
Thirty-Three Years Follow-Up of a Greek Family with Abetalipoproteinemia: Absence of Liver Damage on Long-Term Medium Chain Triglycerides Supplementation

 , , ,
, , , 
Abstract
1. Background
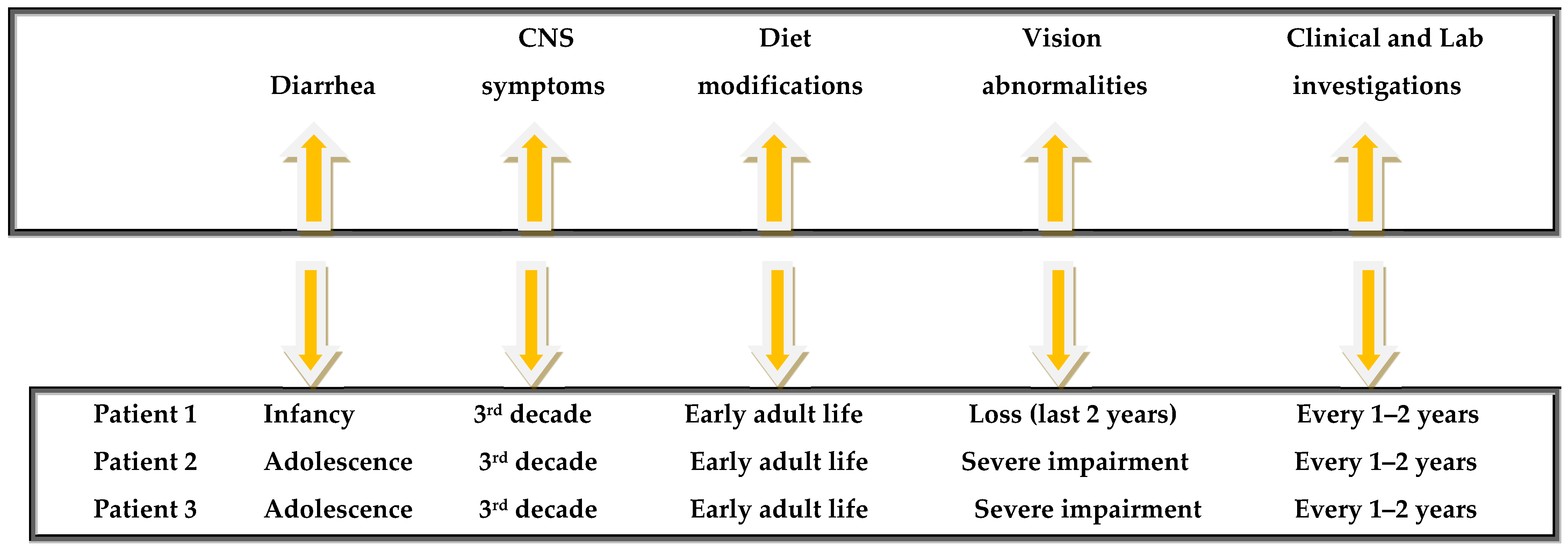
2. Report of Cases
2.1. Long-Term Laboratory Follow-Up
2.1.1. Lipidemic Profile
2.1.2. Liver Function Tests
2.1.3. Serum Vitamin Levels
2.1.4. Linkage Analysis
2.1.5. Mutation Analysis
2.1.6. Cytogenetic Analysis
2.1.7. Fibrinogen Marker
2.1.8. HLA Pattern
2.1.9. Anthropometric Parameters
2.1.10. Current Clinical Situation
3. Discussion
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Takahashi, M.; Okazaki, H.; Ohashi, K.; Ogura, M.; Ishibashi, S.; Okazaki, S.; Hirayama, S.; Hori, M.; Matsuki, K.; Yokoyama, S.; et al. Current Diagnosis and Management of Abetalipoproteinemia. J. Atheroscler. Thromb. 2021, 28, 1009–1019. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Junaid, S.Z.S.; Patel, K. Abetalipoproteinemia. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2025. [Google Scholar] [PubMed]
- Tack, P.; Bourgeois, P.; Devos, E.; Demeester, J. Abetalipoproteinemia or Bassen-Kornz-weig syndrome. Clinical, biochemical and electrophysiological features of two cases. Acta Neurol. Belg. 1988, 88, 229–238. [Google Scholar] [PubMed]
- Zamel, R.; Kand, R.; Pollex, R.L.; Hegele, R.A. Abetalipoproteinemia: Two cases and literature review. Orphanet J. Rare Dis. 2008, 3, 19. [Google Scholar] [CrossRef]
- Nagappa, M.; Bindu, P.S.; Adwani, S.; Seshagiri, S.K.; Saini, J.; Sinha, S.; Taly, A.B. Clinical, hematological, and imaging observations in a 25-year-old woman with abetalipoproteinemia. Ann. Indian Acad. Neurol. 2014, 17, 113–116. [Google Scholar] [CrossRef]
- Paquette, M.; Dufour, R.; Hegele, R.A.; Baass, A. A tale of 2 cousins: An atypical and a typical case of abetalipoproteinemia. J. Clin. Lipidol. 2016, 10, 1030–1034. [Google Scholar] [CrossRef] [PubMed]
- Khatun, I.; Walsh, M.T.; Hussain, M.M. Loss of both phospholipid and triglyceride transfer activities of microsomal triglyceride transfer protein in abetalipoproteinemia. J. Lipid Res. 2013, 54, 1541–1549. [Google Scholar] [CrossRef] [PubMed]
- Walsh, M.T.; Iqbal, J.; Josekutty, J.; Soh, J.; Di Leo, E.; Özaydin, E.; Gündüz, M.; Tarugi, P.; Hussain, M.M. Novel abetalipoproteinemia missense mutation highlights the importance of the n-terminal β-barrel in microsomal triglyceride transfer protein function. Circ. Cardiovasc. Genet. 2015, 8, 677–687. [Google Scholar] [CrossRef] [PubMed]
- Ohashi, K.; Ishibashi, S.; Osuga, J.; Tozawa, R.; Harada, K.; Yahagi, N.; Shionoiri, F.; Iizuka, Y.; Tamura, Y.; Nagai, R.; et al. Novel mutations in the microsomal triglyceride transfer protein gene causing abetalipoproteinemia. J. Lipid Res. 2000, 41, 1199–1204. [Google Scholar] [CrossRef]
- Iqbal, J.; Jahangir, Z.; Al-Qarni, A.A. Microsomal Triglyceride Transfer Protein: From lipid metabolism to metabolic diseases. Adv. Exp. Med. Biol. 2020, 1276, 37–52. [Google Scholar]
- Bredefeld, C.; Peretti, N.; Hussain, M.M. Medical Advisory Panel. New Classification and Management of Abetalipoproteinemia and Related Disorders. Gastroenterology 2021, 160, 1912–1916. [Google Scholar] [CrossRef] [PubMed]
- Welty, F. Hypobetalipoproteinemia and abetalipoproteinemia: Liver disease and cardiovascular disease. Curr. Opin. Lipidol. 2020, 31, 49–55. [Google Scholar] [CrossRef]
- Chardon, L.; Sassolas, A.; Dingeon, B.; Michel-Calemard, L.; Bovier-Lapierre, M.; Moulin, P.; Lachaux, A. Identification of two novel mutations and long-term follow-up in abetalipoproteinemia: A report of four cases. Eur. J. Pediatr. 2009, 168, 983–989. [Google Scholar] [CrossRef] [PubMed]
- Desomer, L.; De Vos, M.; De Looze, D. Fat accumulation in enterocytes: A key to the diagnosis of abetalipoproteinemia or homozygous hypobetalipoproteinemia. Endoscopy 2015, 47 (Suppl. S1), UCTN:E223-4. [Google Scholar] [CrossRef]
- Pai, G.; Sarma, M.S.; Pandey, R. White-Out duodenal mucosa: Clue to a systemic diagnosis. Gastroenterology 2020, 159, e1–e2. [Google Scholar] [CrossRef] [PubMed]
- Bijon, J.; Hussain, M.M.; Bredefeld, C.L.; Boesze-Battaglia, K.; Freund, K.B.; Curcio, C.A. Abetalipoproteinemia with angioid streaks, choroidal neovascularization, atrophy, and extracellular deposits revealed by multimodal retinal imaging. Ophthalmic. Genet. 2024, 45, 583–590. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Cuerq, C.; Henin, E.; Restier, L.; Blond, E.; Drai, J.; Marçais, C.; Di Filippo, M.; Laveille, C.; Michalski, M.-C.; Poinsot, P.; et al. Efficacy of two vitamin E formulations in patients with abetalipoproteinemia and chylomicron retention disease. J. Lipid Res. 2018, 59, 1640–1648. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Le, R.; Zhao, L.; Hegele, R.A. Forty year follow-up of three patients with complete absence of apolipoprotein B-containing lipoproteins. J. Clin. Lipidol. 2022, 16, 155–159. [Google Scholar] [CrossRef] [PubMed]
- Rader, D.J.; Brewer, B.H. Abetalipoproteinemia: New insights into lipoprotein assembly and vitamin E metabolism from a rare genetic disease. JAMA 1993, 270, 865–869. [Google Scholar] [CrossRef]
- Gordon, D.A.; Wetterau, J.R.; Gregg, R.E. Microsomal triglyceride transfer protein: A protein complex required for the assembly of lipoprotein particles. Trends Cell Biol. 1995, 5, 317–321. [Google Scholar] [CrossRef] [PubMed]
- Narcisi, T.M.; Shoulders, C.C.; Chester, S.A.; Read, J.; Brett, D.J.; Harrison, G.B.; Grantham, T.T.; Fox, M.F.; Povey, S.; de Bruin, T.W. Mutations of the microsomal triglyceride-transfer-protein gene in abetalipoproteinemia. Am. J. Hum. Genet. 1995, 57, 1298–1310. [Google Scholar]
- Yang, X.P.; Inazu, A.; Yagi, K.; Kajinami, K.; Koizumi, J.; Mabuchi, H. Abetalipoproteinemia caused by maternal isodisomy of chromosome 4q containing an intron 9 splice acceptor mutation in the microsomal triglyceride transfer protein gene. Arter. Thromb. Vasc. Biol. 1999, 19, 1950–1955. [Google Scholar] [CrossRef]
- Wang, J.; Hegele, R.A. Microsomal triglyceride transfer protein (MTP) gene mutations in Canadian subjects with abetalipoproteinemia. Hum. Mutat. 2000, 15, 294–295. [Google Scholar] [CrossRef]
- Joshi, M.; Hyams, J.; Treem, W.; Ricci, A., Jr. Cytoplasmic vacuolization of enterocytes: An unusual histopathologic finding in juvenile nutritional megaloblastic anemia. Mod. Pathol. 1991, 4, 62–65. [Google Scholar]
- Lausch, E.; Yoshimi, A. Acanthocytosis: A key feature for the diagnosis of abetalipoproteinemia. Blood 2023, 141, 3231. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.O.; Basamh, O.; Alkatan, H.M.; AlBasamh, O. Ophthalmic diagnosis and optical coherence tomography of abetalipoproteinemia, a treatable form of pediatric retinal dystrophy. J. AAPOS 2019, 23, 176–177. [Google Scholar] [CrossRef] [PubMed]
- Burnett, J.R.; Hooper, A.J. Vitamin E and oxidative stress in abetalipoproteinemia and familial hypobetalipoproteinemia. Free. Radic. Biol. Med. 2015, 88 Pt A, 59–62. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, F.; Patel, V.; Matts, S. A successful spontaneous pregnancy in abetalipoproteinemia: Amsterdam or the art of vitamin replacement? BMJ Case Rep. 2014, 2014, bcr2014206754. [Google Scholar] [CrossRef]
- Avigan, M.I.; Ishak, K.G.; Gregg, R.E.; Hoofnagle, J.H. Morphologic features of the liver in abetalipoproteinemia. Hepatology 1984, 4, 1223–1226. [Google Scholar] [CrossRef]
- Partin, J.S.; Partin, J.C.; Schubert, W.K.; McAdams, A.J. Liver ultrastructure in abetalipoproteinemia: Evolution of micronodular cirrhosis. Gastroenterology 1974, 67, 107–109. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Lipidemic Profile/ Year | Patient 1 | Patient 2 | Patient 3 | |||
|---|---|---|---|---|---|---|
| 1990 | 2023 | 1990 | 2023 | 1990 | 2023 | |
| Cholesterol (<200 mg/dL) | 46 | 57 | 65 | 40 | 43 | 39 |
| Triglycerides (<150 mg/dL) | 0 | 0 | 0 | 0 | 0 | 0 |
| High Density Lipoproteins (>60 mg/dL) | 43 | 58 | 61 | 40 | 41 | 46 |
| Low Density Lipoproteins (<100 mg/dL) | 0 | 0 | 0 | 0 | 0 | 0 |
| Apo A-1 Lipoprotein (119–228 mg/dL) | 63 | 70.2 | 76 | 55.3 | 56 | 48 |
| Apo B Lipoprotein (51–165 mg/dL) | 20 | 1.2 | 20 | 2.2 | 20 | 3.1 |
| Lp(a) Lipoprotein (<75 nmol/L) | 1.3 | 1.7 | 0.8 | 1.8 | 1.6 | 1.9 |
| Anthropometric Parameter/Year | Patient 1 1990 | Patient 1 2023 | Patient 2 1990 | Patient 2 2023 | Patient 3 1990 | Patient 3 2023 |
|---|---|---|---|---|---|---|
| Body weight (Kg) | 33.1 | 35.3 | 47.3 | 48.1 | 46.7 | 47.2 |
| Height (cm) | 154 | 153 | 170 | 168 | 165 | 165 |
| Body mass index (BMI) | 13.4 | 15.2 | 15.5 | 16.7 | 15.8 | 16.9 |
| Body fat content (%) | 21.0 | 30.0 | 14.0 | 16.0 | 13.0 | 13.0 |
| Mid arm circumference (cm) | 19.5 | 20 | 21 | 21 | 22.5 | 22.5 |
| Skin fold thickness (SFT) in back of upper arm (triceps) (mm) | 6 | 7 | 4 | 5 | 3 | 3.5 |
| SFT in front of upper arm (biceps) (mm) | 4 | 5 | 2 | 2.5 | 2 | 3 |
| SFT in subscapular (mm) | 5 | 5 | 8 | 8 | 5 | 5.5 |
| SFT in suprailiac (waist) (mm) | 6 | 6 | 3 | 4 | 3 | 3 |
| Total SFT (mm) | 21 | 23 (10.9%) | 17 | 19.5 (14.7%) | 13 | 15 (15.4%) |
| Clinical Parameter | Patient 1 | Patient 2 | Patient 3 |
|---|---|---|---|
| Bowel movements/ abdominal symptoms | Three per day normal or semi formed, absence of abdominal symptoms. | One to three normal abdominal pain, increased bowel sounds. | One to two per day normal, absence of abdominal symptoms. |
| Neurological manifestations | Clinical deterioration, ataxia. | Paraplegia, gait with mechanical help. | Clinical deterioration. |
| Nutritional support | MCT Oil Module 500 mL; various dietetic supplementations, including “Abound 24g”: (powder containing mainly aminoacids such as arginine and glutamine), “Fortimel extra” (containing carbohydrates, aminoacids, and 19% fat), and “Fresubin”. These products were received continuously throughout the follow-up period. | MCT Oil Module 500 mL; various dietetic supplementations including “Abound 24g”: (powder containing mainly aminoacids such as arginine and glutamine), “Fortimel extra” (containing carbohydrates, aminoacids, and 19% fat), and “Fresubin”. These products were received continuously throughout the follow-up period. | MCT Oil Module 500 mL various dietetic supplementations including “Abound 24g”: (powder containing mainly aminoacids such as arginine and glutamine), “Fortimel extra” (containing carbohydrates, aminoacids and 19% fat), and “Fresubin”. These products were received continuously throughout the follow-up period. |
| Vision status | Vision impairment drowsiness, inability to read. | Significant vision impairment. | Almost complete right blindness with only light perception; macular degeneration. |
| Pharmaceutical treatment | Ferrum per os, Vit D. | Metoprolol, Apixaban, Eplerenone, Furosemide, Levothyroxin. | Depakin (valproic acid). |
| Vitamins | Patient 1 | Patient 2 | Patient 3 |
|---|---|---|---|
| A | 100 IU/KgBW/d corresponding to 5000 IU/d (**) | 100 IU/KgBW/d corresponding to 5000 IU/d | 100 IU/KgBW/d corresponding to 5000 IU/d 100 |
| D | 800–1200 IU/d (1 tab 1000 IU/d) | 800–1200 IU/d (1 tab 1000 IU/d) | 800–1200 IU/d (1 tab 1000 IU/d) |
| E | 100 IU/KgBW/d (at least 5000 IU/d) | 100 IU/KgBW/d (at least 5000 IU/d) | 100 IU/KgBW/d (at least 5000 IU/d) |
| K | 5 mg 2 times per week | 5 mg once a week | 5 mg once a week |
| Iron, folate, Vit B12 | Iron and vit b12 and folic acid were supplemented only in this patient, as they had the most severe symptoms. | In the large majority of examinations, the levels were normal. | In the large majority of examinations, the levels were normal. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Triantafillidis, J.K.; Manioti, A.; Pittaras, T.; Kozonis, T.; Kritsotakis, E.; Malgarinos, G.; Pantos, K.; Sfakianoudis, K.; Konstadoulakis, M.M.; Papalois, A.E. Thirty-Three Years Follow-Up of a Greek Family with Abetalipoproteinemia: Absence of Liver Damage on Long-Term Medium Chain Triglycerides Supplementation. J. Pers. Med. 2025, 15, 354. https://doi.org/10.3390/jpm15080354
Triantafillidis JK, Manioti A, Pittaras T, Kozonis T, Kritsotakis E, Malgarinos G, Pantos K, Sfakianoudis K, Konstadoulakis MM, Papalois AE. Thirty-Three Years Follow-Up of a Greek Family with Abetalipoproteinemia: Absence of Liver Damage on Long-Term Medium Chain Triglycerides Supplementation. Journal of Personalized Medicine. 2025; 15(8):354. https://doi.org/10.3390/jpm15080354
Chicago/Turabian StyleTriantafillidis, John K., Areti Manioti, Theodoros Pittaras, Theodoros Kozonis, Emmanouil Kritsotakis, Georgios Malgarinos, Konstantinos Pantos, Konstantinos Sfakianoudis, Manousos M. Konstadoulakis, and Apostolos E. Papalois. 2025. "Thirty-Three Years Follow-Up of a Greek Family with Abetalipoproteinemia: Absence of Liver Damage on Long-Term Medium Chain Triglycerides Supplementation" Journal of Personalized Medicine 15, no. 8: 354. https://doi.org/10.3390/jpm15080354
APA StyleTriantafillidis, J. K., Manioti, A., Pittaras, T., Kozonis, T., Kritsotakis, E., Malgarinos, G., Pantos, K., Sfakianoudis, K., Konstadoulakis, M. M., & Papalois, A. E. (2025). Thirty-Three Years Follow-Up of a Greek Family with Abetalipoproteinemia: Absence of Liver Damage on Long-Term Medium Chain Triglycerides Supplementation. Journal of Personalized Medicine, 15(8), 354. https://doi.org/10.3390/jpm15080354