rus, A.; Straub, V.; Hemmings, R.; Haas, M.; Schlosser-Weber, G.; Stoyanova-Beninska, V.; Mercuri, E.; Muntoni, F.; Sepodes, B.; Vroom, E.; et al. Development of Exon Skipping Therapies for Duchenne Muscular Dystrophy: A Critical Review and a Perspective on the Outstanding Issues. Nucleic Acid Ther. 2017, 27, 251–259. [Google Scholar] [CrossRef] [PubMed]
- Zingariello, C.D.; Kang, P.B. Dollars and antisense for Duchenne muscular dystrophy: Eteplirsen and dystrophin. Neurology 2018. [Google Scholar] [CrossRef] [PubMed]
- Magri, F.; Govoni, A.; D’Angelo, M.G.; Del Bo, R.; Ghezzi, S.; Sandra, G.; Turconi, A.C.; Sciacco, M.; Ciscato, P.; Bordoni, A.; et al. Genotype and phenotype characterization in a large dystrophinopathic cohort with extended follow-up. J. Neurol. 2011, 258, 1610–1623. [Google Scholar] [CrossRef] [PubMed]
- Yokota, T.; Duddy, W.; Echigoya, Y.; Kolski, H. Exon skipping for nonsense mutations in Duchenne muscular dystrophy: Too many mutations, too few patients? Expert Opin. Biol. Ther. 2012, 12, 1141–1152. [Google Scholar] [CrossRef] [PubMed]
- Yokota, T.; Pistilli, E.; Duddy, W.; Nagaraju, K. Potential of oligonucleotide-mediated exon-skipping therapy for Duchenne muscular dystrophy. Expert Opin. Biol. Ther. 2007, 7, 831–842. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Strategy | Study Model | Mutation | Target Molecule | Carrier | Target Regions | Administration | Comments | Ref. |
|---|---|---|---|---|---|---|---|---|
| Skipping exons 45–55 | Primary DMD myotubes | DMD ex46–50 or ex48–50 del. | pre-mRNA | 2 or 12 2′-OMePSs | Ex45–55 | PEI transfection | First published attempt at ex45–55 skipping in vitro using DMD patient cells | [56] |
| mdx52 mice | Dmd ex52 del. | pre-mRNA | 10 vivo-PMOs | Ex45–51, ex53–55 | i.m. (TA) or i.v. | First demonstration of successful in vivo ex45–55 skipping treatment in mice | [42] | |
| mdx52 mice | Dmd ex52 del. | pre-mRNA | 10 vivo-PMOs | Ex45–51, Ex53–55 | i.m. (TA) or i.v. | Showed the long-term systemic efficacy and safety of ex45–55 skipping treatment in mice | [57] | |
| Converted DMD myotubes | DMD ex46–50 del. | pre-mRNA | 6 PMOs | Ex45, Ex51–55 | Endo-porter transfection | Demonstrated the feasibility of ex45–55 skipping in muscle cells transdifferentiated from patient fibroblasts and showed a possibility of tailored cocktail therapy | [58] | |
| Converted DMD myotubes | DMD ex45–50 del. | pre-mRNA | 5 PMOs | Ex51–55 | Endo-porter transfection | |||
| Deleting exons 45–55 | Immortalized DMD myotubes | DMD ex48–50 del. | DNA | CRISPR/Cas9 | Introns44, 55 | Plasmid electroporation: spCas9, 2 gRNAs | First study to successfully delete ex45–55 in vitro; NSG mice transplanted with treated myoblasts showed dystrophin-positive fibers | [59] |
| DMD hiPSCs, hiPSC-derived myotubes, cardiomyocytes | DMD ex46–51 or 46–47 del., or ex50 dup. | DNA | CRISPR/Cas9 | Introns 44, 55 | Plasmid nucleofection: spCas9, 2 gRNA | Restored dystrophin expression/functionality in patient hiPSCs and derivative cell types | [61] | |
| DMD hiPSC-derived skeletal muscle cells | DMD ex46–51 del. | DNA | CRISPR/Cas9 | Introns 44, 55 | Engraftment into NSG-mdx mice TAs | Muscles engrafted with treated hiPSC muscle cells showed proper dystrophin and beta-dystroglycan localization | ||
| hDMD del. 45 mice | DMD ex45 del. | DNA | CRISPR/Cas9 | Introns 44, 55 | Electroporation into FDB muscle | First to show dystrophin restoration in vivo in humanized dystrophic mice following ex45–55 deletion, without cell transplantation | [60] |
| Strategy | Study Model | Mutation | Target Molecule | Carrier | Target Regions | Administration | Comments | Ref. |
|---|---|---|---|---|---|---|---|---|
| Skipping exons 6 & 8 | GRMD dog myotubes | An ASS point mutation in dystrophin intron 6 that results in ex7 removal from mRNA | pre-mRNA | 2 2′OMePSs/PMOs/PPMOs | Ex6, intron 7 | lipofectamine (2′-OMePS) or no agent(PMO/PPMO) | Showed the potential of using a canine DMD model for testing multiple exon skipping therapies | [80] |
| CXMDJ dogs | An ASS mutation in dystrophin intron 6 | pre-mRNA | 3 PMOs | Ex6, 8 | i.m. (TA, ECU) or i.v. | First published in vivo exon skipping study using a canine DMD model | [78] | |
| Converted myotubes of DMD patient or CXMDJ | DMD ex7 del. (patient), an ASS mutation in dystrophin intron 6 (dog) | pre-mRNA | 3 or 4 PMOs | Ex6, 8 | endo-porter transfection | Demonstrated the feasibility of adapting the canine ex6-8 skipping strategy into patients | [81] | |
| GRMD dogs | An ASS mutation in dystrophin intron 6 | pre-mRNA | rAAV6-U7 snRNA construct | Ex6, 8 | transendocardial injection | Reported the long-term efficacy of ex6-8 skipping for rescuing dystrophin and improving cardiac function in vivo | [82] | |
| GRMD dogs | An ASS mutation in dystrophin intron 6 | pre-mRNA | rAAV6-U7 snRNA construct | Ex6, 8 | i.c. or transendocardial injection | Improved transendocardial delivery of ex6-8 skipping snRNAs into the heart using MRI-based injection guidance | [83] | |
| CXMDJ dogs | An ASS mutation in dystrophin intron 6 | pre-mRNA | 4 vivo-PMOs | Ex6, 8 | i.m. (forelimb muscles) | First study to show in vivo dystrophin rescue in a canine DMD model using modified/chemically-conjugated PMOs | [84] | |
| GRMD dogs | An ASS mutation in dystrophin intron 6 | pre-mRNA | rAAV1-U7 snRNA construct | Ex6, 8 | i.m. or transvenous perfusion (forelimb muscles) | Demonstrated improvements in muscular strength following ex6-8 skipping | [85] | |
| GRMD dogs | An ASS mutation in dystrophin intron 6 | pre-mRNA | rAAV8-U7 snRNA construct | Ex6, 8 | transvenous forelimb perfusion | Further supported use of locoregional delivery for rAAV-packaged ex6-8 skipping snRNA therapy in patients | [86] | |
| CXMDJ dogs | An ASS mutation in dystrophin intron 6 | pre-mRNA | 3 PPMOs | Ex6, 8 | i.m. (TA), i.c. or i.v. | Demonstrated the therapeutic utility of PPMO-based exon skipping for ameliorating cardiac conduction defects in vivo | [79] | |
| CXMDJ dogs | An ASS mutation in dystrophin intron 6 | pre-mRNA | 4 PMOs | Ex6, 8 | i.v. | Reported the efficacy of ex6&8 skipping therapy in dystrophic dog neonates, highlighting the need for earlier treatment for a dystrophic pathology | [87] | |
| Deleting exons 3–9 | DMD hiPSCs, hiPSC-derived cardiomyocytes | DMD exons 8–9 del. (induced/native) | DNA | CRISPR/Cas9 | Introns 2, 7 | plasmid nucleofection: spCas9, 2 gRNA | First and only ex3-9 deletion study to date; more functional dystrophin from ex3-9 deleted mRNA than ex6-7 or 7-11-deleted dystrophin; improved cardiomyocyte calcium-handling functions upon treatment | [75] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Echigoya, Y.; Lim, K.R.Q.; Nakamura, A.; Yokota, T. Multiple Exon Skipping in the Duchenne Muscular Dystrophy Hot Spots: Prospects and Challenges. J. Pers. Med. 2018, 8, 41. https://doi.org/10.3390/jpm8040041
Echigoya Y, Lim KRQ, Nakamura A, Yokota T. Multiple Exon Skipping in the Duchenne Muscular Dystrophy Hot Spots: Prospects and Challenges. Journal of Personalized Medicine. 2018; 8(4):41. https://doi.org/10.3390/jpm8040041
Chicago/Turabian StyleEchigoya, Yusuke, Kenji Rowel Q. Lim, Akinori Nakamura, and Toshifumi Yokota. 2018. "Multiple Exon Skipping in the Duchenne Muscular Dystrophy Hot Spots: Prospects and Challenges" Journal of Personalized Medicine 8, no. 4: 41. https://doi.org/10.3390/jpm8040041
APA StyleEchigoya, Y., Lim, K. R. Q., Nakamura, A., & Yokota, T. (2018). Multiple Exon Skipping in the Duchenne Muscular Dystrophy Hot Spots: Prospects and Challenges. Journal of Personalized Medicine, 8(4), 41. https://doi.org/10.3390/jpm8040041