
An eMERGE Clinical Center at Partners Personalized Medicine

 ,
,
Abstract
1. Introduction
2. Infrastructure for a Clinical eMERGE Site at Partners HealthCare
2.1. Partners HealthCare Personalized Medicine
2.2. EMR Infrastructure: The Research Patient Data Registry
2.3. Partners HealthCare Biobank
2.4. Tools for EMR-Based Phenotypic Definition and Validation

{kind=link}
| Steps | Task | Team Member |
|---|---|---|
| 1 | Randomly select 400 subjects with ICD-9 code | Programmer |
| 2 | Review charts, confirm diagnosis for Training Set | Domain expert |
| 3 | Create custom list of concepts relevant to disease | Domain expert |
| 4 | Extract EMR data to create codified variables | Programmer |
| 5 | Create custom list of NLP variables | Domain expert |
| 6 | Map variables UMLS concept unique identifier (CUI) | Informatician |
| 7 | Extract CUIs from narrative text in EMR using NLP | Informatician |
| 8 | Run LASSO regression with codified + NLP variables predicting disease status in Training Set | Statistician |
| 9 | Set specificity at 97%, select predicted probability among Training Set to achieve >90% PPV | Statistician |
| 10 | Apply algorithm to remaining Biobank subjects (excludingTraining Set) | Statistician |
| 11 | Randomly select 100 subjects for Validation Set | Programmer |
| 12 | Perform chart review in Test Set, define PPV | Domain expert |
3. Scientific Aims of the eMERGE Clinical Center at Partners Personalized Medicine
3.1. Discovery: Detecting Association of Common and Rare Variants with EMR-Based Phenotypes
3.2. Evaluating Penetrance and Pleiotropy of Rare Variants
3.2.1. Cardiovascular Genes
3.2.2. Neuropsychiatric Genes
3.2.3. Selection of Immune Disease Genes
3.3. Implementation Research: Impact of Return of Genetic Results on Health Outcomes
3.3.1. Prior Experience with Return of Genetic Results at the Partners Site
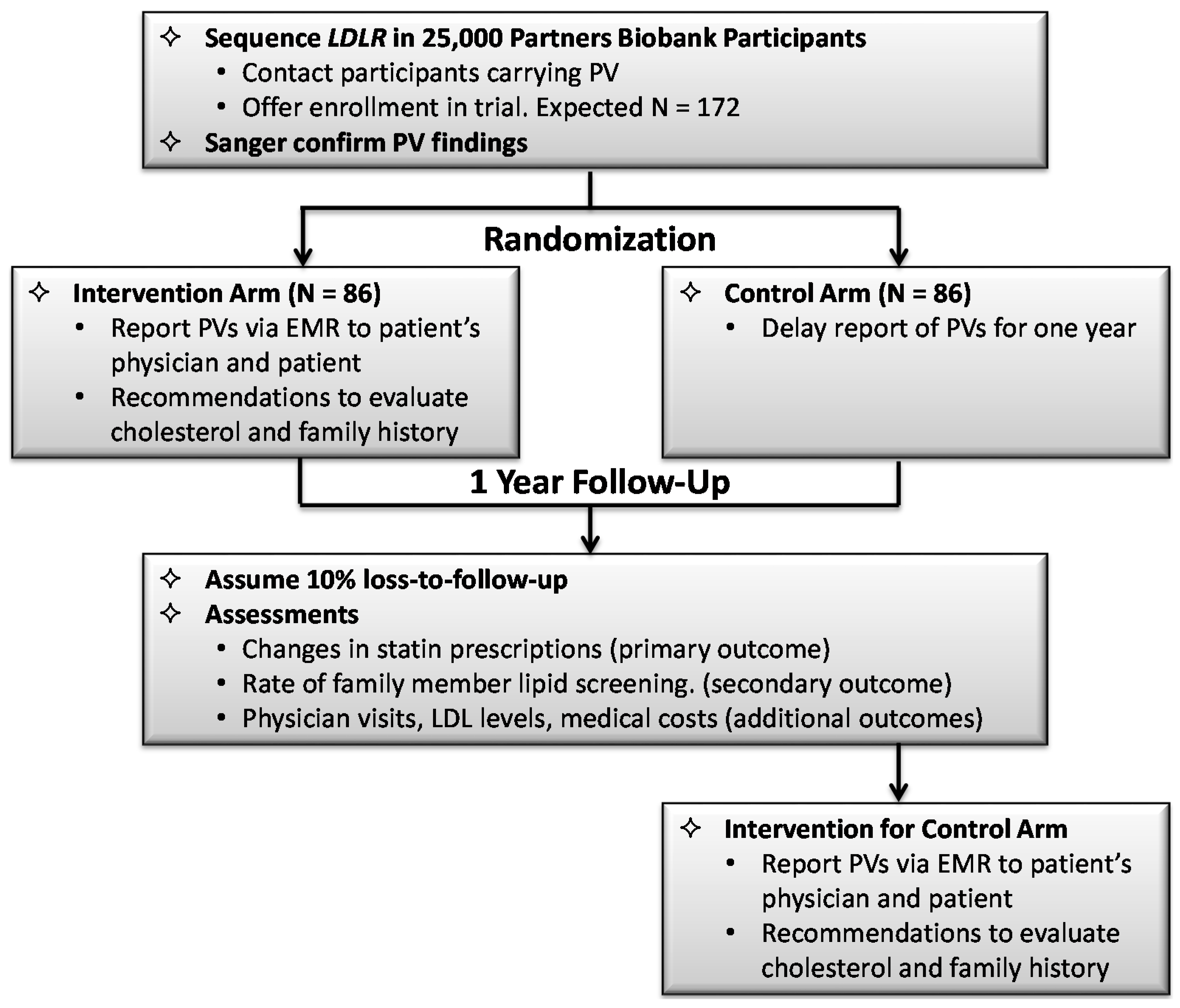
3.3.2. Design of the Randomized Implementation Trial
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Kohane, I.S. Using electronic health records to drive discovery in disease genomics. Nat. Rev. Genet. 2011, 12, 417–428. [Google Scholar] [CrossRef] [PubMed]
- Gottesman, O.; Kuivaniemi, H.; Tromp, G.; Faucett, W.A.; Li, R.; Manolio, T.A.; Sanderson, S.C.; Kannry, J.; Zinberg, R.; Basford, M.A.; et al. The electronic medical records and genomics (emerge) network: Past, present, and future. Genet. Med. 2013, 15, 761–771. [Google Scholar] [CrossRef] [PubMed]
- Crawford, D.C.; Crosslin, D.R.; Tromp, G.; Kullo, I.J.; Kuivaniemi, H.; Hayes, M.G.; Denny, J.C.; Bush, W.S.; Haines, J.L.; Roden, D.M.; et al. eMERGEing progress in genomics—The first seven years. Front. Genet. 2014. [Google Scholar] [CrossRef] [PubMed]
- Aronson, S.; Mahanta, L.; Ros, L.L.; Clark, E.; Babb, L.; Oates, M.; Rehm, H.; Lebo, M. Information technology support for clinical genetic testing within an academic medical center. Partners HealthCare Personalized Medicine, Cambridge, MA, USA. J. Pers. Med. 2016. [Google Scholar] [CrossRef]
- Karlson, E.W.; Boutin, N.T.; Hoffnagle, A.G.; Allen, N.L. Building the Partners HealthCare Biobank at Partners Personalized Medicine: Informed Consent, Return of Research Results, Recruitment Lessons and Operational Considerations. J. Pers. Med. 2016. [Google Scholar] [CrossRef]
- Kohane, I.S.; Churchill, S.E.; Murphy, S.N. A translational engine at the national scale: Informatics for integrating biology and the bedside. J. Am. Med. Inform. Assoc. 2012, 19, 181–185. [Google Scholar] [CrossRef] [PubMed]
- Liao, K.P.; Cai, T.; Gainer, V.; Goryachev, S.; Zeng-treitler, Q.; Raychaudhuri, S.; Szolovits, P.; Churchill, S.; Murphy, S.; Kohane, I.; et al. Electronic medical records for discovery research in rheumatoid arthritis. Arthritis Care Res. 2010, 62, 1120–1127. [Google Scholar] [CrossRef] [PubMed]
- Castro, V.M.; Minnier, J.; Murphy, S.N.; Kohane, I.; Churchill, S.E.; Gainer, V.; Cai, T.; Hoffnagle, A.G.; Dai, Y.; Block, S.; et al. Validation of electronic health record phenotyping of bipolar disorder cases and controls. Am. J. Psychiatry 2015, 172, 363–372. [Google Scholar] [CrossRef] [PubMed]
- Green, R.C.; Berg, J.S.; Grody, W.W.; Kalia, S.S.; Korf, B.R.; Martin, C.L.; McGuire, A.L.; Nussbaum, R.L.; O’Daniel, J.M.; Ormond, K.E.; et al. Acmg recommendations for reporting of incidental findings in clinical exome and genome sequencing. Genet. Med. 2013, 15, 565–574. [Google Scholar] [CrossRef] [PubMed]
- Solovieff, N.; Cotsapas, C.; Lee, P.H.; Purcell, S.M.; Smoller, J.W. Pleiotropy in complex traits: Challenges and strategies. Nat. Rev. Genet. 2013, 14, 483–495. [Google Scholar] [CrossRef] [PubMed]
- Denny, J.C.; Bastarache, L.; Ritchie, M.D.; Carroll, R.J.; Zink, R.; Mosley, J.D.; Field, J.R.; Pulley, J.M.; Ramirez, A.H.; Bowton, E.; et al. Systematic comparison of phenome-wide association study of electronic medical record data and genome-wide association study data. Nat. Biotechnol. 2013, 31, 1102–1110. [Google Scholar] [CrossRef] [PubMed]
- Cross Disorder Group of the Psychiatric GWAS Consortium. Identification of risk loci with shared effects on five major psychiatric disorders: A genome-wide analysis. Lancet 2013, 381, 1371–1379. [Google Scholar]
- Talkowski, M.E.; Rosenfeld, J.A.; Blumenthal, I.; Pillalamarri, V.; Chiang, C.; Heilbut, A.; Ernst, C.; Hanscom, C.; Rossin, E.; Lindgren, A.M.; et al. Sequencing chromosomal abnormalities reveals neurodevelopmental loci that confer risk across diagnostic boundaries. Cell 2012, 149, 525–537. [Google Scholar] [CrossRef] [PubMed]
- Trowsdale, J.; Knight, J.C. Major histocompatibility complex genomics and human disease. Annu. Rev. Genom. Hum. Genet. 2013, 14, 301–323. [Google Scholar] [CrossRef] [PubMed]
- Beaudoin, M.; Goyette, P.; Boucher, G.; Lo, K.S.; Rivas, M.A.; Stevens, C.; Alikashani, A.; Ladouceur, M.; Ellinghaus, D.; Torkvist, L.; et al. Deep resequencing of gwas loci identifies rare variants in card9, il23r and rnf186 that are associated with ulcerative colitis. PLoS Genet. 2013, 9, e1003723. [Google Scholar] [CrossRef] [PubMed]
- Jostins, L.; Ripke, S.; Weersma, R.K.; Duerr, R.H.; McGovern, D.P.; Hui, K.Y.; Lee, J.C.; Schumm, L.P.; Sharma, Y.; Anderson, C.A.; et al. Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature 2012, 491, 119–124. [Google Scholar] [CrossRef] [PubMed]
- Kilic, S.S.; Hacimustafaoglu, M.; Boisson-Dupuis, S.; Kreins, A.Y.; Grant, A.V.; Abel, L.; Casanova, J.L. A patient with tyrosine kinase 2 deficiency without hyper-ige syndrome. J. Pediatr. 2012, 160, 1055–1057. [Google Scholar] [CrossRef] [PubMed]
- De Jager, P.L.; Jia, X.; Wang, J.; de Bakker, P.I.; Ottoboni, L.; Aggarwal, N.T.; Piccio, L.; Raychaudhuri, S.; Tran, D.; Aubin, C.; et al. Meta-analysis of genome scans and replication identify cd6, irf8 and tnfrsf1a as new multiple sclerosis susceptibility loci. Nat. Genet. 2009, 41, 776–782. [Google Scholar] [CrossRef] [PubMed]
- Kuehn, H.S.; Ouyang, W.; Lo, B.; Deenick, E.K.; Niemela, J.E.; Avery, D.T.; Schickel, J.N.; Tran, D.Q.; Stoddard, J.; Zhang, Y.; et al. Immune dysregulation in human subjects with heterozygous germline mutations in ctla4. Science 2014, 345, 1623–1627. [Google Scholar] [CrossRef] [PubMed]
- Green, R.C.; Roberts, J.S.; Cupples, L.A.; Relkin, N.R.; Whitehouse, P.J.; Brown, T.; Eckert, S.L.; Butson, M.; Sadovnick, A.D.; Quaid, K.A.; et al. Disclosure of apoe genotype for risk of alzheimer's disease. N. Engl. J. Med. 2009, 361, 245–254. [Google Scholar] [CrossRef] [PubMed]
- Green, R.C.; Christensen, K.D.; Cupples, L.A.; Relkin, N.R.; Whitehouse, P.J.; Royal, C.D.; Obisesan, T.O.; Cook-Deegan, R.; Linnenbringer, E.; Butson, M.B.; et al. A randomized noninferiority trial of condensed protocols for genetic risk disclosure of alzheimer's disease. Alzheimers Dement. 2015, 11, 1222–1230. [Google Scholar] [CrossRef] [PubMed]
- Christensen, K.D.; Roberts, J.S.; Whitehouse, P.J.; Royal, C.D.M.; Obisesan, T.O.; Cupples, L.A.; Vernarelli, J.A.; Bhatt, D.L.; Linnenbringer, E.; Butson, M.B.; et al. A randomized trial of disclosing incidental pleiotropic genetic risk information. Ann. Intern. Med. in press.
- Carere, D.A.; Couper, M.P.; Crawford, S.D.; Kalia, S.S.; Duggan, J.R.; Moreno, T.A.; Mountain, J.L.; Roberts, J.S.; Green, R.C.; Group, P.G.S. Design, methods, and participant characteristics of the impact of personal genomics (PGen) study, a prospective cohort study of direct-to-consumer personal genomic testing customers. Genome Med. 2014. [Google Scholar] [CrossRef] [PubMed]
- Carere, D.A.; VanderWeele, T.; Moreno, T.A.; Mountain, J.L.; Roberts, J.S.; Kraft, P.; Green, R.C.; Group, P.G.S. The impact of direct-to-consumer personal genomic testing on perceived risk of breast, prostate, colorectal, and lung cancer: Findings from the pgen study. BMC Med. Genom. 2015. [Google Scholar] [CrossRef] [PubMed]
- Meisel, S.F.; Carere, D.A.; Wardle, J.; Kalia, S.S.; Moreno, T.A.; Mountain, J.L.; Roberts, J.S.; Green, R.C.; Group, P.G.S. Explaining, not just predicting, drives interest in personal genomics. Genome Med. 2015. [Google Scholar] [CrossRef] [PubMed]
- Ostergren, J.E.; Gornick, M.C.; Carere, D.A.; Kalia, S.S.; Uhlmann, W.R.; Ruffin, M.T.; Mountain, J.L.; Green, R.C.; Roberts, J.S.; Group, P.G.S. How well do customers of direct-to-consumer personal genomic testing services comprehend genetic test results? Findings from the impact of personal genomics study. Public Health Genom. 2015, 18, 216–224. [Google Scholar] [CrossRef] [PubMed]
- Vassy, J.L.; Lautenbach, D.M.; McLaughlin, H.M.; Kong, S.W.; Christensen, K.D.; Krier, J.; Kohane, I.S.; Feuerman, L.Z.; Blumenthal-Barby, J.; Roberts, J.S.; et al. The medseq project: A randomized trial of integrating whole genome sequencing into clinical medicine. Trials 2014, 15, 85–97. [Google Scholar] [CrossRef] [PubMed]
- McLaughlin, H.M.; Ceyhan-Birsoy, O.; Christensen, K.D.; Kohane, I.S.; Krier, J.; Lane, W.J.; Lautenbach, D.; Lebo, M.S.; Machini, K.; MacRae, C.A.; et al. A systematic approach to the reporting of medically relevant findings from whole genome sequencing. BMC Med. Genet. 2014. [Google Scholar] [CrossRef] [PubMed]
- Vassy, J.L.; McLaughlin, H.M.; MacRae, C.A.; Seidman, C.E.; Lautenbach, D.; Krier, J.B.; Lane, W.J.; Kohane, I.S.; Murray, M.F.; McGuire, A.L.; et al. A one-page summary report of genome sequencing for the healthy adult. Public Health Genom. 2015, 18, 123–129. [Google Scholar] [CrossRef] [PubMed]
- Biesecker, L.G.; Green, R.C. Diagnostic clinical genome and exome sequencing. N. Engl. J. Med. 2014, 370, 2418–2425. [Google Scholar] [CrossRef] [PubMed]
- Green, R.C.; Lupski, J.R.; Biesecker, L.G. Reporting genomic sequencing results to ordering clinicians: Incidental, but not exceptional. JAMA 2013, 310, 365–366. [Google Scholar] [CrossRef] [PubMed]
- Wilfond, B.S.; Fernandez, C.V.; Green, R.C. Disclosing secondary findings from pediatric sequencing to families: Considering the “benefit to families”. J. Law Med. Ethics 2015, 43, 552–558. [Google Scholar]
- Wolf, S.M.; Branum, R.; Koenig, B.A.; Petersen, G.M.; Berry, S.A.; Beskow, L.M.; Daly, M.B.; Fernandez, C.V.; Green, R.C.; LeRoy, B.S.; et al. Returning a research participant’s genomic results to relatives: Analysis and recommendations. J. Law Med. Ethics 2015, 43, 440–463. [Google Scholar]
- Wolf, S.M.; Crock, B.N.; van Ness, B.; Lawrenz, F.; Kahn, J.P.; Beskow, L.M.; Cho, M.K.; Christman, M.F.; Green, R.C.; Hall, R.; et al. Managing incidental findings and research results in genomic research involving biobanks and archived data sets. Genet. Med. 2012, 14, 361–384. [Google Scholar] [CrossRef] [PubMed]
- Aronson, S.J.; Clark, E.H.; Babb, L.J.; Baxter, S.; Farwell, L.M.; Funke, B.H.; Hernandez, A.L.; Joshi, V.A.; Lyon, E.; Parthum, A.R.; et al. The geneinsight suite: A platform to support laboratory and provider use of DNA-based genetic testing. Hum. Mutat. 2011, 32, 532–536. [Google Scholar] [CrossRef] [PubMed]
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Smoller, J.W.; Karlson, E.W.; Green, R.C.; Kathiresan, S.; MacArthur, D.G.; Talkowski, M.E.; Murphy, S.N.; Weiss, S.T. An eMERGE Clinical Center at Partners Personalized Medicine. J. Pers. Med. 2016, 6, 5. https://doi.org/10.3390/jpm6010005
Smoller JW, Karlson EW, Green RC, Kathiresan S, MacArthur DG, Talkowski ME, Murphy SN, Weiss ST. An eMERGE Clinical Center at Partners Personalized Medicine. Journal of Personalized Medicine. 2016; 6(1):5. https://doi.org/10.3390/jpm6010005
Chicago/Turabian StyleSmoller, Jordan W., Elizabeth W. Karlson, Robert C. Green, Sekar Kathiresan, Daniel G. MacArthur, Michael E. Talkowski, Shawn N. Murphy, and Scott T. Weiss. 2016. "An eMERGE Clinical Center at Partners Personalized Medicine" Journal of Personalized Medicine 6, no. 1: 5. https://doi.org/10.3390/jpm6010005
APA StyleSmoller, J. W., Karlson, E. W., Green, R. C., Kathiresan, S., MacArthur, D. G., Talkowski, M. E., Murphy, S. N., & Weiss, S. T. (2016). An eMERGE Clinical Center at Partners Personalized Medicine. Journal of Personalized Medicine, 6(1), 5. https://doi.org/10.3390/jpm6010005