Molecular Therapeutic Advances in Personalized Therapy of Melanoma and Non-Small Cell Lung Cancer


Abstract
:1. Introduction
2. Molecular Therapeutics and Non-Small Cell Lung Cancer
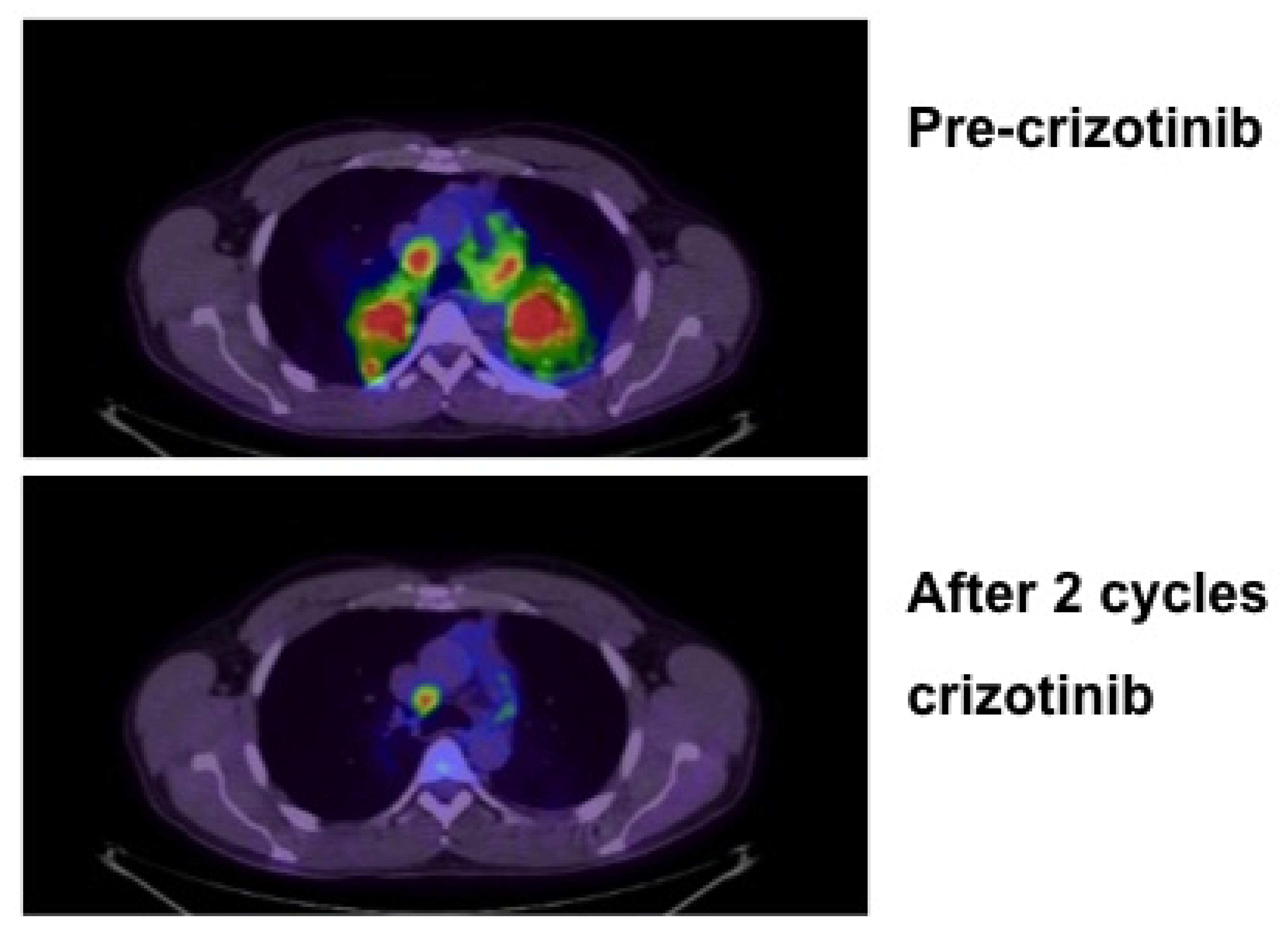

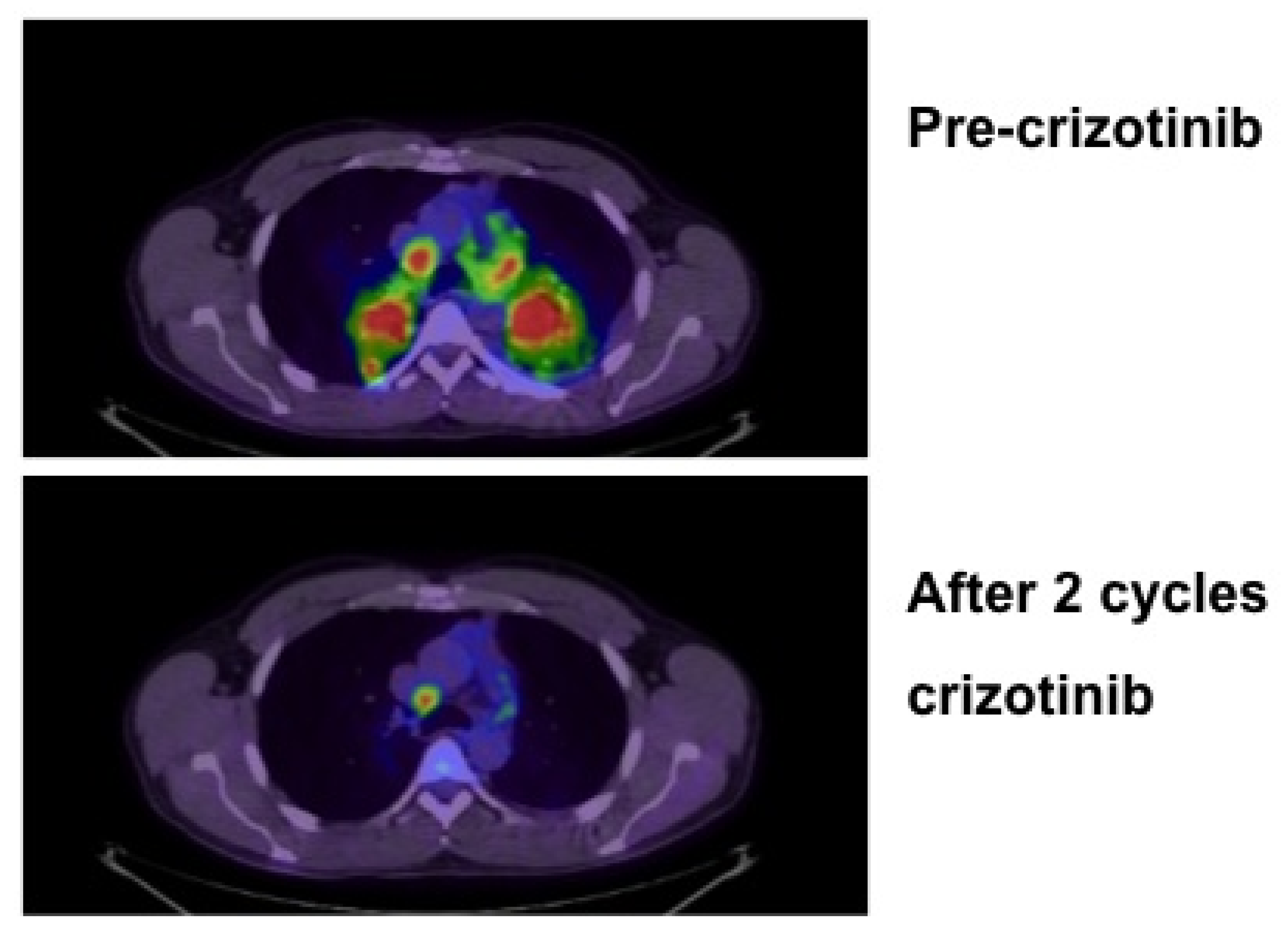{kind=link}
| Histologic Subtype | Molecular Sub Category (Frequency) |
|---|---|
| Adenocarcinoma (~50% of NSCLC) | |
| EGFR tyrosine kinase domain mutations (10–35%) | |
| KRAS mutations (15–25%) | |
| ALK Gene Rearrangements (3–5%) | |
| Her2 mutations | |
| BRAF mutations (3%) | |
| MET amplification (1%) | |
| Squamous Cell Carcinoma (~30% of NSCLC) | |
| FGFR1 Amplification (20%) | |
| DDR2 mutation (4%) | |
| PIK3CA mutations (1–3%) | |
| EGFRvIII mutations (<5%) |
3. Melanoma
| Gene | Frequency (%) | Gene | Frequency (%) |
|---|---|---|---|
| BRAF | 45 | GNAQ | 8 |
| CDKN2A | 29 | CTNNB1 | 6 |
| NRAS | 19 | NF2 | 5 |
| TP53 | 17 | PDGFRA | 4 |
| PTEN | 17 | PIK3CA | 2 |
| STK11 | 10 | HRAS | 2 |
| FGFR2 | 9 | KRAS | 2 |
| KIT | 8 | GNA11 | 2 |
| Melanoma | Implicated genes (approximate frequencies) |
|---|---|
| Arising from skin without chronic sun damage | Mutant BRAF (59%), mutant RAS (22%) KIT mutations or increased copy number (0%) |
| Arising from skin with chronic sun damage | Mutant BRAF (11%), mutant RAS (15%), mutant or increased copy number KIT (28%) |
| Arising from mucosal surfaces | Mutant BRAF (11%), mutant RAS (5%) mutant or increased copy number KIT (38%) |
| Arising from acral surfaces | Mutant BRAF (23%), mutant RAS (10%) mutant or increased copy number KIT (36%) |
| Uveal melanomas | Mutations in GNAQ or GNA11 (83%) |
4. Uveal Melanoma
5. Molecular Drug Resistance and the Emergence of Drug Resistant Tumor Clones
6. Acquired Resistance in NSCLC
7. Acquired Resistance in Melanoma
8. Conclusions
Conflict of Interest
References
- Ferlay, J.; Shin, H.; Bray, F.; Forman, D.; Mathers, C.; Parkin, D. Estimates of worldwide burden of cancer in 2008: GLOBOCAN 2008. Int. J. Cancer 2010, 127, 2893–2917. [Google Scholar] [CrossRef]
- Parkin, D.M.; Bray, F.; Ferlay, J.; Pisani, P. Global cancer statistics, 2002. CA Cancer J. Clin. 2005, 55, 74–108. [Google Scholar] [CrossRef]
- Devesa, S.; Bray, F.; Vascaino, A.; Parkin, D. International Lung Cancer trends by histologic type: male:femaile differences diminishing and adenocarcinoma rates rising. Int. J. Cancer 2005, 117, 294–299. [Google Scholar] [CrossRef]
- Fukouka, M.; Yano, S.; Giaccone, G.; Tamura, T.; Nakagawa, K.; Douillard, J. Multi-institutional randomized phase II trial of gefitinib for previously treated patients with advanced non-small-cell lung cancer. J. Clin. Oncol. 2003, 21, 2237–2246. [Google Scholar]
- Kris, M.G.; Natale, R.B.; Herbst, R.S.; Lynch, T.J., Jr.; Prager, D.; Belani, C.P.; Schiller, J.H.; Kelly, K.; Spiridonidis, H.; Sandler, A.; et al. Efficacy of gefitinib, an inhibitor of the epidermal growth factor receptor tyrosine kinase, in symptomatic patients with non-small cell lung cancer: a randomized trial. JAMA 2003, 290, 2149–2158. [Google Scholar]
- Thatcher, N.; Chang, A.; Parikh, P.; Rodrigues Pereira, J.; Ciuleanu, T.; von Pawel, J.; Thongprasert, S.; Tan, E.H.; Pemberton, K.; Archer, V.; et al. Gefitinib plus best supportive care in previously treated patients with refractory advanced non-small-cell lung cancer: results from a randomised, placebo-controlled, multicentre study (Iressa Survival Evaluation in Lung Cancer). Lancet 2005, 366, 1527–1537. [Google Scholar]
- Shepherd, F.A.; Rodrigues Pereira, J.; Ciuleanu, T.; Tan, E.H.; Hirsh, V.; Thongprasert, S.; Campos, D.; Maoleekoonpiroj, S.; Smylie, M.; Martins, R.; et al. Erlotinib in previously treated non-small-cell lung cancer. N. Engl. J. Med. 2005, 353, 123–132. [Google Scholar]
- Lynch, T.; Bell, D.; Sordella, R; Gurubhagavatula, S.; Okimoto, R.; Branningan, B. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N. Engl. J. Med. 2004, 350, 2129. [Google Scholar] [CrossRef]
- Guillermo Paez, J.; Janne, P.A.; Lee, J.C.; Tracy, S.; Greulich, H.; Gabriel, S.; Herman, P.; Kaye, F.J.; Lindeman, N.; Boggon, T.J.; et al. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science 2004, 304, 1497–1500. [Google Scholar]
- Pao, W.; Miller, V.; Zakowski, M.F.; Doherty, J.; Politi, K.A.; Sarkaria, I. EGF receptor gene mutations are common in lung cancers from "never smokers" and are associated with sensitivity of tumors to gefitinib and erlotinib. Proc. Natl. Acad. Sci. USA 2004, 101, 13306–13311. [Google Scholar]
- Rosell, R.; Moran, T.; Queralt, C.; Porta, R.; Cardenal, F.; Camps, C.; Majem, M.; Lopez-Vivanco, G.; Isla, D.; Provencio, M.; et al. Screening for epidermal growth factor receptor mutations in lung cancer. N. Engl. J. Med. 2009, 361, 958–967. [Google Scholar]
- Mok, T.S.; Wu, Y.L.; Thongprasert, S.; Yang, C.H.; Chu, D.T.; Saijo, N.; Sunpaweravong, P.; Han, B.; Margono, B.; Ichinose, Y.; et al. Gefitinib or carboplatin-paclitaxel in pulmonary adenocarcinoma. N. Engl. J. Med. 2009, 361, 947–957. [Google Scholar] [CrossRef]
- Lee, J.S.; Park, K.; Kim, S.W.; Lee, D.H.; Kim, H.T.; Han, J.-Y.; Yun, T.; Ahn, M.J.; Ahn, J.S.; Suh, C.; Lee, J.S.; Han, J.H.; Yu, S.Y.; Lee, J.W.; Jo, S. A randomized phase III study of gefitinib versus standard chemotherapy (gemcitabine plus cisplatin) as a first-line treatment for never-smokers with advanced or metastatic adenocarcinoma of the lung. J. Thorac. Oncol. 2009, 4 Suppl 1, Issue 9. S283 (abstr PRS.4). [Google Scholar]
- Mitsudomi, T.; Morita, S.; Yatabe, Y.; Negoro, S.; Okamoto, I.; Tsurutani, J.; Seto, T.; Satouchi, M.; Tada, H.; Hirashima, T.; et al. Gefitinib versus cisplatin plus docetaxel in patients with non-small-cell lung cancer harbouring mutations of the epidermal growth factor receptor (WJTOG3405): an open label, randomised phase 3 trial. Lancet Oncol. 2010, 11, 121–128. [Google Scholar] [CrossRef]
- Maemondo, M.; Inoue, A.; Kobayashi, K.; Sugawara, S.; Oizumi, S.; Isobe, H.; Gemma, A.; Harada, M.; Yoshizawa, H.; Kinoshita, I.; et al. Gefitinib or chemotherapy for non-small-cell lung cancer with mutated EGFR. N. Engl. J. Med. 2010, 362, 2380–2388. [Google Scholar] [CrossRef]
- Zhou, C. Erlotinib versus chemotherapy as first-line treatment for patients with advanced EGFR mutation-positive non-small-cell lung cancer (OPTIMAL, CTONG-0802): a multicentre, open-label, randomised, phase 3 study. Lancet Oncol. 2011, 12, 735–742. [Google Scholar] [CrossRef]
- Rosell, R.; Gervais, A.; Vergnenegre, B.; Massuti, E.; Felip, F.; Cardenal, R.; Garcia, G.; Pallares, C.; Sanchez, M.; Porta, R.; et al. Spanish Lung Cancer Group; Erlotinib versus chemotherapy (CT) in advanced non-small cell lung cancer (NSCLC) patients (p) with epidermal growth factor receptor (EGFR) mutations: Interim results of the European Erlotinib versus Chemotherapy (EURTAC) phase III randomized trial. J. Clin. Oncol. 2011, 29 (suppl.), abstr 7503. [Google Scholar]
- Keedy, V.L.; Temin, S.; Somerfield, M.R.; Beasley, M.B.; Johnson, D.H.; McShane, L.M.; Milton, D.T.; Strawn, J.R.; Wakelee, H.A.; Giaccone, G. American Society of Clinical Oncology provisional clinical opinion: epidermal growth factor receptor (EGFR) Mutation testing for patients with advanced non-small-cell lung cancer considering first-line EGFR tyrosine kinase inhibitor therapy. J. Clin. Oncol. 2011, 29, 2121–2127. [Google Scholar]
- Soda, M.; Choi, Y.L.; Enomoto, M.; Takada, S.; Yamashita, Y.; Ishikawa, S.; Fujiwara, S.; Watanabe, H.; Kurashina, K.; Hatanaka, H.; et al. Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature 2007, 448, 561–566. [Google Scholar]
- Rikova, K.; Guo, A.; Zeng, Q.; Possemato, A.; Yu, J.; Haack, H.; Nardone, J.; Lee, K.; Reeves, C.; Li, Y.; et al. Global survey of phosphotyrosinesignaling identifies oncogenickinases in lung cancer. Cell 2007, 131, 1190–1203. [Google Scholar] [CrossRef]
- Shaw, A.T.; Solomon, B. Targeting anaplastic lymphoma kinase in lung cancer. Clin. Cancer Res. 2011, 17, 2081–2086. [Google Scholar] [CrossRef]
- Kwak, E.L.; Bang, Y.J.; Camidge, D.R.; Shaw, A.T.; Solomon, B.; Maki, R.G.; Ou, S.H.; Dezube, B.J.; Janne, P.A.; Costa, D.B.; et al. Anaplastic lymphoma kinase inhibition in non-small-cell lung cancer. N. Engl. J. Med. 2010, 363, 1693–1703. [Google Scholar] [CrossRef]
- Camidge, D.R.; Kono, S.A.; Lu, X.; Okuyama, S.; Baron, A.E.; Oton, A.B.; Davies, A.M.; Varella-Garcia, M.; Franklin, W.; Doebele, R.C. Anaplastic lymphoma kinase gene rearrangements in non-small cell lung cancer are associated with prolonged progression-free survival on pemetrexed. J. Thorac. Oncol. 2011, 6, 774–780. [Google Scholar] [CrossRef]
- Ding, L.; Getz, G.; Wheeler, D.A.; Mardis, E.R.; McLellan, M.D.; Cibulskis, K.; Sougnez, C.; Greulich, H.; Muzny, D.M.; Morgan, M.B.; et al. Somatic mutations affect key pathways in lung adenocarcinoma. Nature 2008, 455, 1069–1075. [Google Scholar]
- Weir, B. Characterizing the cancer genome in lung adenocarcinoma. Nature 2008, 450, 893–898. [Google Scholar]
- Paik, P.; Arcila, M.; Fara, M. Clinical characteristics of patients with lung adenocarcinomasharboring BRAF mutations. J. Clin. Oncol. 2011, 29, 2046–2059. [Google Scholar] [CrossRef]
- Buttitta, F.; Barassi, F.; Fresu, G.; Felicioni, L.; Chella, A.; Paolizzi, D.; Lattanzio, G.; Salvatore, S.; Camplese, P.P.; Rosini, S.; et al. Mutational analysis of the HER2 gene in lung tumors from Caucasian patients: mutations are mainly present in adenocarcinomas with bronchioloalveolar features. Int. J. Cancer 2006, 119, 2586–2591. [Google Scholar]
- Shigematsu, H.; Takahashi, T.; Nomura, M.; Majmudar, K.; Suzuki, M.; Lee, H.; Wistuba, I.I.; Fong, K.M.; Toyooka, S.; Shimizu, N.; et al. Somatic mutations of the HER2 kinase domain in lung adenocarcinomas. Cancer Res. 2005, 65, 1642–1646. [Google Scholar]
- Stephens, P.; Hunter, C.; Bignell, G.; Edkins, S.; Davies, H.; Teague, J.; Stevens, C.; O'Meara, S.; Smith, R.; Parker, A.; et al. Lung cancer: intragenic ERBB2 kinase mutations in tumours. Nature 2004, 431, 525–526. [Google Scholar]
- Riely, G.J.; Kris, M.G.; Rosenbaum, D.; Marks, J.; Li, A.; Chitale, D.A.; Nafa, K.; Riedel, E.R.; Hsu, M.; Pao, W.; et al. Frequency and distinctive spectrum of KRAS mutations in never smokers with lung adenocarcinoma. Clin. Cancer Res. 2008, 14, 5731–5734. [Google Scholar] [CrossRef]
- Sun, Y.; Ren, Y.; Fang, Z.; Li, C.; Fang, R.; Gao, B.; Han, X.; Tian, W.; Pao, W.; Chen, H.; et al. Lung adenocarcinoma from East Asian never-smokers is a disease largely defined by targetable oncogenic mutant kinases. J. Clin. Oncol. 2010, 28, 4616–4620. [Google Scholar] [CrossRef]
- Weiss, J.; Sos, M.L.; Seidel, D.; Peifer, M.; Zander, T.; Heuckmann, J.M.; Ullrich, R.T.; Menon, R.; Maier, S.; Soltermann, A.; et al. Frequent and focal FGFR1 amplification associates with therapeutically tractable FGFR1 dependency in squamous cell lung cancer. Sci. Transl. Med. 2010, 2, 62ra93. [Google Scholar] [CrossRef]
- Dutt, A.; Ramos, A.H.; Hammerman, P.S.; Mermel, C.; Cho, J.; Sharifnia, T.; Chande, A.; Tanaka, K.E.; Stransky, N.; Greulich, H.; et al. Inhibitor-Sensitive FGFR1 Amplification in Human Non-Small Cell Lung Cancer. PLoS One 2011, 6, e20351. [Google Scholar] [Green Version]
- Hammerman, P.; Sos, M.; Ramos, A.; Xu, C. Mutations in the DDR2 Kinase Gene Identify a Novel Therapeutic Target in Squamous Cell Lung Cancer. Cancer Discovery 2011, 1, 78–79. [Google Scholar] [CrossRef]
- Ji, H.; Zhao, X.; Yuza, Y.; Shimamura, T.; Li, D.; Protopopov, A.; Jung, B.L.; McNamara, K.; Xia, H.; Glatt, K.A.; et al. Epidermal growth factor receptor variant III mutations in lung tumorigenesis and sensitivity to tyrosine kinase inhibitors. Proc. Natl. Acad. Sci. USA 2006, 103, 7817–7822. [Google Scholar]
- Yamamoto, H.; Shigematsu, H.; Nomura, M.; Lockwood, W.W.; Sato, M.; Okumura, N.; Soh, J.; Suzuki, M.; Wistuba, I.I.; Fong, K.M.; et al. PIK3CA mutations and copy number gains in human lung cancers. Cancer Res. 2008, 68, 6913–6921. [Google Scholar] [CrossRef]
- Romano, E.; Schwartz, G.K.; Chapman, P.B.; Wolchock, J.D.; Carvajal, R.D. Treatment implications of the emerging molecular classification system for melanoma. Lancet Oncol. 2011, 12, 913–922. [Google Scholar] [CrossRef]
- Curtin, J.A.; Busam, K.; Pinkel, D.; Bastian, B.C. Somatic activation of KIT in distinct subtypes of melanoma. J. Clin. Oncol. 2006, 24, 4340–4346. [Google Scholar] [CrossRef]
- Davies, H.; Bignell, G.R.; Cox, C.; Stephens, P.; Edkins, S.; Clegg, S.; Teague, J.; Woffendin, H.; Garnett, M.J.; Bottomley, W.; et al. Mutations of the BRAF gene in human cancer. Nature 2002, 417, 949–954. [Google Scholar]
- Flaherty, K.T.; Puzanov, I.; Kim, K.B.; Ribas, A.; McArthur, G.A.; Sosman, J.A.; O'Dwyer, P.J.; Lee, R.J.; Grippo, J.F.; Nolop, K.; et al. Inhibition of mutated, activated BRAF in metastatic melanoma. N. Engl. J. Med. 2010, 363, 809–819. [Google Scholar]
- Hatzivassiliou, G.; Song, K.; Yen, I.; Brandhuber, B.J.; Anderson, D.J.; Alvarado, R.; Ludlam, M.J.; Stokoe, D.; Gloor, S.L.; Vigers, G.; Morales, T.; Aliagas, I.; Liu, B.; Sideris, S.; Hoeflich, K.P.; Jaiswal, B.S.; Seshagiri, S.; Koeppen, H.; Belvin, M.; Friedman, L.S.; Malek, S. RAF inhibitors prime wild-type RAF to activate the MAPK pathway and enhance growth. Nature 2010, 464, 431–435. [Google Scholar]
- Poulikakos, P.I.; Zhang, C.; Bollag, G.; Shokat, K.M.; Rosen, N. RAF inhibitors transactivate RAF dimers and ERK signalling in cells with wild-type BRAF. Nature 2010, 464, 427–430. [Google Scholar]
- Heidorn, S.J.; Milagre, C.; Whittaker, S.; Nourry, A.; Niculescu-Duvas, I.; Dhomen, N.; Hussain, J.; Reis-Filho, J.S.; Springer, C.J.; Pritchard, C.; Marais, R. Kinase-dead BRAF and oncogenic RAS cooperate to drive tumor progression through CRAF. Cell 2010, 140(2), 209–221. [Google Scholar] [CrossRef]
- Oberholzer, P.; Kee, D.; Dziunycz, P.; Sucker, A.; Kamsukom, N.; Jones, R.; Roden, C.; Chalk, C.; Ardie, K.; Palescandolo, E.; et al. RAS Mutations Are Associated with the Development of CutaneousSquamous Cell Tumors in Patients treated with RAF inhibitors. J. Clin. Oncol. 2011, 30, 316–321. [Google Scholar]
- Su, F.; Viros, A.; Milagre, C.; Trunzer, K.; Bollag, G.; Spleiss, O.; Reis-Filho, J.; Kong, X.; Koya, R.; Flaherty, K.; et al. RAS mutations in cutaneous squamous cell carcinomas with BRAF inhibitors. N. Engl. J. Med. 2012, 366(3), 207–215. [Google Scholar] [CrossRef]
- Guo, J.; Si, L.; Kong, Y.; Flaherty, K.T.; Xu, X.; Zhu, Y.; Corless, C.L.; Li, L.; Li, H.; Sheng, X.; et al. Phase II, open-label, single-arm trial of imatinibmesylate in patients with metastatic melanoma harboring c-Kit mutation or amplification. J. Clin. Oncol. 2011, 29, 2904–2909. [Google Scholar]
- Curtin, J.A.; Fridlyand, J.; Kageshita, T.; Patel, H.N.; Busam, K.J.; Kutzner, H.; Cho, K.H.; Aiba, S.; Brocker, E.B.; LeBoit, P.E.; et al. Distinct sets of genetic alterations in melanoma. N. Engl. J. Med. 2005, 353, 2135–2147. [Google Scholar] [CrossRef]
- Van Raamsdonk, C.D.; Bezrookove, V.; Green, G.; Bauer, J.; Gaugler, L.; O'Brien, J.M.; Simpson, E.M.; Barsh, G.S.; Bastian, B.C. Frequent somatic mutations of GNAQ in uveal melanoma and blue naevi. Nature 2009, 457, 599–602. [Google Scholar]
- van Raamsdonk, C.D.; Fitch, K.R.; Fuchs, H.; de Angelis, M.H.; Barsh, G.S. Effects of G-protein mutations on skin color. Nat. Genet. 2004, 36, 961–968. [Google Scholar] [CrossRef]
- van Raamsdonk, C.D.; Griewank, K.G.; Crosby, M.B.; Garrido, M.C.; Vemula, S.; Wiesner, T.; Obenauf, A.C.; Wackernagel, W.; Green, G.; Bouvier, N.; et al. Mutations in GNA11 in uveal melanoma. N. Engl. J. Med. 2010, 363, 2191–2199. [Google Scholar]
- Kobayashi, S.; Boggon, T.J.; Dayaram, T.; Janne, P.A.; Kocher, O.; Meyerson, M.; Johnson, B.E.; Eck, M.J.; Tenen, D.G.; Halmos, B. EGFR mutation and resistance of non-small-cell lung cancer to gefitinib. N. Engl. J. Med. 2005, 352, 786–792. [Google Scholar] [CrossRef]
- Pao, W.; Miller, V.A.; Politi, K.A.; Riely, G.J.; Somwar, R.; Zakowski, M.F.; Kris, M.G.; Varmus, H. Acquired resistance of lung adenocarcinomas to gefitinib or erlotinib is associated with a second mutation in the EGFR kinase domain. PLoSMed. 2005, 2, e73. [Google Scholar]
- Yun, C.H.; Mengwasser, K.E.; Toms, A.V.; Woo, M.S.; Greulich, H.; Wong, K.K.; Meyerson, M.; Eck, M.J. The T790M mutation in EGFR kinase causes drug resistance by increasing the affinity for ATP. Proc. Natl. Acad. Sci. USA 2008, 105, 2070–2075. [Google Scholar]
- Bean, J.; Brennan, C.; Shih, J.Y.; Riely, G.; Viale, A.; Wang, L.; Chitale, D.; Motoi, N.; Szoke, J.; Broderick, S.; et al. MET amplification occurs with or without T790M mutations in EGFR mutant lung tumors with acquired resistance to gefitinib or erlotinib. Proc. Natl. Acad. Sci. USA 2007, 104, 20932–20937. [Google Scholar]
- Engelman, J.A.; Zejnullahu, K.; Mitsudomi, T.; Song, Y.; Hyland, C.; Park, J.O.; Lindeman, N.; Gale, C.M.; Zhao, X.; Christensen, J.; et al. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science 2007, 316, 1039–1043. [Google Scholar] [CrossRef]
- Turke, A.B.; Zejnullahu, K.; Wu, Y.L.; Song, Y.; Dias-Santagata, D.; Lifshits, E.; Toschi, L.; Rogers, A.; Mok, T.; Sequist, L.; et al. Preexistence and clonal selection of MET amplification in EGFR mutant NSCLC. Cancer Cell 2010, 17, 77–88. [Google Scholar] [CrossRef]
- Arcila, M.E.; Oxnard, G.R.; Nafa, K.; Riely, G.J.; Solomon, S.B.; Zakowski, M.F.; Kris, M.G.; Pao, W.; Miller, V.A.; Ladanyi, M. Rebiopsy of lung cancer patients with acquired resistance to EGFR inhibitors and enhanced detection of the T790M mutation using a locked nucleic acid-based assay. Clin. Cancer Res. 2011, 17, 1169–1180. [Google Scholar]
- Sequist, L.V.; Waltman, B.A.; Dias-Santagata, D.; Digumarthy, S.; Turke, A.B.; Fidias, P.; Bergethon, K.; Shaw, A.T.; Gettinger, S.; Cosper, A.K.; et al. Genotypic and histological evolution of lung cancers acquiring resistance to EGFR inhibitors. Sci. Transl. Med. 2011, 3, 75ra26. [Google Scholar]
- Choi, Y.L.; Soda, M.; Yamashita, Y.; Ueno, T.; Takashima, J.; Nakajima, T.; Yatabe, Y.; Takeuchi, K.; Hamada, T.; Haruta, H.; et al. EML4-ALK mutations in lung cancer that confer resistance to ALK inhibitors. N. Engl. J. Med. 2010, 363, 1734–1739. [Google Scholar] [CrossRef]
- Sasaki, T.; Koivunen, J.; Ogino, A.; Yanagita, M.; Nikiforow, S.; Zheng, W.; Lathan, C.; Marcoux, J.P.; Du, J.; Okuda, K.; et al. A novel ALK secondary mutation and EGFR signaling cause resistance to ALK kinase inhibitors. Cancer Res. 2011, 71, 6051–6060. [Google Scholar]
- Katayama, R.; Khan, T.M.; Benes, C.; Lifshits, E.; Ebi, H.; Rivera, V.M.; Shakespeare, W.C.; Iafrate, A.J.; Engelman, J.A.; Shaw, A.T. Therapeutic strategies to overcome crizotinib resistance in non-small cell lung cancers harboring the fusion oncogene EML4-ALK. Proc. Natl. Acad. Sci. USA 2011, 108, 7535–7540. [Google Scholar]
- Nazarian, R.; Shi, H.; Wang, Q.; Kong, X.; Koya, R.C.; Lee, H.; Chen, Z.; Lee, M.K.; Attar, N.; Sazegar, H.; et al. Melanomas acquire resistance to B-RAF(V600E) inhibition by RTK or N-RAS upregulation. Nature 2010, 468, 973–977. [Google Scholar]
- Johannessen, C.M.; Boehm, J.S.; Kim, S.Y.; Thomas, S.R.; Wardwell, L.; Johnson, L.A.; Emery, C.M.; Stransky, N.; Cogdill, A.P.; Barretina, J.; et al. COT drives resistance to RAF inhibition through MAP kinase pathway reactivation. Nature 2010, 468, 968–972. [Google Scholar] [CrossRef]
- Horn, L.; Pao, W. EML4-ALK: honing in on a new target in non-small-cell lung cancer. J. Clin. Oncol. 2009, 27, 4232–4235. [Google Scholar] [CrossRef]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Kelleher, F.C.; Solomon, B.; McArthur, G.A. Molecular Therapeutic Advances in Personalized Therapy of Melanoma and Non-Small Cell Lung Cancer. J. Pers. Med. 2012, 2, 35-49. https://doi.org/10.3390/jpm2020035
Kelleher FC, Solomon B, McArthur GA. Molecular Therapeutic Advances in Personalized Therapy of Melanoma and Non-Small Cell Lung Cancer. Journal of Personalized Medicine. 2012; 2(2):35-49. https://doi.org/10.3390/jpm2020035
Chicago/Turabian StyleKelleher, Fergal C., Benjamin Solomon, and Grant A. McArthur. 2012. "Molecular Therapeutic Advances in Personalized Therapy of Melanoma and Non-Small Cell Lung Cancer" Journal of Personalized Medicine 2, no. 2: 35-49. https://doi.org/10.3390/jpm2020035
APA StyleKelleher, F. C., Solomon, B., & McArthur, G. A. (2012). Molecular Therapeutic Advances in Personalized Therapy of Melanoma and Non-Small Cell Lung Cancer. Journal of Personalized Medicine, 2(2), 35-49. https://doi.org/10.3390/jpm2020035